

Abstract
Synaptogenesis involves the transformation of a growth cone into synaptic boutons specialized for transmitter release. In Drosophila embryos lacking the α2δ-3 subunit of presynaptic, voltage-dependent Ca2+ channels, we find that motor neuron terminals fail to develop synaptic boutons and cytoskeletal abnormalities arise, including the loss of ankyrin2. Nevertheless, functional presynaptic specializations are present and apposed to clusters of postsynaptic glutamate receptors. Heretofore, the α2δ-3 protein has been thought to function strictly as an auxiliary subunit of the Ca2+ channel, but the phenotype of α2δ-3 mutations cannot be explained by a channel defect: embryos lacking the pore-forming α1 subunit cacophony form boutons. The synaptogenic function of α2δ-3 requires only the α2 peptide, whose expression suffices to rescue bouton formation. Our results indicate that α2δ proteins have functions independent of their roles in the biophysics and localization of Ca2+ channels, and synaptic architecture depends on these novel functions.
Introduction
Synaptic terminals are formed through the transformation of distal axons and growth cones into a set of rounded swellings or boutons. This process requires a remodeling of the cytoskeleton, the organization of presynaptic proteins into active zones, and the accumulation of synaptic vesicles at axon-target contacts1. Signaling between the target and developing synapse is thought to guide this process and is likely to involve proteins on the surface of the nerve ending. One universal component of presynaptic membranes is voltage-dependent Ca2+ channels2. Whereas the large α1 subunits of these channels have a well-known function in triggering synaptic transmission, the α2δ subunits of these channels are less well understood. These subunits protrude far into the extracellular space3 and α2δ subunits thus have the potential to contribute to the signaling events that coordinate synaptic development4.
Although the α1 subunit is the largest subunit of the channel and contains the channel pore and voltage sensors5, channel properties and expression levels are influenced by the auxiliary subunits α2δ, β and γ (Fig. 1a)6. The cytoplasmic β subunit can influence the biophysical properties of the channel and mediate regulation by intracellular signaling pathways7. In contrast, the α2δ subunit is primarily on the extracellular surface and less is known about its functional significance.
Figure 1. α2δ-3 null embryos exhibit a defect in bouton morphogenesis.

a. Schematic representation of a voltage-dependent Ca2+ channel, after Arikkath and Campbell6. Stars mark the positions of stop codons in the null allele α2δ-3106 and the truncated allele α2δ-3196. The Von Willebrand Factor A (VWFa) homology domain is marked in orange. b–d. Neuronal membranes of embryonic NMJs, 21h AEL, labeled with anti-HRP. b. Wildtype NMJs have rounded boutons (arrows), averaging 17.8 ± 0.6 (SEM, N = 29 NMJs) boutons per A2–A5 hemisegment on embryonic muscles 6, 7, 12 and 13. c. Boutons do not form in α2δ-3106/Df(2R)Ex7128 (arrowheads point to neuronal endings); none were observed in over 100 embryos. d. Neuronal expression of α2δ-3 in the null background rescues bouton formation (arrows; genotype C155elav-gal4/+; α2δ-3106/Df(2R)Ex7128; UAS-HA-α2δ-3/+). Muscles are indicated by numbers and boundary lines. Scale bar = 5 µm. e, f. HA-tagged α2δ-3 expressed neuronally at a 3rd instar (e) and embryonic (f) NMJs (genotype: C155elav-gal4/+;; UAS-HA-α2δ-3/+). Scale bars = 5 µm.
Both the fly and mammalian genomes contains four genes predicted to encode an α2δ subunit8, 9 (In the fly: CG4587, CG12455, CG12295 (α2δ-3), and CG16868). The protein product of an α2δ gene is post-translationally cleaved to form the α2 and δ peptides, which remain linked by a disulfide bond10. The δ subunit contains a C-terminal transmembrane domain and almost no intracellular residues, while the α2 portion is entirely extracellular11. This large, extracellular region is heavily glycosylated and contains several potential protein-protein interacting domains. These include 1) a Von Willebrand factor-A (VWFa) domain akin to extracellular matrix-binding regions of integrins12, and 2) a poorly understood cache domain13. The transmembrane segment of δ and some extracellular regions of α2 are thought to associate with the α1 subunit14, 15, but α2 is also well-situated to interact with other extracellular or cell surface proteins.
α2δ expression can increase surface expression of α1 both in vitro and in vivo16–21. α2δ is therefore thought to be involved in the trafficking, retention, or localization of the α1 subunit. Consistent with this hypothesis, mutations in the murine α2δ-2 subunit decrease the amplitude of Ca2+ currents in Purkinje cells22, 23. In Drosophila, weak mutant alleles of α2δ-3 reduce the expression of CaV2-type α1 subunits at the active zones of NMJs and consequently reduce evoked excitatory junctional potentials20, 24. Together these studies indicate that α2δ is needed for proper localization and function of the α1 subunit, but leave open the possibility of additional functions.
Some indications of a broader developmental role for α2δ have emerged from genetic studies. Purkinje cells lacking α2δ-2 have abnormal dendrites23 and loss of the α2δ-4 subunit causes abnormalities in the outer plexiform layer of the murine retina25. It is unknown whether these anatomical changes are a consequence of the reduced expression of the α1 subunit or reflect a direct influence of α2δ on neuronal development.
Our previous study of a hypomorphic allele of α2δ-3 revealed its role in α1 localization at the Drosophila NMJ20. The present examination of null mutant embryos revealed an unexpected and severe neuromuscular phenotype indicative of a role for α2δ- 3 in synaptic morphogenesis. This developmental function for α2δ-3 is independent of its role in localizing synaptic Ca2+ channels.
Results
α2δ-3 is required for morphological development of the NMJ
A forward genetic screen for mutants in synaptic transmission uncovered mutations in the gene encoding α2δ-320. The α2δ-3106 allele contains a very early stop codon (R192Stop), and is therefore a likely null allele. α2δ-3106 mutants died as late embryos that were fully formed but failed to hatch from their egg cases. We examined the NMJs of α2δ-3106 null alleles (either as homozygotes or in trans with two different deficiencies that remove the locus) at 21h after egg-laying (AEL), when control embryos have fully formed NMJs with a characteristic morphology of rounded boutons separated by short sections of neurites (Fig. 1b). In contrast to this “beads on a string” morphology, the α2δ-3 nulls completely lacked boutons; throughout the bodywall musculature, only thin neurites extended on the muscle surface (Fig. 1c).
Expression of an HA-tagged α2δ-3 transgene (UAS-HA-α2δ-3), by means of either a neuron-specific (C155elav-GAL4) or ubiquitous (da-GAL4) driver, rescued bouton formation in α2δ-3 null genotypes (Fig. 1d). This rescue confirms that the “bouton-less” phenotype is indeed due to the absence of α2δ-3 and moreover demonstrates that α2δ-3 is required presynaptically in the neuron. Neuronally expressed UAS-HA-α2δ-3 appeared in the synaptic boutons of both embryonic and larval NMJs (Fig. 1e,f), consistent with a function at the nerve terminals.
