

Abstract
The estrogen receptor (ER) is a major prognostic and therapeutic marker that is expressed in nearly 75% of breast tumors. We have previously shown that the presence of inflammatory mediators can alter the genomic function of the ER in a gene-specific manner. In particular, 17β-estradiol (E2) works in combination with the pro-inflammatory cytokines to enhance the expression of a number of pro-survival factors, including the inhibitor of apoptosis (IAP) family member, cIAP2. Here, we confirm that mRNA and protein levels for cIAP2, but not the related family members cIAP1 and XIAP, are highly up-regulated in MCF-7 breast cancer cells by E2 and cytokines. Similar regulation of cIAP2 is evident in other ER-positive but not ER-negative cell lines. In agreement with its role as a pro-survival factor, cIAP2 is highly expressed in a subset of invasive breast carcinomas but not in normal breast tissue or ductal carcinoma in situ. Antagonizing IAPs with mimetics of Smac, which is a known endogenous IAP antagonist, or knockdown of IAPs by siRNA led to greater cell death by TNFα and prevented E2 from promoting cell survival. In addition, a Smac mimetic reversed TNFα resistance in ER-positive breast cancer cells that express high levels of endogenous IAPs. In summary, our findings indicate a new mechanism by which E2 allows breast cancer cells to evade cell death and suggest that an antagonist of IAPs may be a potential therapeutic option for a subset of ER-positive breast tumors.
Electronic supplementary material
The online version of this article (doi:10.1007/s12672-010-0018-6) contains supplementary material, which is available to authorized users.
Keywords: Estrogen, IAPs, Cytokine, Breast cancer, Cell survival
Introduction
Estrogen drives the growth of estrogen receptor (ER)-positive breast tumors through its ability to stimulate both proliferation and survival of breast cancer cells. The effects of estrogens, such as 17β-estradiol (E2), are mediated by the estrogen receptors, ERα and ERβ. The ERs are ligand-dependent transcription factors that mediate expression of target genes through direct DNA binding at cognate response elements or tethering to other transcription factors [1]. In breast cancer, the gene networks and transcriptional mechanisms for ERα-dependent cell proliferation have been well described [2, 3], however, the pro-survival mechanisms of ERα in the breast are less clear. While ERα is known to regulate the expression of genes that are involved in the maintenance of mitochondrial membrane integrity, such as Bcl-2 and Bcl-xL [4, 5], gene expression profiling has revealed a wider range of E2-dependent cell survival targets, including up-regulated anti-apoptotic factors and down-regulated pro-apoptotic factors [6]. However, the role of many of these ER targets in E2-mediated breast cancer cell survival has yet to be confirmed by functional assays.
In a recent gene-profiling study, we found that activation of the transcription factor NFκB, which is also a strong pro-survival mediator in the breast [7], cooperates with E2 in an ER-dependent manner to synergistically up-regulate anti-apoptotic genes and promote cell survival [8]. One of the most highly up-regulated ER and NFκB target genes identified in the profiling study was the inhibitor of apoptosis (IAP) family member, cIAP2 (also known as BIRC3), which can protect a variety of cancer cell types from apoptotic stimuli [9]. However, the function of cIAP2 has not been well studied in breast cancer cells.
There are eight IAP family members, which are distinguished by the presence of baculoviral IAP repeat (BIR) domains. cIAP1 and cIAP2 are highly homologous [10] and are similar in structure to XIAP [11]. These members have three BIR domains that are involved in caspase binding and a C-terminal RING finger with ubiquitin ligase activity. XIAP is the most potent inhibitor of caspase activity, whereas cIAP1 and cIAP2 are recognized more for their important roles in ubiquitin-mediated signaling and participation in the activation of NFκB by TNFα [12–16]. An endogenous cellular inhibitor of IAPs, called Smac for second mitochondria derived activator of caspases, is released into the cytoplasm after mitochondrial membrane permeabilization. Smac promotes apoptosis by interacting with the BIR domain of IAPs, thereby preventing the IAP-caspase interaction and allowing apoptosis to proceed [17]. Various small molecule Smac mimetics (SM) that can mimic the Smac-IAP interaction have been developed and shown to promote apoptosis by antagonizing IAP activity. More specifically, SMs can prevent XIAP from interacting with caspases, rapidly induce degradation of cIAP1 and cIAP2, and modulate autocrine TNFα production and activity [18–21]. SMs have also been shown to sensitize cancer cells to cytotoxic drugs and Her2/epidermal growth factor receptor (EGFR) inhibitors [22, 23]. Thus, by promoting apoptosis of cancer cells directly or by acting synergistically with other known therapeutic agents, SMs represent a promising new class of anti-cancer drugs, several of which are currently in clinical trials [24].
Here, we demonstrate that IAPs are key mediators of E2-dependent breast cancer cell survival. We show that members of the IAP family have distinct expression patterns in breast cancer cells co-treated with E2 and the proinflammatory cytokine, TNFα. In particular, cIAP2 is highly up-regulated, whereas cIAP1 is down-regulated, and XIAP is not affected. E2 can no longer protect cells against TNFα- and TNF-related apoptosis inducing ligand (TRAIL)-induced death in the presence of SMs or IAP knockdown, indicating that IAPs are essential for the ability of E2 to promote breast cancer cell survival. In agreement with its role as a pro-survival factor, we find cIAP2 to be expressed in a subset of invasive breast carcinomas but not in normal breast tissue or ductal carcinoma in situ (DCIS). Taken together, these results indicate that IAPs may represent an important new therapeutic target in ER positive breast tumors.