To determine whether this boutonless phenotype resulted from an early arrest in synaptic morphogenesis or degeneration of normally formed synapses, we looked at earlier developmental stages (Fig. 2). Synaptic morphogenesis is the process by which axons, after reaching their targets, transform their terminal from a motile growth cone to a mature synaptic ending optimized for neurotransmission. This process entails cytoskeletal rearrangements and the proper localization of synaptic components. At the Drosophila NMJ, motor neuron growth cones are flat with long filopodia (Fig. 2a,d). At 17h AEL the growth cones thicken, forming “prevaricosities”26, and establish the characteristic branching pattern of the NMJ (Fig. 2b). Between 17–19h AEL, the prevaricosities constrict into the rounded boutons that characterize larval endings26. In α2δ-3 nulls, motor neurons successfully navigated to and halted on their target muscles at the appropriate time, and growth cones appeared normally shaped and sized (Fig. 2d). However, as early as 16h AEL, during the prevaricosity stage, NMJs became abnormal with thinner neurites and atypical branching (Fig. 2e). Although muscles 6, 7, 12, and 13 were each contacted by the neurons, the nerve often did not end by bifurcating in a “T” at the boundary between muscles 12 and 13, but bifurcated instead more ventrally, on muscle 6 in a “Y” that extended to the other muscles. Other embryonic development continued appropriately (e.g. gut folding, ventral nerve cord retraction, trachea airfilling), but the NMJ never progressed past the prevaricosity stage and no boutons ever arose (Fig. 2f). As late as 24h AEL, when wildtype larvae would have already hatched, no boutons had formed in the null. Thus the phenotype appears to be a specific arrest of morphogenesis rather than a delay of neuronal developmental.
Figure 2. α2δ-3 null NMJs exhibit an arrest of synaptic morphogenesis.

Developing NMJs of wildtype (a–c) and mutant (d–f) embryos. Neuronal membranes are labeled with anti-HRP. Wildtype motorneuron growth cones reach their target muscles at 13h AEL26, 45–47 and, as shown at 14h AEL (a), look flat and filamentous. α2δ-3106/106 motorneurons (d) also reach their targets at this time, and appear indistinguishable from wildtype. By 16h AEL, wildtype endings (b) branch in a characteristic pattern, with long neurites extending along the boundary between muscles 6 and 7 (arrow) and a shorter branch forming between muscles 12 and 13 (arrow head). Prevaricosities, still somewhat flat and filamentous, begin to form. At this time, α2δ-3106/106 NMJs are noticeably abnormal, branching in the wrong place (*), lacking neurites along the 12/13 border, and lacking prevaricosities. Just before hatching, wildtype endings (c) have formed rounded boutons but α2δ-3106/106 endings (f) remain as they were at 16h. Scale bar = 5 µm.
α2δ-3 mutant terminals lack ankyrin2-XL
The severe morphological defect at the α2δ-3 null NMJs suggested that their cytoskeleton might be disrupted. We therefore examined the microtubule-binding protein futsch, a homolog of mammalian MAP1B, and the actin/spectrin -adaptor protein ankyrin. Futsch influences the size and number of boutons at the larval NMJ27 by regulating microtubule structure. Ankyrin2-L and ankyrin2-XL, two presynaptic isoforms, stabilize NMJ synapses28, 29. Ankyrins can organize the spectrin cytoskeleton and anchor cell surface proteins within membrane domains, including synaptic terminals30.
Ank2-XL staining was almost completely absent from α2δ-3 motor neuron terminals, though it persisted in other neuronal compartments (Fig. 3), and could be restored to terminals by neuronal expression of the UAS-HA-α2δ-3 construct (Fig. 3g). Futsch staining was also decreased (Fig. 3b), but more variably, being sometimes completely absent, occasionally at wild-type levels, and in most cases appearing discontinuous in the branches, unlike wildtype NMJs wherein it consistently extended throughout them. We also assayed several cell-adhesion molecules such as Drosophila N-cadherin, as well as known interactors of ankyrin such as fasII and the Drosophila L1-CAM homolog neuroglian. These proteins, however, were present at α2δ-3 mutant endings, despite the absence of ankyrin (data not shown). Similarly, the cell-surface antigens recognized by anti-HRP were unchanged in intensity of immunostaining in the α2δ-3 mutant endings (Fig. 3). Conceivably, the lack of detectable ank2-XL might arise indirectly from the small branch diameter of mutants. However, embryos lacking the kinesin motor imac, have a similar boutonless morphology but show no reduction in ank2-XL staining (Fig. 3c). Nor is the absence of ankyrin2 likely to cause the boutonless phenotype because ank2 null embryos form boutons (Supplementary Fig. S1 online). Thus the absence of ank2-XL from the α2δ-3 mutant endings is a specific feature of the phenotype but cannot on its own account for the absence of boutons.
Figure 3. Ankyrin2-XL is absent at the α2δ-3 null NMJ.
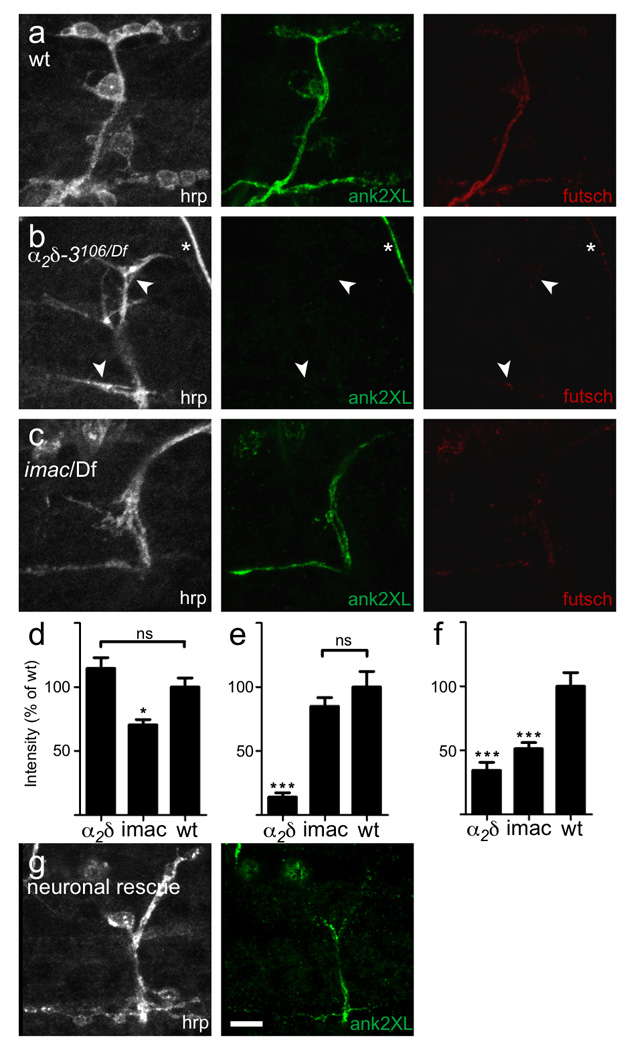
a. A wildtype NMJ immunostained with anti-HRP, anti-ank2-XL and anti-futsch. b. An α2δ-3 null (α2δ-3106/Df(2R)Ex7128) NMJ. Ank2-XL immunostaining was almost completely lacking from these synaptic terminals (arrowheads), and what little did remain was only visible by increasing the gain on the microscope. Loss of ank2-XL staining was specific to synaptic terminals, as wildtype levels of ank2-XL immunoreactivity could be seen in other neuronal compartments, including in the nearby axon of a sensory neuron (asterisk). Futsch staining was also greatly reduced in α2δ-3 mutants. c. imac null mutants, which also lack boutons, nonetheless retained ank-2XL staining. Thus the lack of detectable ank2-XL in α2δ-3 null endings was not simply a result of the morphological abnormality. In comparison, futsch staining was reduced in imac to a similar extent as in α2δ-3. d, e, f. Quantification of staining intensity for hrp (d), ank2-XL (e) and futsch (f). α2δ-3 null mutants exhibited an 86% reduction in ank2-XL immunoreactivity (N = 10 embryos/genotype, p < 0.001, one way ANOVA with Tukey post-hoc test) and a 66% reduction in futsch immunoreactivity (p < 0.001) at the NMJ terminal. imac mutants had no reduction in ank2-XL immunoreactivity, but exhibited a 50% reduction in futsch immunoreactivity (p < 0.001). g. Neuronal expression of α2δ-3 in the mutant background partially restored ank2-XL immunostaining at the endings. Scale bar = 5 µm.