Materials and Methods
Cell Culture and Reagents
The ER-positive breast cancer cell lines, MCF-7, and T47D, were cultured as previously described [8]. BT-474 cells, which are both ER and Her2 positive, were obtained from Dr. Debra Tonetti (University of Illinois at Chicago, Chicago, IL, USA) and cultured in RPMI-1640 supplemented with 10 μg/ml of insulin and 10% fetal bovine serum. Prior to treatments, all cell lines were cultured in phenol red-free media supplemented with 5% charcoal-dextran stripped serum for 3 days. E2 was from Sigma, ICI 182,780 from Tocris, and TNFα from R&D Systems. The Smac mimetics were generous gifts; compound 3 was provided by Dr. Xiaodong Wang (University of Texas Southwestern Medical Center, Dallas, TX, USA) and MV1 and BV6 by Dr. Domagoj Vucic (Genentech Inc., South San Francisco, CA, USA).
RNA Isolation and QPCR
RNA isolation and quantitative polymerase chain reaction (QPCR) were carried out as previously described [8]. Fold change was determined by the ΔΔCt method using 36B4 as the internal control. QPCR primer sequences are available upon request.
Western Blots
Whole cell extracts were prepared by direct lysis of cells in boiling 2× electrophoresis buffer (100 mM Tris buffer, 3.3% SDS, 7% β-mercaptoethanol, 15% glycerol, 0.02% bromophenol blue). Cell lysate was sonicated (Sonic Dismembrator, Fisher Scientific) three times at level 2 for 5 s, with the sample in ice/water and 15 s cooling periods between bursts. Parallel plates were processed using M-Per Reagent (Thermo Scientific) so that the protein concentration could be measured by the BCA method (Thermo Scientific) according to the manufacturer’s instructions. Cell lysates were separated by SDS-PAGE on 10% Tris-HCl pre-cast gels (Bio-Rad) and transferred onto nitrocellulose membranes (Thermo Scientific). Membranes were blocked for 1 h with 5% non-fat dry milk (Lab Scientific) in TBS-Tween 20 (0.05%, Fisher Scientific) and probed with primary antibody overnight at 4°C. Secondary antibodies were conjugated to horseradish peroxidase (Pierce Biotechnology) and signal was developed using SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific). Images were captured by the Molecular Imager Chemidoc XRS (Bio-Rad) and densitometry was performed using Quantity One Software (Bio-Rad). The following primary antibodies were used: cIAP1 (#AF8181, R&D Systems), cIAP2 (#552782, BD Pharmingen,), XIAP (#610717, BD Biosciences), and Bcl-2 (#MS-123-P0, Thermo Scientific). β-actin (#A5441, Sigma) was used as the loading control.
Tissue Microarray Construction
cIAP2 protein expression in breast cancer patients was assessed using a tissue microarray (TMA) that consisted of 43 normal breast, 38 DCIS, and 63 invasive breast carcinoma specimens. Normal samples were from cases of benign breast disease. The TMA contained two tissue cores of 2 mm in size from each patient’s formalin-fixed and paraffin-embedded tissue. An experienced surgical pathologist (E.W.) evaluated H&E-stained slides for all specimens prior to construction of the TMA in order to identify representative normal and tumor areas.
Immunohistochemistry
The TMA was used to assess cIAP2 expression by immunohistochemistry (IHC). Slides containing formalin-fixed, paraffin-embedded cores were deparaffinized and hydrated in graduated alcohol concentrations. Antigen retrieval was performed by submerging the slides in a low pH Antigen Unmasking Solution (Vector Laboratories) and incubating for 5 min in a Decloaking Chamber electric pressure cooker at 20 psi and 120°C (Biocare Medical). Endogenous peroxidase was quenched in 0.3% H2O2 in methanol for 30 min and tissue was blocked in 5% goat serum. Slides were probed with primary cIAP2 antibody (#ab32059, Abcam) overnight at 4°C and subsequently probed with secondary antibody for 30 min at room temperature. Slides were then incubated with avidin-biotin peroxidase complex (Vector Elite ABC Kit, Vector Laboratories) for 30 min. Color was developed by immersing slides in 3,3’-diaminobenzidine and counterstained with hematoxylin. As a negative control, the primary antibody was omitted. TMA staining was independently reviewed and scored by two pathologists. cIAP2 positivity was defined as clumped or distinct granular staining, whereas a diffuse light brown color or no brown color was considered negative.
Cell Viability
Cell viability was assessed by two methods. The MTS assay (CellTiter 96 AQueous One Solution Cell Proliferation Assay, Promega) was carried out according to the manufacturer’s instructions. For siRNA studies, cell viability was determined by methylene blue staining [25]. Briefly, each well was rinsed once with phosphate-buffered saline (PBS) and methylene blue staining solution (Hanks’ Balanced Salt Solution + 1.25% glutaraldehyde + 0.6% methylene blue) was added to each well. Following 1-h incubation at 37 °C, methylene blue staining solution was removed, and the plates were gently rinsed three times in ddH2O. Elution solution (50% ethanol + 49% PBS + 1% acetic acid) was added to each well and subsequently incubated for 20 min at room temperature with gentle agitation. Absorbance was read on a microplate reader (Bio-Tek Synergy HT) at a wavelength of 562 nm and viability was calculated as a percentage of control cells.
Gene Silencing with Small Interfering RNAs
siRNA oligonucleotides were purchased from Ambion/Applied Biosystems (Austin, TX, USA). The following validated siRNAs were used: cIAP1 (#4390824, s1448 Ambion), XIAP (#4390824, s1455 Ambion) and a Silencer Select Negative Control #1 (siNeg, #4390843, Ambion). The cIAP2 siRNA sequence was purchased through Ambion as a Custom Select siRNA based on a validated sequence [26]. All siRNAs were reconstituted according to the manufacturer’s instructions. When MCF-7 cells reached ~30-40% confluency, they were transfected with 10 nM siRNA for cIAP2 and XIAP, 50 nM siRNA for cIAP1, or 70 nM siNeg, using DharmaFECT1 (Thermo Scientific), based on the manufacturer’s instructions. Briefly, siRNA and DharmaFECT were incubated separately with serum-free, antibiotic-free media, with each mix containing a total volume of 100 μL per well of a six-well plate. Following a 5-min incubation, siRNA and DharmaFECT mixes were combined and incubated together for 20 min. At this time, cells were rinsed twice with PBS. An antibiotic-free, complete media containing 5% charcoal-dextran stripped serum was added to each well followed by addition of the siRNA/DharmaFECT mixture. Following 24 h of transfection, wells were rinsed once with PBS and complete, antibiotic-free media was added to each well. After 48 h of transfection, cells were treated with hormone and cytokine for an additional 24 h.
Statistics
Data shown are reported as the mean ± SEM from at least three replicates. Cell viability and QPCR data were analyzed by one-way or two-way ANOVA followed by the post-hoc Tukey or Bonferroni tests, as appropriate. For the TMA, Fisher’s exact test was used to identify significant correlations between cIAP2 expression and clinical parameters.
Results
The Combination of E2 and TNFα Differentially Regulates Expression of IAP mRNA and Protein Levels in Breast Cancer Cells
In a previous study, we found that the IAP family of anti-apoptotic proteins may play a role in mediating E2-dependent breast cancer cell survival in response to TNFα [8]. Furthermore, we found that one member of the IAP family, cIAP2, was up-regulated by the combination of E2 + TNFα in an ER- and NFκB-dependent manner. In this current study, we sought to determine if other members of the IAP family, notably cIAP1 and XIAP, are also expressed and regulated and what role these family members may play in E2-stimulated cell survival. We found that E2, in the absence of TNFα, has little to no effect on the expression of any of the three IAP family members, whereas cIAP1 and cIAP2 mRNA levels are both up-regulated by TNFα (Fig. 1a), as previously described [27]. As our gene expression profiling studies suggested [8], cIAP2 mRNA is highly up-regulated by co-treatment with E2 + TNFα, and here we show that this enhanced expression is sustained throughout a 24-h treatment period (Fig. 1a). We also found a similar pattern of cIAP2 up-regulation in response to E2 + TNFα in two other ER-positive cell lines, BT-474 and T47D, but not in an ER-negative cell line, MDA-MB-231, indicating that this may be a common regulatory mechanism in multiple hormone-dependent breast cancer models (Fig. 1b). In contrast to cIAP2, the combination of E2 + TNFα had little effect on cIAP1 when compared to TNFα alone and none of the treatments had any significant effect on XIAP mRNA levels (Fig. 1a).
Fig. 1.
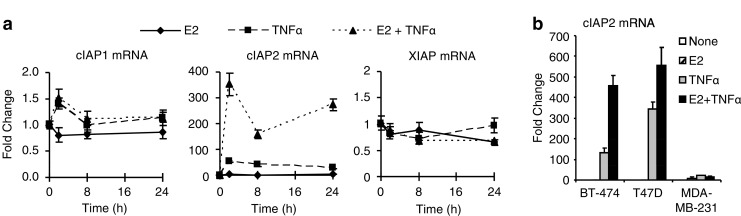
Regulation of IAP mRNA by E2 and TNFα in MCF-7 Cells. a Time course analysis of IAP mRNA expression following treatment of MCF-7 cells with E2 (10 nM), TNFα (10 ng/ml), or both was carried out by QPCR. Data shown are fold change relative to untreated controls using the ΔΔCt method with 36B4 as an internal control. b cIAP2 mRNA was examined by QPCR in ER positive (BT-474, T47D) and ER negative (MDA-MB-231) breast cancer cell lines following treatment with E2, TNFα or both for 24 h.
To examine the regulation of IAPs at the protein level, Western blots were carried out after treatment of MCF-7 cells with E2 + TNFα for up to 24 h (Fig. 2, left panel and Supplemental Fig. 1) or with E2, TNFα or both for 24 h (Fig. 2, right panel and Supplemental Fig. 1). Regulation of cIAP2 protein levels corresponded to regulation of mRNA with a major increase in cIAP2 expression seen over time. The regulation of cIAP2 protein appears to be specific for the combination of E2 + TNFα compared to either E2 or TNFα alone. In contrast, cIAP1 protein, unlike its mRNA transcript, was rapidly decreased by the combination of E2 + TNFα, suggesting a post-transcriptional regulatory mechanism for this IAP family member. It appears that the down-regulation of cIAP1 occurs in response to TNFα alone or the combination of E2 + TNFα, indicating a TNFα-dependent mechanism, as has been suggested previously [28]. There was no effect on XIAP protein levels by any of the treatments, in accordance with mRNA levels. Together, these findings show that E2 is able to greatly potentiate TNFα-regulated expression of cIAP2, suggesting that cIAP2 may be a crucial player in IAP-dependent cell survival by E2.
Fig. 2.