Most synaptic proteins localize normally in α2δ-3 mutants
The α2δ-3106 phenotypes raised the question of whether the appropriate accumulation of pre- and post-synaptic proteins had properly arisen. We therefore immunostained the NMJs of 21h AEL mutant embryos with antibodies to the active zone component bruchpilot (brp, a homolog of the vertebrate protein CAST)31, the synaptic vesicle proteins synaptotagmin I and cysteine string protein, the glutamate receptor subunits GluRIIA and GluRIIC, and the postsynaptic density protein discs large (Fig. 4 and data not shown). Each of these proteins was concentrated at the nerve-muscle contacts and glutamate receptors appeared to be appropriately clustered and closely apposed to the Brp puncta. This organization suggested that, despite the absence of normal bouton structures, active zones had formed and were aligned with postsynaptic receptor clusters (Fig. 4c,d), and that vesicle release sites might be functional. The reduced size and complexity of the nerve-muscle contacts, however, was reflected in fewer active zones and receptor clusters per muscle (compare Fig. 4a and b), and slightly smaller active zones. However, this reduction paralleled the decrease in total surface area due to the lack of boutons. Thus the percent of the contact surface occupied by active zones, and the alignment of those active zones with postsynaptic glutamate receptors was relatively normal, despite the absence of rounded boutons.
Figure 4. α2δ-3 null embryos retain synaptic components at their terminals.

a, b. Wildtype (a) and α2δ-3 null (α2δ-3106/Df(2R)Ex7128) (b) 21h embryos express the active zone protein bruchpilot (anti-brp, red), and glutamate receptors (GluRIIC subunit; green). Neuronal membranes are immunostained with anti-hrp (white). The intensity of individual puncta of BRP did not consistently differ between mutant and wildtype, but α2δ-3106 NMJs had only 45% of the total number of colocalized brp and GluRIIC puncta compared to wildtype (average puncta number ± SEM in wt was 63 ± 8, N = 5 NMJs; in α2δ-3106/Df7128, 29 ± 3, N = 6 NMJs). This reduction parallels the finding that their nerve-muscle contact area was only 39% of that in wildtype NMJs by anti-hrp staining (wt: 160 ± 17 µm2, N = 12 NMJs; α2δ-3106/Df7128: 63 ± 4 µm2, N = 9 NMJs). Moreover, active zones appeared slightly smaller: the total area of colocalized brp and GluRIIC puncta was only 30% of that in wildtype (wt: 14 ± 2 µm2, N = 5 NMJs; α2δ-3106/Df7128: 4.3 ± 0.4 µm2, N = 6 NMJs). c, d. Details of (a) and (b) above; brp and GluRIIC puncta are closely apposed in both wildtype (c) and α2δ-3 (d) embryos. e, f. Synaptotagmin1 (green) is also present in both wildtype (e) and α2δ-3 null (f) embryos. Scale bars = 5 µm (a, b, e, f) and 1 µm (c, d).
In electron micrographs of α2δ-3 mutant NMJs, long thin processes containing active zones were found in place of the large round profiles of boutons (Supplementary Fig. S2 online). As expected from light microscopy, wildtype muscles had boutons with diameters of up to 3 µm. In α2δ-3 null embryos, neuronal processes averaged only 0.6 µm across (± 0.03 µm SEM, N = 98 profiles from 5 animals), a decrease of almost 50% from wildtype, and profiles wider than 1.3 µm were never encountered.
Despite these morphological changes, the active zones within α2δ-3 null endings looked normal, exhibiting closely apposed pre- and post-synaptic densities of similar sizes, clusters of synaptic vesicles, and the electron-dense presynaptic specializations known as T-bars (Supplementary Fig. S2 online). Thus in the absence of the α2δ-3 subunit, synapses with normal ultrastructure form within the morphologically abnormal nerve-muscle contacts.
α2δ-3 is required for α1 subunit localization at the NMJ
In contrast to the presence of brp and other pre- and post-synaptic components, one active zone component was not detectable at the α2δ-3 null NMJs: the Ca2+ channel α1 subunit encoded by the cacophony (cac) locus. This subunit, also known as Dmca1A, is the homolog of mammalian N/P/Q-type channels (CaV2) and is the α1 subunit that predominates at the presynaptic terminal in Drosophila32–36. Channel localization was assessed using a GFP-tagged transgene (cacGFP)34. We and others have previously shown that cacGFP is reduced at the active zones of third-instar larval NMJs in weak alleles of α2δ-3 20, 24. Wildtype embryonic NMJs have less cacGFP fluorescence than third-instar larvae, probably because active zones are smaller and contain fewer channels, and when co-stained with anti-brp, the amount of colocalizing cacGFP varied considerably among the active zones. Nonetheless, some cacGFP was detectable at most wildtype active zones. In contrast, cacGFP was not detected at any α2δ-3 null synapses (Fig. 5).
Figure 5. Ca2+ channel α1 subunits are not detectable in α2δ-3 null embryonic active zones.

a, b. Heterozygous control (Df(2R)Ex7128/+; elavGAL4/UAScacGFP), and c, d. α2δ-3 null (α2δ-3106/Df(2R)Ex7128; elavGAL4;UAScacGFP) NMJs. Anti-HRP staining (blue) marks neuronal membranes and anti-brp (red) labels active zones. Arrows in (a) point to cacGFP fluorescent puncta that colocalize with brp in wildtype. (b) and (d) are enlargements of regions of (a) and (c), respectively. Scale bar = 5 µm in (a, c); 1 µm in (b, d).
Evoked but not spontaneous vesicle release requires α2δ-3
To determine the extent to which synaptic function could develop in α2δ-3 null embryos, despite the absence of boutons and the apparent absence of the α1 subunit, we examined the electrophysiological properties of their NMJs and those of another allele, α2δ-3196, which has a late stop codon and is predicted to lack the δ peptide. Spontaneous miniature excitatory junctional currents (minis) of normal amplitude were observed in both null (α2δ-3106/106 and α2δ-3106/Df(2R)Ex7128) and α2δ-3196 (α2δ-3196/196 and α2δ-3196/Df(2R)Ex7128) embryos (Fig. 6a–c), indicating that functional release sites were properly aligned with functional receptors, despite the absence of boutons. Mini frequency was diminished by 64%, however (Fig. 6a,b,c), suggesting the presence of fewer release sites and consistent with the 70% reduction in active zone area in the mutants, as defined by the area of colocalized brp and GluRCII, and with the 61% decrease in the area of nerve-muscle contacts (Fig. 4). α2δ-3196/196 alleles exhibited normal mini frequency (Fig. 6a,c), a finding consistent with the persistence of boutons in that genotype (see below).
Figure 6. α2δ-3 null embryos have spontaneous minis, but no evoked synaptic transmission.