Regulation of IAP protein by E2 and TNFα in MCF-7 cells. MCF-7 cells were treated with the combination of E2 (10 nM) and TNFα (10 ng/ml) for up to 24 h (left) or individually with E2 (10 nM) or TNFα (10 ng/ml) or the combination of both for 24 h (right). Western Blot for cIAP1, cIAP2 and XIAP were carried out with β-actin serving as a loading control.
cIAP2 Expression is Detected in a Subset of Invasive Breast Tumors
We next examined expression of cIAP2 in a TMA composed of normal breast, DCIS, and invasive breast carcinoma tissue samples. We observed that only one of 43 normal breast cores and one of 38 DCIS cores were positive for cIAP2 (not shown). In contrast, eight of 63 (12.7%) invasive breast carcinomas were strongly positive for cIAP2, indicating cIAP2 is expressed in a subset of invasive breast cancer (Fig. 3). Although we have only a small number of tumors staining positive for cIAP2, a significant correlation with EGFR status was observed but the relevance of this finding requires further investigation in a larger cohort of tumors with additional clinical information (Table 1).
Fig. 3.

Expression of cIAP2 in invasive breast carcinoma. A TMA containing paraffin embedded breast tissue was examined for cIAP2 expression by IHC. Positive staining for cIAP2 (a at 10×, b at 40×) was observed in ~13% of invasive breast tumors while ~87% of tumors were negative (c at 10×, d at 40×).
Table 1.
Association of cIAP2 staining with clinical parameters in invasive breast tumors
| Positive for cIAP2 (n = 8) | Negative for cIAP2 (n = 55) | ||
|---|---|---|---|
| ER | Pos | 1 | 28 |
| Neg | 5 | 15 | |
| Unknown | 2 | 12 | |
| PR | Pos | 0 | 13 |
| Neg | 6 | 28 | |
| Unknown | 2 | 14 | |
| EGFR | Pos | 6* | 13 |
| Neg | 2 | 41 | |
| Unknown | 0 | 1 | |
| Her2/ErbB2 | Pos | 3 | 9 |
| Neg | 3 | 34 | |
| Unknown | 2 | 12 | |
| p53 | Pos | 5 | 18 |
| Neg | 1 | 19 | |
| Unknown | 2 | 18 | |
| Lymph node involvement | Pos | 3 | 31 |
| Neg | 2 | 3 | |
| Unknown | 3 | 21 | |
*P < 0.01
IAPs are Required for E2-mediated Cell Survival in Response to TNFα
To study the role of IAPs in influencing breast cancer cell survival, we carried out dose responses for three small molecule antagonists of IAPs, called SMs: Compound 3 (Cmpd3), MV1, and BV6. Each was designed to mimic the IAP-interacting domain of the mature Smac protein and can thus neutralize IAP action [20, 29]. In the absence of SMs, treatment of MCF-7 cells with a high dose of TNFα for 24 h reduced cell viability to 20-50% of that seen in untreated control cells and this was prevented by the addition of E2 (Fig. 4, gray vs. black bars). Previous work from our lab and others has demonstrated that the effect of TNFα on cell viability is the result of increased apoptosis and that E2 reverses the effect of TNFα specifically by inhibiting apoptosis [8, 28]. We found that all three SMs are able to potentiate TNFα-induced death with cell viability significantly reduced compared to TNFα alone. In addition, all SMs significantly reduced the ability of E2 to protect cells against TNFα-induced death (black bars). A similar effect of SM was seen when TRAIL was used to induce apoptosis (data not shown). In contrast, the SMs had no effect on untreated control cells (not shown) or on E2-treated cells (white bars) in the absence of TNFα or TRAIL. For both Cmpd 3 and MV1, a significant reduction in E2-stimulated cell viability was observed at doses of 1-10 nM (Fig. 4a and b). However, MCF-7 cells are especially sensitive to BV6 with significantly increased cell death observed at concentrations as low as 0.01 nM (Fig. 4c). In addition, we confirmed that SMs are capable of rapidly down-regulating expression of cIAP1 and cIAP2 but not XIAP, or the unrelated anti-apoptotic protein, Bcl-2 (Fig. 5 and data not shown). These data indicate that IAPs not only play a protective role in breast cancer cells by limiting TNFα-induced death but are also essential for the ability of E2 to promote cell survival in response to TNFα and TRAIL.
Fig. 4.
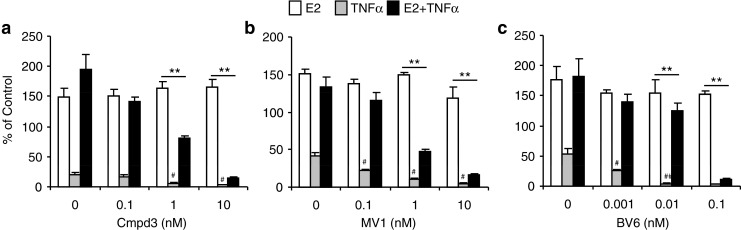
Smac mimetics sensitize MCF-7 cells to TNFα-induced cell death and block E2-mediated cell survival. MCF-7 cells were treated for 24 h with E2 (10 nM), TNFα (50 ng/ml), or both in the absence or presence of increasing doses of three different SMs, Cmpd3 (a), MV1 (b), and BV6 (c), and cell viability was assessed by a MTS assay. **P < 0.001 for E2 + TNFα compared to E2 alone at the same dose of SM, # P < 0.01 for TNFα with a SM compared to TNFα alone in the absence of SMs.
Fig. 5.

Smac mimetics down-regulate cIAP1 and cIAP2 protein levels. WCE were prepared from MCF-7 cells treated with or without the combination of E2 (10 nM) and TNFα (10 ng/ml) for 24 h followed by a 1-h treatment with vehicle or BV6 (5 μM). Western Blot for cIAP1, cIAP2, XIAP, and Bcl-2 were carried out with β-actin as the loading control.
We next examined the effect of a SM in BT-474 cells, which are known to be resistant to TNFα-induced cell death [30] and which we confirm in Fig. 6a. However, treating BT-474 cells with Cmpd3 to block IAP activity greatly sensitized this cell line to TNFα-induced death as well as blocked E2 from promoting cell survival (Fig. 6a). Analysis of IAP mRNA levels revealed that BT-474 cells have high levels of intrinsic IAP transcripts in the absence of treatment when compared to IAP levels found in MCF-7 cells (Fig. 6b), suggesting that IAPs may be key pro-survival factors in multiple ER positive breast cancer cell lines and may mediate resistance to known killing agents.
Fig. 6.

Inhibition of IAPs sensitizes BT-474 Cells to TNFα. a BT-474 cells were treated for 24 h with E2, TNFα (50 ng/ml), or both in the presence of Cmpd 3 (10 nM) and cell viability was determined by MTS assay. b The basal levels of IAP mRNA in untreated BT-474 were compared to that found in untreated MCF-7 cells by QPCR.
To examine the individual roles of IAPs in mediating breast cancer cell survival, siRNA was used to knock down each IAP individually or in combination in MCF-7 cells. As shown in Fig. 7a, an 87-99% knockdown of IAP protein was achieved using this strategy with no effect on Bcl-2 expression. The effect on E2-mediated cell survival was measured following a 24-h treatment with E2 + TNFα. Individual knockdown of cIAP1 or cIAP2 reduced E2-dependent cell survival to 30-50% of that seen with the negative control (siNeg), whereas knockdown of XIAP had little effect on E2-mediated cell survival (Fig. 7b). Knockdown of both cIAP1 and cIAP2 together, either with or without XIAP siRNA, further reduced E2-dependent cell survival to ~10% of that seen with siNeg. These findings suggest that both cIAP1 and cIAP2, but not XIAP, are involved in E2-mediated breast cancer cell survival.
Fig. 7.

siRNA for IAPs Prevent E2-mediated Cell Survival. a MCF-7 cells were transfected with siRNA for each IAP or a negative control (siNeg) for 48 h. Cells were then treated with the combination of E2 (10 nM) and TNFα (10 ng/ml) for an additional 24 h for cIAP2 detection whereas cIAP1, XIAP and Bcl-2 detection was carried out in untreated cells. Protein expression was examined by Western Blot. Percent knockdown for each siRNA relative to siNeg was determined by densitometry and normalized to β-actin. b MCF-7 cells were transfected with siRNA for single, double, and triple knockdowns of IAPs for 48 h before cells were treated for an additional 24 h with the combination of E2 (10 nM) and TNFα (10 ng/mL). Viability was measured by methylene blue assay. Survival is reported as percent of control (siNeg) for the mean and standard error of three biological replicates, with four technical replicates for each sample. *P < 0.05 compared to siNeg.
Discussion
In this study, we have identified a novel mechanism by which E2 is able to promote survival of breast cancer cells. While previous studies have focused on the role of Bcl-2 family members, here we demonstrate that the IAP family members, cIAP1 and cIAP2, are also essential mediators of E2-dependent breast cancer cell survival.
Initially, our studies suggested an important role for cIAP2 in breast cancer cell survival since it is highly up-regulated at both the mRNA and protein level in response to E2 + TNFα, whereas a major reduction in cIAP1 protein was seen in response to the same agents. Although both IAPs are known to be up-regulated by TNFα through the NFκB pathway [27], we find that E2 specifically enhances the regulation of cIAP2, but not cIAP1, by TNFα. This is of interest because E2 is known to repress NFκB activity on many classical proinflammatory NFκB target genes [31]. However, a recent study from our lab indicated that ER and NFκB can also work together to enhance the expression of a specific subset of target genes and that these target genes, including cIAP2, are enriched in the more aggressive ER positive tumors of the Luminal B intrinsic subtype [8].
Based on these regulation patterns of cIAP1 and cIAP2, our original hypothesis was that cIAP2 may be the more important regulator of E2-dependent breast cancer cell survival. We confirmed that it does play a role, given that E2 was less able to protect against TNFα-induced death when cIAP2 was knocked down by siRNA. However, knockdown of cIAP1 also impacted E2 action and simultaneous knockdown of both cIAP1 and cIAP2 caused the greatest reduction in the ability of E2 to promote cell survival. These findings suggested that both cIAP1 and cIAP2 are essential regulators of E2-dependent cell survival. Alternatively, it is possible that cIAP1 and cIAP2 may compensate for one another in the knockdown experiments. cIAP1 knockout mice had no discernable phenotype potentially because elevated cIAP2 levels were detected [32]. The increase in cIAP2 was explained by the finding that cIAP1 can target cIAP2 for proteasomal degradation. In cIAP2 knockout animals, a specialized role for cIAP2 in LPS-induced macrophage survival was uncovered [33]. More recently, it was found that cIAP1 and cIAP2 have both redundant and specialized roles in TNFα-mediated activation of NFκB [15]. In either case, it is clear from our studies that elimination of both cIAP1 and cIAP2, rather than either alone, was more effective in reducing the pro-survival effect of E2 in breast cancer cells.