Whole-cell patch recordings from embryonic muscles 6 and 7 were performed in voltage-clamp in a modified HL-3 saline containing 1 mM Ca2+ (see Methods). a. Representative traces of spontaneous miniature junctional currents in wildtype, α2δ-3106/106 null and α2δ-3196/196 (“δ-less”) alleles. Average amplitude ± SEM in pA was 85.3 ± 0.6 for wt, 72.0 ± 0.5 for α2δ-3106/106, and 81.5 ± 1.1 for α2δ-3196/196 alleles; N (embryos) = 8 wt, 11 α2δ-3106/106, 4 α2δ-3196/196. Scale bar: 30 pA, 10 s. b. Representative individual minis. Scale bar: 30 pA, 20 ms. c. Mini amplitudes are normal in α2δ-3 mutants, but mini frequency is reduced in α2δ-3106/106 NMJs, from 0.11 ± 0.03 minis/s in wt to 0.04 ± 0.01 minis/s in α2δ-3106/106 nulls. α2δ-3196/196 alleles exhibited normal mini frequency (0.12 ± 0.06 minis/s. p < 0.01, ANOVA with Dunnett posthoc test. d. Overlayed representative evoked responses (10 traces) in 5 mM Ca2+. α2δ-3106/Df(2R)Ex7128 NMJs have no evoked response, while α2δ-3196/Df(2R)Ex7128 NMJs have greatly reduced responses. e. Average evoked responses were obtained from 4 (wildtype), 8 (α2δ-3106/Df(2R)Ex7128), and 4 (α2δ-3196/Df(2R)Ex7128) recordings from different embryos, and 10–40 stimuli per recording. Average amplitude ± SEM in pA was 802 ± 113 for wt, and 76 ± 23 for α2δ-3196/Df(2R)Ex7128; N (embryos) = 4 wt, 4 α2δ-3196/Df(2R)Ex7128. Scale bar = 250 pA, 10 ms in (d, e). Stimulus indicated by black bar above trace.
In contrast to the persistence of minis, however, stimulation of motor axons in α2δ-3106 null mutants never evoked a postsynaptic response. Because the average wildtype response was approximately 10 quanta/stimulus in 1 mM Ca2+, we estimate that the probability of release was reduced at least 1000-fold in the mutants. To maximize the likelihood of observing any residual nerve-evoked transmission, we recorded in a 5 mM Ca2+ saline, but nerve-evoked transmission was still undetectable (Fig. 6d,e). The inability to evoke release in these null mutants indicates the absence of cac α1 subunits from these terminals.
Unlike the α2δ-3106 allele, the α2δ-3196 “δ-less” allele had a small evoked response in the high Ca2+ saline (9% of wildtype; Fig. 6d,e), implying some Ca2+ channels remained in that allele. Overexpression of cacGFP rescues the embryonic lethality of the “δ-less” allele, but not of the null genotypes of α2δ-3, and the fluorescence of these GFP-tagged subunits can be detected at reduced levels at third-instar active zones of α2δ-3196 20. Thus the phenotypes of this allele are consistent with a few functional α1 subunits remaining at the synapse if the α2 peptide is present, even in the absence of the δ portion and its transmembrane domain.
α2δ’s synaptogenic function does not require the δ peptide
To gain additional insight into the developmental function of α2δ-3, we examined the other alleles of α2δ-3 that arose in our screen (Supplementary Fig. S4e online). In addition to the α2δ-3196 (Q989Stop, or “δ-less”) allele, they include two missense mutations (α2δ-3173, G226D; α2δ-3174, R234C) and a splice acceptor mutation (α2δ-3131). These alleles also died as embryos, but formed some synaptic boutons, although many of these boutons were morphologically abnormal (Supplementary Fig. S4 online).
Notably, embryos homozygous for α2δ-3196 had the mildest anatomical phenotype, with normally shaped boutons (Fig. 7b) and ank2-XL (Fig. 7d). Because the stop codon in α2δ-3196 should prevent transcription of the δ peptide, it appears that proper synaptic morphogenesis requires only the α2 portion. To verify this result, we engineered a “delta-less” α2-only construct that was reintroduced into the α2δ-3 null mutant background. This α2 construct could rescue bouton formation (Fig. 7c), although the degree of rescue was less consistent and effective than rescue with full length α2δ.
Figure 7. Bouton formation does not require the δ subunit.

Embryonic NMJs on muscles 6, 7, 12, and 13, labeled with anti-HRP (unless otherwise indicated). The α2δ-3 null (α2δ-3106/Df(2R)Ex7128) (a) lacks boutons but they are present (arrows) in the “δ-less” allele (α2δ-3196/196) (b) and when an α2-only construct is expressed in the null mutant background (C155elav-gal4/+; α2δ-3106/Df(2R)Ex7128); UAS-HA-α2/+) (c). d. Ankyrin2-XL is present in the “δ-less” allele. Scale bar = 5 µm.
Physiological defects of α2δ-3 nulls resemble those of cac
The cac α1 subunit it is presumed to elicit transmitter release at the embryonic NMJ because cac alleles are embryonic lethal32, cac is present at the larval and embryonic NMJ (Fig. 5 and 34), and viable alleles of cac diminish transmission in third-instar larvae33, 35, 36. The hypothesis had not been demonstrated directly by recording from the embryonic NMJs of null alleles until recently37. To characterize their synaptic transmission and to verify that the cac null alleles are indeed functionally null, we recorded from cacL13HC129 (Fig. 8) and cacL1320–3 (data not shown), as homozygous or hemizygous embryos. Both alleles are late embryonic lethal, resemble a deficiency in complementation tests32, and can be rescued by neuronal expression of a cac transgene34. Moreover, cacL1320–3 has a frame-shifting 341 bp deletion in the 3rd homology domain (T. Littleton, personal communication). Both alleles had normal amplitude minis (Fig. 8a,b and data not shown) but severely reduced synaptic transmission. In 1 mM Ca2+ saline, no evoked response could be detected, compared to an average response of over 1 nA in wildtype embryos (Fig. 8c,d). This represents, at a minimum, an approximately 1000-fold decrease in release probability compared to control. This is consistent with cac serving as the sole α1 subunit at embryonic active zones, though other α1 subunits may be present elsewhere in the axons and endings (see Supplementary Fig. S3 online and 37).
Figure 8. Bouton formation does not require the cac α1 subunit.

a–e. Recordings from wildtype and cac null embryonic NMJs indicate that cac encodes the α1 subunit that mediates evoked transmitter release at embryonic active zones. a. Representative traces depicting spontaneous miniature junctional currents (minis) in wildtype and cacL1320-3 nulls. Scale bar: 30 pA, 10 s. b. Representative individual minis. Scale bar: 30 pA, 20 ms. Mini amplitude and frequency are similar between wildtype and cac nulls. c. Average evoked responses ± SEM in pA was 1051 ± 37 for wildtype in 1 mM Ca2+. No evoked responses are detectable in cac null NMJs. Recordings were obtained from 6 wildtype and 7 cacL13 embryos, with 10–40 stimuli per recording. d. Ten representative evoked responses in 1 mM Ca2+e–g. Synaptic boutons form at NMJs of cac null embryos. NMJs on muscles 6, 7, 12, and 13, labeled with anti-HRP or anti-ank2-XL as indicated. cac nulls (cacL1320–3) (e) develop normal boutons (arrows) and have normal ank2-XL staining (f). cacL1320–3; α2δ-3196/196 double mutants (g) also develop boutons (arrows), although their morphology is slightly abnormal. Scale bar = 5 µm. h. Localization of neuronally expressed cacGFP and HA-tagged α2δ-3. HA-α2δ-3 is not exclusively colocalized at active zones with cacGFP (genotype: C155elav-gal4, UAScacGFP;;UAS-HA-α2δ-3). Scale bar = 2 µm.
Synaptogenesis defects in α2δ-3 are not due to lack of cac
The absence of the cac α1 subunit at the NMJs of α2δ-3 null embryos raised the possibility that the morphological and cytoskeletal phenotype in α2δ-3 arises from a requirement for the cac α1 subunit in synaptic development. This could arise either through a need for Ca2+ entry in bouton formation or through a requirement for the α1 subunit to organize other synaptic components, as reported at mammalian NMJs4. We therefore examined synaptogenesis in both cac null alleles, cacL13HC129 and cacL1320–3. Unlike α2δ-3106, both cac null alleles developed boutons (Fig. 8e and data not shown), as others have observed35, and had normal ank2-XL localization (Fig. 8f). Thus the developmental defect of α2δ-3 cannot be attributed to its role in localizing α1 channel subunits to the active zone and the synaptogenic defect in α2δ-3 mutants is independent of its role in Ca2+ channel function or localization. In addition, double mutants of α2δ-3106 and cac nulls look indistinguishable from α2δ-3106 single mutants (data not shown), indicating that the complete absence of the cacophony channel has no modifying effect on the α2δ-3 mutant phenotype. Even double mutants of cac and α2δ-3196 are able to form boutons (Fig. 8g), indicating that the α2 subunit can still promote synaptic morphogenesis even in the absence of both the α1 and δ subunits. Moreover, UAS-HA-α2δ-3, when immunolocalized in boutons of third instar larvae, resided in bright puncta that generally did not overlap with cacGFP puncta (Fig. 8h), other active zone markers such as brp (Supplementary Fig. S5 online) or periactivezone markers such as fasII (Supplementary Fig. S5 online). Thus α2δ-3 localization is consistent with multiple functions for this protein, including functions that are neither at active zones nor in association with α1 subunits.
Discussion
The α2δ subunit of voltage-gated Ca2+ channels has been considered to play a largely modulatory role, subtly modifying Ca2+ channel biophysics and promoting cell surface expression through an interaction with the α1 subunit10. Here we have shown that α2δ-3 subunits in the nerve terminal also function in synaptic morphogenesis and cytoskeletal organization, and that this role is independent of their function in α1 subunit localization and physiology. This novel role for α2δ, and more specifically for the α2 portion, offers an attractive explanation for the presence of the large extracellular segment of this subunit, with protein-binding domains that are highly conserved between species and between α2δ isoforms. α2δ-3 is likely to be the primary presynaptic α2δ isoform mediating morphological development of the Drosophila NMJ, since null alleles have such a large effect on NMJ development and abolish all action-potential evoked transmission.
The severe effect of eliminating α2δ-3 on synapse morphology at the NMJ is in stark contrast to the modest effects on current amplitude and biophysical properties attributed to α2δ subunits in expression systems6, 38. Indeed, even the severity of the electrophysiological defect could not be fully anticipated from the phenomena described in expression systems. Whereas the absence of α2δ in oocytes or cell lines caused a 2–10 fold drop in current amplitude16, 21, 38–40, our null mutations had no residual evoked responses even in high Ca2+ saline, and no detectable GFP-tagged α1 subunits at active zones. The localization of functional α1 subunits at terminals is therefore absolutely dependent on the presence of α2δ. Appreciating the full impact of the α2δ subunit for synaptogenesis and channel biology therefore required analysis in vivo in the context of the cell biology of the synapse.
The NMJ of Drosophila shares many features with excitatory synapses of the vertebrate central nervous system, including glutamatergic transmission, discrete synaptic boutons, and graded, sub-threshold postsynaptic responses that undergo summation41. Because of these similarities, the development and plasticity of the fly NMJ have received intensive scrutiny and there are many reported mutants that alter the guidance of motor axons, the branching pattern of the axon on the muscle surface, or exhibit a change in the size, shape, or number of synaptic boutons. The α2δ-3 null mutants described here, however, stand out for the severity and penetrance of their defects. On all muscle fibers and in every animal, no synaptic boutons remain.
To our knowledge, only one other mutant shares this severe phenotype, the kinesin-3 mutant imac42. In imac mutants an axonal transport defect leads to the absence of synaptic vesicles from the nerve terminals in addition to the failure of bouton formation. In contrast, α2δ-3 mutant terminals contain functional synaptic vesicles, as assayed by immunocytochemistry, electron microscopy, and electrophysiology, demonstrating that bouton formation is independent of the presence or absence of synaptic vesicles. Instead, we have hypothesized that boutons fail to form in imac due to a failure to transport unknown factors that are critical for triggering the cytoskeletal changes involved in synaptic morphogenesis. While it is tempting to speculate that α2δ-3 is one of those components whose transport is dependent on imac, this seems unlikely because ank2-XL is present at imac endings but is lacking in α2δ-3 mutants. The interplay of the mechanisms revealed by the α2δ-3 and imac phenotypes will be of interest to explore in future studies.
The α2δ-3 null phenotype also demonstrates that the morphological transformation of a growth cone into a synaptic bouton is mechanistically separable from the formation of functional release sites and the alignment of pre- and post-synaptic components. Although no boutons formed in these mutants and no voltage-dependent Ca2+-channels were present at the active zones, active zones looked otherwise relatively normal by electron microscopy, with presynaptic densities, T-bars, and adjacent clusters of vesicles. The active zones contained functional vesicle release sites that were apposed to glutamate receptors clusters that gave rise to minis of normal amplitude. Besides the absence of action-potential evoked release, the only detected consequence that coincided with the absence of boutons was a decrease in active zone number and mini-frequency that was proportional to the decrease in surface area of the nerve-muscle contacts. The molecular signals that drive synapse formation on the level of individual active zones are thus distinct from those that cause the morphological changes necessary for bouton formation.
The morphological defect in our α2δ-3 null alleles led us to investigate whether a perturbation in the cytoskeleton could be the cause of this phenotype. The severe reduction in ankyrin2 staining and abnormal futsch staining in α2δ-3 null alleles indicated that cytoskeletal defects were indeed present. Two findings, however, argue against a direct causal relationship between the loss of Ankyrin2 and the failure to form boutons: imac mutants retain ankyrin2 at their endings but still fail to form boutons, and boutons are present in ank2 null mutants. Instead we hypothesize that α2δ-3 may have multiple effects on the cytoskeleton of the NMJ, that include but are not limited to, the loss of ankyrin2. Moreover, since ankyrin2 staining is normal in the α2δ-3196 allele that lacks the transmembrane domain, and can be partially rescued by neuronal expression of the entirely extracellular α2-only construct, it is unlikely that α2δ-3 interacts directly with ankyrin2. Instead we hypothesize that α2δ-3 may be interacting with an additional transmembrane protein that is responsible for localizing ankyrin to the endings. Two candidate proteins that might interact with ankyrin, L1 CAM neuroglian43 and fasII28, were investigated but were not mislocalized in α2δ-3 mutant NMJs and mutants in those genes formed boutons at the embryonic NMJ (data not shown). At present, therefore, the link between α2δ-3 and the ankyrin cytoskeleton remains unidentified.
There is precedent for a role of a Ca2+ channel subunit in synaptogenesis; the CaV2 mammalian α1 subunit contributes to the organization of the active zone by binding directly to the extracellular matrix protein lamininβ4. This interaction enhances synaptogenesis by clustering Ca2+ channel α1 subunits in vitro and thereby induces clusters of other synaptic markers; in vivo the interaction appeared to promote the maintenance of active zones. The α2-dependent phenotype reported here suggests that a different mechanism, independent of active zone formation, may be responsible for morphological maturation.