Many IAPs have been found to be overexpressed in a number of different cancers. For example, XIAP expression was associated with patient outcome in acute myeloid leukemia [34], whereas in a separate study, cIAP2 expression along with cIAP1 and XIAP were associated with progression of chronic lymphocytic leukemia [35]. The results of our TMA study showed that cIAP2 expression was present in a subset of invasive breast tumor cores (12.7%) as opposed to DCIS (2.6%) and normal breast (2.3%). A previous report demonstrated by Western Blot that cIAP2 is indeed expressed in very few breast cancer cell lines but was actually higher in normal tissue than tumors [23]. It is possible that the different detection methods used in these studies may account for the different findings. A previous study of cIAP1 found that, unlike cIAP2, it is expressed at relatively equivalent levels in breast tumors vs. normal tissues, as well as in a number of breast cancer cell lines, and is relatively constant across a breast tumor progression panel [23]. Taken together, cIAP1 and cIAP2 appear to be expressed in breast tumors but whether they are co-expressed is not known. Larger, more definitive studies are also needed to determine whether their expression levels correlate with disease progression or patient outcome. This is further warranted by our finding that BT-474 cells, which express high levels of IAPs, are resistant to TNFα- induced cell death but can be sensitized by antagonizing IAP activity.
Perhaps the most exciting finding from our studies is the ability of SMs to completely prevent E2 from rescuing breast cancer cells from TNFα- and TRAIL-induced cell death. SMs were originally designed to target XIAP, the most well studied and most potent inhibitor of apoptosis [13]. However, XIAP does not appear to play a role in regulating MCF-7 cell death in response to E2 and TNFα. Although cIAP1 and cIAP2 are less potent at inhibiting caspase activity, SMs can facilitate autoubiquitination and degradation of cIAP1 and cIAP2 [20]. In combination with the siRNA data, our findings suggest that SMs are most likely acting through this mechanism, rather than inhibition of XIAP activity, to block E2-dependent cell survival. While SMs have been shown to synergize with a number of anticancer agents, including Her2 and EGFR inhibitors [23], to kill breast cancer cells, this is the first study showing that SMs can also counteract the pro-survival function of E2 in response to specific apoptotic stimuli, such as TNFα and TRAIL. These findings further suggest that without the IAP pathway intact, activation of other prosurvival signals, such as Bcl2, by E2 are unable to prevent apoptosis.
In conclusion, our findings indicate that the unique pattern of expression and regulation of IAPs contributes to the ability of E2 to prevent apoptosis of breast cancer cells. Furthermore, the effect of SMs in preventing the pro-survival effect of E2, as well as in sensitizing resistant cells to apoptotic agents, suggests that SMs may prove to be a valuable therapeutic option in patients with hormone-dependent breast cancer.
Electronic supplementary material
Below is the link to the electronic supplementary material.
MCF-7 cells treated with 10 nM E2, 10 ng/mL TNFα, or both for up to 24 h followed by a SMAC mimetic, BV6 (5 uM), for 1 h. Cell lysate was collected by Direct Lysis method using a 6X Electrophoresis Buffer. Western Blot was performed using the samples as follows: 1 Precision Plus Protein Standard Bio-Rad #161-0376, 2 control 0 h, 3 E2 + TNFα 2 h, 4 E2 + TNFα 6 h, 5 E2 + TNFα 24 h, 6 control 24 h, 7 E2 24 h, 8 TNFα 24 h, 9 E2 + TNFα 24 h, 10 control 24 h + BV6 1 h, with the exception of cIAP2 blot where 10 is E2 + TNFα 24 h + BV6 1 h (PPT 559 kb)
Acknowledgments
The authors would like to thank Drs. X. Wang and D. Vucic for generously providing SMAC mimetics/IAP antagonists. We are also grateful to Dana Felice, Shaungping Zhao, and Madhumita Pradhan for their constructive discussions and critical reading of the manuscript. This work was funded by the National Institute of Health HL07692-19 (AS) and CA130932-2 (JF) and the Illinois Dept. of Public Health (JF).
Disclosure Statement
The authors have nothing to disclose.
Footnotes
Financial support: T32 HL07692-19 (AS), NIH R01 CA130932-2 (JF), Illinois Dept. of Public Health (JF)
References
- 1.Bjornstrom L, Sjoberg M. Mechanisms of estrogen receptor signaling: convergence of genomic and nongenomic actions on target genes. Mol Endocrinol. 2005;19:833–842. doi: 10.1210/me.2004-0486. [DOI] [PubMed] [Google Scholar]
- 2.Doisneau-Sixou SF, Sergio CM, Carroll JS, Hui R, Musgrove EA, Sutherland RL. Estrogen and antiestrogen regulation of cell cycle progression in breast cancer cells. Endocr Relat Cancer. 2003;10:179–186. doi: 10.1677/erc.0.0100179. [DOI] [PubMed] [Google Scholar]
- 3.Eeckhoute J, Carroll JS, Geistlinger TR, Torres-Arzayus MI, Brown M. A cell type-specific transcriptional network required for estrogen regulation of cyclin D1 and cell cycle progression in breast cancer. Genes Dev. 2006;20:2513–2526. doi: 10.1101/gad.1446006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gompel A, Somai S, Chaouat M, Kazem A, Kloosterboer HJ, Beusman I, Forgez P, Mimoun M, Rostene W. Hormonal regulation of apoptosis in breast cells and tissues. Steroids. 2000;65:593–598. doi: 10.1016/S0039-128X(00)00172-0. [DOI] [PubMed] [Google Scholar]
- 5.Teixeira C, Reed JC, Pratt MA. Estrogen promotes chemotherapeutic drug resistance by a mechanism involving Bcl-2 proto-oncogene expression in human breast cancer cells. Cancer Res. 1995;55:3902–3907. [PubMed] [Google Scholar]
- 6.Frasor J, Danes JM, Komm B, Chang KC, Lyttle CR, Katzenellenbogen BS. Profiling of estrogen up- and down-regulated gene expression in human breast cancer cells: insights into gene networks and pathways underlying estrogenic control of proliferation and cell phenotype. Endocrinology. 2003;144:4562–4574. doi: 10.1210/en.2003-0567. [DOI] [PubMed] [Google Scholar]
- 7.Biswas DK, Martin KJ, McAlister C, Cruz AP, Graner E, Dai SC, Pardee AB. Apoptosis caused by chemotherapeutic inhibition of nuclear factor-kappaB activation. Cancer Res. 2003;63:290–295. [PubMed] [Google Scholar]
- 8.Frasor J, Weaver A, Pradhan M, Dai Y, Miller LD, Lin CY, Stanculescu A. Positive cross-talk between estrogen receptor and NF-kappaB in breast cancer. Cancer Res. 2009;69:8918–8925. doi: 10.1158/0008-5472.CAN-09-2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Miura K, Karasawa H, Sasaki I. cIAP2 as a therapeutic target in colorectal cancer and other malignancies. Expert Opin Ther Targets. 2009;13:1333–1345. doi: 10.1517/14728220903277256. [DOI] [PubMed] [Google Scholar]
- 10.Rothe M, Pan MG, Henzel WJ, Ayres TM, Goeddel DV. The TNFR2-TRAF signaling complex contains two novel proteins related to baculoviral inhibitor of apoptosis proteins. Cell. 1995;83:1243–1252. doi: 10.1016/0092-8674(95)90149-3. [DOI] [PubMed] [Google Scholar]
- 11.Srinivasula SM, Ashwell JD. IAPs: what’s in a name? Mol Cell. 2008;30:123–135. doi: 10.1016/j.molcel.2008.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bertrand MJ, Milutinovic S, Dickson KM, Ho WC, Boudreault A, Durkin J, Gillard JW, Jaquith JB, Morris SJ, Barker PA. cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Mol Cell. 2008;30:689–700. doi: 10.1016/j.molcel.2008.05.014. [DOI] [PubMed] [Google Scholar]
- 13.Eckelman BP, Salvesen GS. The human anti-apoptotic proteins cIAP1 and cIAP2 bind but do not inhibit caspases. J Biol Chem. 2006;281:3254–3260. doi: 10.1074/jbc.M510863200. [DOI] [PubMed] [Google Scholar]
- 14.Hu S, Yang X. Cellular inhibitor of apoptosis 1 and 2 are ubiquitin ligases for the apoptosis inducer Smac/DIABLO. J Biol Chem. 2003;278:10055–10060. doi: 10.1074/jbc.M207197200. [DOI] [PubMed] [Google Scholar]
- 15.Mahoney DJ, Cheung HH, Mrad RL, Plenchette S, Simard C, Enwere E, Arora V, Mak TW, Lacasse EC, Waring J, Korneluk RG. Both cIAP1 and cIAP2 regulate TNFalpha-mediated NF-kappaB activation. Proc Natl Acad Sci USA. 2008;105:11778–11783. doi: 10.1073/pnas.0711122105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Varfolomeev E, Goncharov T, Fedorova AV, Dynek JN, Zobel K, Deshayes K, Fairbrother WJ, Vucic D. c-IAP1 and c-IAP2 are critical mediators of tumor necrosis factor alpha (TNFalpha)-induced NF-kappaB activation. J Biol Chem. 2008;283:24295–24299. doi: 10.1074/jbc.C800128200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Du C, Fang M, Li Y, Li L, Wang X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell. 2000;102:33–42. doi: 10.1016/S0092-8674(00)00008-8. [DOI] [PubMed] [Google Scholar]
- 18.Wu H, Tschopp J, Lin SC. Smac mimetics and TNFalpha: a dangerous liaison? Cell. 2007;131:655–658. doi: 10.1016/j.cell.2007.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Petersen SL, Wang L, Yalcin-Chin A, Li L, Peyton M, Minna J, Harran P, Wang X. Autocrine TNFalpha signaling renders human cancer cells susceptible to Smac-mimetic-induced apoptosis. Cancer Cell. 2007;12:445–456. doi: 10.1016/j.ccr.2007.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Varfolomeev E, Blankenship JW, Wayson SM, Fedorova AV, Kayagaki N, Garg P, Zobel K, Dynek JN, Elliott LO, Wallweber HJ, Flygare JA, Fairbrother WJ, Deshayes K, Dixit VM, Vucic D. IAP antagonists induce autoubiquitination of c-IAPs, NF-kappaB activation, and TNFalpha-dependent apoptosis. Cell. 2007;131:669–681. doi: 10.1016/j.cell.2007.10.030. [DOI] [PubMed] [Google Scholar]