One unexpected conclusion from our studies is that the morphological effect of α2δ-3 is not mediated by the absence of α1 subunits from the active zone, because the α1 mutant cacophony had normal bouton morphogenesis. Our studies indicated that transmitter release at the embryonic NMJ is mediated by the CaV2 channel encoded by cacophony, consistent with previous studies of fly NMJs. 33, 35, 36 20, 24, 34, 37. L-type α1 subunits may also be present in endings, but are more distant from the release sites (Supplementary Fig. S3 online). Thus, although the phenotype of the α2δ-3 nulls indicates that this subunit is needed both for the synaptic localization of the cac α1 subunit and for the morphological development of the NMJ, the latter function does not depend on the former; the morphological role of the α2δ protein can be carried out in the complete absence of cac and is thus independent of its conventional role as an auxiliary channel subunit. Moreover, the cac subunit cannot be required as either a tether or signal transducer for the α2δ-3 subunit. The α2 portion of the subunit was shown to be sufficient for bouton morphogenesis by analysis of the allele α2δ-3196, in which a stop codon occurs just before the putative δ-peptide cleavage site, and by reintroducing an α2 construct into the null allele. Because the δ peptide promotes the association of α2 with α116, this further supports the independence of the morphogenic function from channel regulatory functions.
If α2δ-3 influences synapse development independently of its function as a Ca2+-channel subunit, it may do so either by serving as a ligand or a receptor on the cell surface, or by acting as a chaperone that promotes the localization of a signaling protein to the synaptic membrane. The latter hypothesis invokes a function that would be parallel to the known ability of α2δ-3 to promote α1 subunit localization. On the other hand, the α2 portion of the subunit has been shown by rotary shadow electron microscopy of the channel complex to protrude extensively into the extracellular space3 and contains several protein domains that may play a role in signaling with extracellular ligands. These include a VWFa domain, which is a well-studied protein-protein interaction domain occurring fairly commonly in adhesive proteins and receptors12, 44, and a cache domain with homology to bacterial receptors13. If α2δ-3 functions as a receptor that induces the nerve terminal to mature, the δ subunit cannot be responsible for the transmembrane transduction of a signal as it is dispensable for α2 function in synaptic development. Even in combination with cac null alleles, boutons formed in α2δ-3196 embryos. Since α2 does not have a transmembrane domain of its own, this suggests that an as yet unknown mechanism, possibly a co-receptor if α2 is acting as a receptor, must exist for tethering α2 to the neuronal surface and for transducing its influence to the intracellular apparatus of the nerve terminal that produces synaptic maturation.
Morphological defects also occur in Purkinje cells of mice lacking α2δ-223 and mice lacking α2δ-4 have reduced retinal outer plexiform layers25, suggesting a defect in synapse formation that may parallel what we have observed in α2δ-3 mutants. Thus the crucial developmental function of α2δ described at the fly NMJ may well be representative of a widespread role for this protein that transcends its conventional function as an accessory subunit of Ca2+ channels.
Methods
Drosophila stocks and genetics
α2δ-3 alleles were isolated from a previously described forward genetic screen20, 48. The FRT42D chromosome49 used in the screen, the elav-GAL4, mhc-GAL4, and daughterless-GAL4 drivers, Df(2R)Ex7128, DfBSC9, and all other stocks were obtained from the Bloomington Stock Center (Bloomington, IN) unless otherwise noted. cacL13HC129 and cacL1320–3 null alleles, as well as the UAS-cacGFP transgenic flies34 were generously provided by R. Ordway (Penn. State). Dmca1DX10 flies were generous donated by L. Hall (Functional Insect Genomics Institute). Unless otherwise indicated, “wildtype” refers to the y,w; FRT42D stock, which is the genetic background of our α2δ-3 alleles.
Generation of UAS-HA-α2δ-3 and UAS-HA-α2 transgenic flies
An expressed sequence tag construct (SD03196) containing the entire open reading frame of α2δ-3 cDNA was obtained from Invitrogen (Carlsbad, CA). A UAS-α2δ3 construct with an internal HA-tag (at amino acid 246) was cloned into pUASTattB vector (kindly provided by Dr. K. Basler, University of Zurich) using standard molecular biology techniques. This construct was then injected into the fly line 9732 (PBac{yellow[+]-attP-9A}VK00013, 76A2) to create transgenic flies using the FlyC31 site-directed recombination system50 at BestGene, Inc.
Immunocytochemistry
21hr embryos were dechorionated in 50% bleach and dissected on Sylgard slides with Nexaband glue (Abbot Laboratories) in ice cold PBS (or HL-3 for cacGFP imaging), fixed Bouin’s fixative for 5 min (or 4% paraformaldehyde for 20 min for cacGFP and anti-HA), stained, and mounted in Vectashield (Vector Laboratories, Burlingame, CA). 14h AEL embryos were dissected on polylysine-covered slides and otherwise treated the same. Antibody staining was performed in PBS, containing 0.1% Triton-X-100 and 5% normal goat or donkey serum. The following primary and secondary antibodies were used: mouse anti-brp (1:100; Dr. E. Buchner, Germany and the Developmental Studies Hybridoma Bank, DSHB; Wagh et al., 2006); rabbit anti-Ank2XL (1:1000; H. Aberle, Universität Münster); mouse anti-Futsch (22C10, 1:100; DSHB); mouse anti–discs large (4F3, 1:1000; DSHB); mouse anti-FasII (1D4, 1:20; DSHB); rabbit anti-synaptotagmin-I (1:4000; N. Reist, Colorado State University); rabbit anti-GluRIIC (1:2000; A. DiAntonio, Washington University); rat anti-HA (1:100; Roche); FITC- or Cy3-conjugated secondary antibodies (1:100; Jackson ImmonuResearch, West Grove, PA); Cy5-, Cy3-, or FITC-conjugated anti-HRP (1:100; Jackson ImmunoResearch). Embryos were imaged with an LSM 510 laser scanning confocal microscope (Zeiss, Oberkochen, Germany) and 63× 1.4 NA objective using separate channels and processed using the LSM software or Adobe Photoshop (San Jose, California). For immunoreactivity intensity quantification (Fig. 3), embryos were stained in parallel and images were collected at the same gain settings. The hrp image was thresholded and a mask was created around the NMJ that was transferred to the other channels and used for intensity measurements. Background intensity was subtracted and the total intensity for each channel was normalized to neuronal area and is shown as a percentage of wt.
Electrophysiology
21 hr embryos grown at 25°C were dechorionated in 50% bleach and dissected in modified Ca2+-free HL-3 saline with the following ionic concentrations in mM: NaCl 70, KCl 5, MgCl2 5 (for evoked responses) or 10 (for mini analysis), NaHCO3 5, sucrose 115, Trehalose 5, HEPES (pH 7.2) 5. Dissected embryos were treated with 1 mg/ml Collagenase IV (Sigma) for 1 min. Evoked responses were measured in either 1 mM or 5 mM CaCl2 and 5 mM MgCl2. When used, the L-type Ca2+ channel blocker diltiazem (Sigma) was added at a concentration of 1 mM to recording solution containing 5 mM CaCl2 and 5 mM MgCl2. Recording electrodes of 3–8 ohm resistance were used to patch onto muscle 6 or 7 in abdominal segments A2 through A4. Recording electrodes were filled with an internal solution of, in mM: NaCl 1, KCl 155, MgCl2 1, HEPES (pH 7.2) 10, ATP 1. For evoked responses, the nerve was sucked into a stimulating electrode (filled with bath solution), and stimulated 10 times at 0.1 Hz using a 2 ms stimulus. For wildtype embryos, the stimulus strength was slowly raised until a response was elicited, and then raised slightly higher to ensure stimulation of both motor neuron axons. For mutants, stimulus intensities of 2–5 times that of wildtype were used. Data were collected using a Digidata and Axopatch 200B amplifier. pClamp 9 (Molecular Devices, Union City, CA), miniAnalysis (Synaptosoft, Decatur, GA) and Igor Pro (Wavemetrics, Portland, OR) were used for data analysis.
Electron Microscopy
21 hr embryos grown at 25°C were dechorionated in 50% bleach and either dissected in 0.1 M cacodylate buffer (pH 7.4) and then fixed overnight at 4°C or injected directly with fixative (5% glutaraldehyde, 2.5% paraformaldehyde, 0.06% picric acid in a 0.1 M cacodylate buffer, pH 7.4). After injection, the anterior and posterior tips of the embryos were cut off, and embryos were placed in fixative overnight at 4°C. After several washes in 0.1 M cacodylate washing buffer, embryos were incubated in 1% osmium tetroxide (in washing buffer) for 1h at RT, washed, and then stained with 2% uranyl acetate for 0.5h at RT. Embryos were dehydrated with sequential ethanol solutions (10 min each at RT), culminating with 2× 15 min in propylene oxide and an overnight incubation in 1:1 propyleneoxide/Araldite resin. Embryos were embedded in Araldite/Embed812 resin (Electron Microscopy Sciences) and sections were cut either in cross-section (for injected embryos) or parallel to the muscle field (for dissected embryos) and stained with 0.2% lead citrate and were viewed using a Tecnai G2 Spirit BioTWIN microscope and imaged with an AMT 2k CCD camera. In parallel sections of wildtype muscle, a string of rounded boutons could sometimes be seen within a section (Supplementary Fig. S2 online). Only parallel sections from dissected embryos were used for quantification. The following measurements were taken of each motor neuron profile found on the muscle surface: total membrane length; diameter measured by a line perpendicular to the edge of the muscle at the widest part of the neuron profile; total active zone length; number of active zones; number of T-bars.
Supplementary Material
Acknowledgements
This work was supported by National Institutes of Health Grants RO1 NS041062 and MH075058 (T.L.S.) and National Defense Science and Engineering Graduate Fellowship (P.T.K.). We thank H. Kazama for assistance with the electrophysiology; R. Ordway, J. T. Littleton, L. Hall, H. Aberle, K. Basler and the Bloomington Stock Center for stocks and reagents; D. Featherstone, T. Littleton, N. Reese and A. Goldstein for helpful discussions; and L. Bu and M. Liana of the MRDDRC Imaging and Histology Cores, the Harvard Medical School EM facility, and E. Pogoda for assistance.
Footnotes
Author Contributions
P.T.K performed the experiments. A.O.M. designed and generated the HA-tagged α2δ-3 construct, collaborated in the design of experiments involving that construct, and assisted with manuscript editing. P.T.K. and T.L.S. designed experiments and wrote the paper.
References
- 1.Lardi-Studler B, Fritschy JM. Matching of pre- and postsynaptic specializations during synaptogenesis. Neuroscientist. 2007;13:115–126. doi: 10.1177/1073858406296803. [DOI] [PubMed] [Google Scholar]
- 2.Robitaille R, Adler EM, Charlton MP. Strategic location of calcium channels at transmitter release sites of frog neuromuscular synapses. Neuron. 1990;5:773–779. doi: 10.1016/0896-6273(90)90336-e. [DOI] [PubMed] [Google Scholar]
- 3.Wolf M, Eberhart A, Glossmann H, Striessnig J, Grigorieff N. Visualization of the domain structure of an L-type Ca2+ channel using electron cryo-microscopy. J Mol Biol. 2003;332:171–182. doi: 10.1016/s0022-2836(03)00899-4. [DOI] [PubMed] [Google Scholar]
- 4.Nishimune H, Sanes JR, Carlson SS. A synaptic laminin-calcium channel interaction organizes active zones in motor nerve terminals. Nature. 2004;432:580–587. doi: 10.1038/nature03112. [DOI] [PubMed] [Google Scholar]
- 5.De Waard M, Gurnett CA, Campbell KP. Structural and functional diversity of voltage-activated calcium channels. Ion Channels. 1996;4:41–87. doi: 10.1007/978-1-4899-1775-1_2. [DOI] [PubMed] [Google Scholar]
- 6.Arikkath J, Campbell KP. Auxiliary subunits: essential components of the voltage-gated calcium channel complex. Curr Opin Neurobiol. 2003;13:298–307. doi: 10.1016/s0959-4388(03)00066-7. [DOI] [PubMed] [Google Scholar]
- 7.Dolphin AC. Beta subunits of voltage-gated calcium channels. J Bioenerg Biomembr. 2003;35:599–620. doi: 10.1023/b:jobb.0000008026.37790.5a. [DOI] [PubMed] [Google Scholar]
- 8.Littleton JT, Ganetzky B. Ion channels and synaptic organization: analysis of the Drosophila genome. Neuron. 2000;26:35–43. doi: 10.1016/s0896-6273(00)81135-6. [DOI] [PubMed] [Google Scholar]
- 9.Qin N, Yagel S, Momplaisir ML, Codd EE, D’Andrea MR. Molecular cloning and characterization of the human voltage-gated calcium channel alpha(2)delta-4 subunit. Mol Pharmacol. 2002;62:485–496. doi: 10.1124/mol.62.3.485. [DOI] [PubMed] [Google Scholar]
- 10.Klugbauer N, Marais E, Hofmann F. Calcium channel alpha2delta subunits: differential expression, function, and drug binding. J Bioenerg Biomembr. 2003;35:639–647. doi: 10.1023/b:jobb.0000008028.41056.58. [DOI] [PubMed] [Google Scholar]
- 11.Jay SD, et al. Structural characterization of the dihydropyridine-sensitive calcium channel alpha 2-subunit and the associated delta peptides. J Biol Chem. 1991;266:3287–3293. [PubMed] [Google Scholar]
- 12.Whittaker CA, Hynes RO. Distribution and evolution of von Willebrand/integrin A domains: widely dispersed domains with roles in cell adhesion and elsewhere. Mol Biol Cell. 2002;13:3369–3387. doi: 10.1091/mbc.E02-05-0259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Anantharaman V, Aravind L. Cache - a signaling domain common to animal Ca(2+)-channel subunits and a class of prokaryotic chemotaxis receptors. Trends Biochem Sci. 2000;25:535–537. doi: 10.1016/s0968-0004(00)01672-8. [DOI] [PubMed] [Google Scholar]
- 14.Gurnett CA, Felix R, Campbell KP. Extracellular interaction of the voltage-dependent Ca2+ channel alpha2delta and alpha1 subunits. J Biol Chem. 1997;272:18508–18512. doi: 10.1074/jbc.272.29.18508. [DOI] [PubMed] [Google Scholar]
- 15.Canti C, et al. The metal-ion-dependent adhesion site in the Von Willebrand factor-A domain of alpha2delta subunits is key to trafficking voltage-gated Ca2+ channels. Proc Natl Acad Sci U S A. 2005;102:11230–11235. doi: 10.1073/pnas.0504183102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gurnett CA, De Waard M, Campbell KP. Dual function of the voltage-dependent Ca2+ channel alpha 2 delta subunit in current stimulation and subunit interaction. Neuron. 1996;16:431–440. doi: 10.1016/s0896-6273(00)80061-6. [DOI] [PubMed] [Google Scholar]
- 17.Wiser O, et al. The alpha 2/delta subunit of voltage sensitive Ca2+ channels is a single transmembrane extracellular protein which is involved in regulated secretion. FEBS Lett. 1996;379:15–20. doi: 10.1016/0014-5793(95)01475-6. [DOI] [PubMed] [Google Scholar]