- 21.Vince JE, Wong WW, Khan N, Feltham R, Chau D, Ahmed AU, Benetatos CA, Chunduru SK, Condon SM, McKinlay M, Brink R, Leverkus M, Tergaonkar V, Schneider P, Callus BA, Koentgen F, Vaux DL, Silke J. IAP antagonists target cIAP1 to induce TNFalpha-dependent apoptosis. Cell. 2007;131:682–693. doi: 10.1016/j.cell.2007.10.037. [DOI] [PubMed] [Google Scholar]
- 22.Bockbrader KM, Tan M, Sun Y. A small molecule Smac-mimic compound induces apoptosis and sensitizes TRAIL- and etoposide-induced apoptosis in breast cancer cells. Oncogene. 2005;24:7381–7388. doi: 10.1038/sj.onc.1208888. [DOI] [PubMed] [Google Scholar]
- 23.Foster FM, Owens TW, Tanianis-Hughes J, Clarke RB, Brennan K, Bundred NJ, Streuli CH. Targeting inhibitor of apoptosis proteins in combination with ErbB antagonists in breast cancer. Breast Cancer Res. 2009;11:R41. doi: 10.1186/bcr2328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Flygare JA, Fairbrother WJ. Small-molecule pan-IAP antagonists: a patent review. Expert Opin Ther Pat. 2010;20:251–267. doi: 10.1517/13543770903567077. [DOI] [PubMed] [Google Scholar]
- 25.Felice DL, Sun J, Liu RH. A modified methylene blue assay for accurate cell counting. J Funct Foods. 2009;1:109–118. doi: 10.1016/j.jff.2008.09.014. [DOI] [Google Scholar]
- 26.Wang L, Du F, Wang X. TNF-alpha induces two distinct caspase-8 activation pathways. Cell. 2008;133:693–703. doi: 10.1016/j.cell.2008.03.036. [DOI] [PubMed] [Google Scholar]
- 27.Wang CY, Mayo MW, Korneluk RG, Goeddel DV, Baldwin AS., Jr NF-kappaB antiapoptosis: induction of TRAF1 and TRAF2 and c-IAP1 and c-IAP2 to suppress caspase-8 activation. Science. 1998;281:1680–1683. doi: 10.1126/science.281.5383.1680. [DOI] [PubMed] [Google Scholar]
- 28.Messmer UK, Pereda-Fernandez C, Manderscheid M, Pfeilschifter J. Dexamethasone inhibits TNF-alpha-induced apoptosis and IAP protein downregulation in MCF-7 cells. Br J Pharmacol. 2001;133:467–476. doi: 10.1038/sj.bjp.0704093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li L, Thomas RM, Suzuki H, De Brabander JK, Wang X, Harran PG. A small molecule Smac mimic potentiates TRAIL- and TNFalpha-mediated cell death. Science. 2004;305:1471–1474. doi: 10.1126/science.1098231. [DOI] [PubMed] [Google Scholar]
- 30.Rivas MA, Tkach M, Beguelin W, Proietti CJ, Rosemblit C, Charreau EH, Elizalde PV, Schillaci R. Transactivation of ErbB-2 induced by tumor necrosis factor alpha promotes NF-kappaB activation and breast cancer cell proliferation. Breast Cancer Res Treat. 2009;122(1):111–124. doi: 10.1007/s10549-009-0546-3. [DOI] [PubMed] [Google Scholar]
- 31.De Bosscher K, Vanden Berghe W, Haegeman G. Cross-talk between nuclear receptors and nuclear factor kappaB. Oncogene. 2006;25:6868–6886. doi: 10.1038/sj.onc.1209935. [DOI] [PubMed] [Google Scholar]
- 32.Conze DB, Albert L, Ferrick DA, Goeddel DV, Yeh WC, Mak T, Ashwell JD. Posttranscriptional downregulation of c-IAP2 by the ubiquitin protein ligase c-IAP1 in vivo. Mol Cell Biol. 2005;25:3348–3356. doi: 10.1128/MCB.25.8.3348-3356.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Conte D, Holcik M, Lefebvre CA, Lacasse E, Picketts DJ, Wright KE, Korneluk RG. Inhibitor of apoptosis protein cIAP2 is essential for lipopolysaccharide-induced macrophage survival. Mol Cell Biol. 2006;26:699–708. doi: 10.1128/MCB.26.2.699-708.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tamm I, Kornblau SM, Segall H, Krajewski S, Welsh K, Kitada S, Scudiero DA, Tudor G, Qui YH, Monks A, Andreeff M, Reed JC. Expression and prognostic significance of IAP-family genes in human cancers and myeloid leukemias. Clin Cancer Res. 2000;6:1796–1803. [PubMed] [Google Scholar]
- 35.Grzybowska-Izydorczyk O, Cebula B, Robak T, Smolewski P. Expression and prognostic significance of the inhibitor of apoptosis protein (IAP) family and its antagonists in chronic lymphocytic leukaemia. Eur J Cancer. 2010;46:800–810. doi: 10.1016/j.ejca.2009.11.023. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
MCF-7 cells treated with 10 nM E2, 10 ng/mL TNFα, or both for up to 24 h followed by a SMAC mimetic, BV6 (5 uM), for 1 h. Cell lysate was collected by Direct Lysis method using a 6X Electrophoresis Buffer. Western Blot was performed using the samples as follows: 1 Precision Plus Protein Standard Bio-Rad #161-0376, 2 control 0 h, 3 E2 + TNFα 2 h, 4 E2 + TNFα 6 h, 5 E2 + TNFα 24 h, 6 control 24 h, 7 E2 24 h, 8 TNFα 24 h, 9 E2 + TNFα 24 h, 10 control 24 h + BV6 1 h, with the exception of cIAP2 blot where 10 is E2 + TNFα 24 h + BV6 1 h (PPT 559 kb)