- 18.Felix R. Molecular regulation of voltage-gated Ca2+ channels. J Recept Signal Transduct Res. 2005;25:57–71. doi: 10.1081/rrs-200068102. [DOI] [PubMed] [Google Scholar]
- 19.Bernstein GM, Jones OT. Kinetics of internalization and degradation of N-type voltage-gated calcium channels: Role of the alpha(2)/delta subunit. Cell Calcium. 2006 doi: 10.1016/j.ceca.2006.04.010. [DOI] [PubMed] [Google Scholar]
- 20.Dickman DK, Kurshan PT, Schwarz TL. Mutations in a Drosophila alpha2delta voltage-gated calcium channel subunit reveal a crucial synaptic function. J Neurosci. 2008;28:31–38. doi: 10.1523/JNEUROSCI.4498-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dolphin AC, et al. The effect of alpha2-delta and other accessory subunits on expression and properties of the calcium channel alpha1G. J Physiol. 1999;519(Pt 1):35–45. doi: 10.1111/j.1469-7793.1999.0035o.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Barclay J, et al. Ducky mouse phenotype of epilepsy and ataxia is associated with mutations in the Cacna2d2 gene and decreased calcium channel current in cerebellar Purkinje cells. J Neurosci. 2001;21:6095–6104. doi: 10.1523/JNEUROSCI.21-16-06095.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brodbeck J, et al. The ducky mutation in Cacna2d2 results in altered Purkinje cell morphology and is associated with the expression of a truncated alpha 2 delta-2 protein with abnormal function. J Biol Chem. 2002;277:7684–7693. doi: 10.1074/jbc.M109404200. [DOI] [PubMed] [Google Scholar]
- 24.Ly CV, Yao CK, Verstreken P, Ohyama T, Bellen HJ. straightjacket is required for the synaptic stabilization of cacophony, a voltage-gated calcium channel alpha1 subunit. J Cell Biol. 2008;181:157–170. doi: 10.1083/jcb.200712152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wycisk KA, et al. Structural and functional abnormalities of retinal ribbon synapses due to Cacna2d4 mutation. Invest Ophthalmol Vis Sci. 2006;47:3523–3530. doi: 10.1167/iovs.06-0271. [DOI] [PubMed] [Google Scholar]
- 26.Yoshihara M, Rheuben MB, Kidokoro Y. Transition from growth cone to functional motor nerve terminal in Drosophila embryos. J Neurosci. 1997;17:8408–8426. doi: 10.1523/JNEUROSCI.17-21-08408.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Roos J, Hummel T, Ng N, Klambt C, Davis GW. Drosophila Futsch regulates synaptic microtubule organization and is necessary for synaptic growth. Neuron. 2000;26:371–382. doi: 10.1016/s0896-6273(00)81170-8. [DOI] [PubMed] [Google Scholar]
- 28.Pielage J, et al. A presynaptic giant ankyrin stabilizes the NMJ through regulation of presynaptic microtubules and transsynaptic cell adhesion. Neuron. 2008;58:195–209. doi: 10.1016/j.neuron.2008.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Koch I, et al. Drosophila ankyrin 2 is required for synaptic stability. Neuron. 2008;58:210–222. doi: 10.1016/j.neuron.2008.03.019. [DOI] [PubMed] [Google Scholar]
- 30.Bennett V, Chen L. Ankyrins and cellular targeting of diverse membrane proteins to physiological sites. Curr Opin Cell Biol. 2001;13:61–67. doi: 10.1016/s0955-0674(00)00175-7. [DOI] [PubMed] [Google Scholar]
- 31.Wagh DA, et al. Bruchpilot, a protein with homology to ELKS/CAST, is required for structural integrity and function of synaptic active zones in Drosophila. Neuron. 2006;49:833–844. doi: 10.1016/j.neuron.2006.02.008. [DOI] [PubMed] [Google Scholar]
- 32.Smith LA, et al. A Drosophila calcium channel alpha1 subunit gene maps to a genetic locus associated with behavioral and visual defects. J Neurosci. 1996;16:7868–7879. doi: 10.1523/JNEUROSCI.16-24-07868.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kawasaki F, Felling R, Ordway RW. A temperature-sensitive paralytic mutant defines a primary synaptic calcium channel in Drosophila. J Neurosci. 2000;20:4885–4889. doi: 10.1523/JNEUROSCI.20-13-04885.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kawasaki F, Zou B, Xu X, Ordway RW. Active zone localization of presynaptic calcium channels encoded by the cacophony locus of Drosophila. J Neurosci. 2004;24:282–285. doi: 10.1523/JNEUROSCI.3553-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rieckhof GE, Yoshihara M, Guan Z, Littleton JT. Presynaptic N-type calcium channels regulate synaptic growth. J Biol Chem. 2003;278:41099–41108. doi: 10.1074/jbc.M306417200. [DOI] [PubMed] [Google Scholar]
- 36.Kuromi H, Honda A, Kidokoro Y. Ca2+ influx through distinct routes controls exocytosis and endocytosis at drosophila presynaptic terminals. Neuron. 2004;41:101–111. doi: 10.1016/s0896-6273(03)00815-8. [DOI] [PubMed] [Google Scholar]
- 37.Hou J, Tamura T, Kidokoro Y. Delayed synaptic transmission in Drosophila cacophonynull embryos. J Neurophysiol. 2008;100:2833–2842. doi: 10.1152/jn.90342.2008. [DOI] [PubMed] [Google Scholar]
- 38.Felix R, Gurnett CA, De Waard M, Campbell KP. Dissection of functional domains of the voltage-dependent Ca2+ channel alpha2delta subunit. J Neurosci. 1997;17:6884–6891. doi: 10.1523/JNEUROSCI.17-18-06884.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bangalore R, Mehrke G, Gingrich K, Hofmann F, Kass RS. Influence of L-type Ca channel alpha 2/delta-subunit on ionic and gating current in transiently transfected HEK 293 cells. Am J Physiol. 1996;270:H1521–H1528. doi: 10.1152/ajpheart.1996.270.5.H1521. [DOI] [PubMed] [Google Scholar]
- 40.Shistik E, Ivanina T, Puri T, Hosey M, Dascal N. Ca2+ current enhancement by alpha 2/delta and beta subunits in Xenopus oocytes: contribution of changes in channel gating and alpha 1 protein level. J Physiol. 1995;489(Pt 1):55–62. doi: 10.1113/jphysiol.1995.sp021029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ruiz-Canada C, Budnik V. Introduction on the use of the Drosophila embryonic/larval neuromuscular junction as a model system to study synapse development and function, and a brief summary of pathfinding and target recognition. Int Rev Neurobiol. 2006;75:1–31. doi: 10.1016/S0074-7742(06)75001-2. [DOI] [PubMed] [Google Scholar]
- 42.Pack-Chung E, Kurshan PT, Dickman DK, Schwarz TL. A Drosophila kinesin required for synaptic bouton formation and synaptic vesicle transport. Nat Neurosci. 2007;10:980–989. doi: 10.1038/nn1936. [DOI] [PubMed] [Google Scholar]
- 43.Hortsch M, et al. A conserved role for L1 as a transmembrane link between neuronal adhesion and membrane cytoskeleton assembly. Cell Adhes Commun. 1998;5:61–73. doi: 10.3109/15419069809005599. [DOI] [PubMed] [Google Scholar]
- 44.Bork P, Rohde K. More von Willebrand factor type A domains? Sequence similarities with malaria thrombospondin-related anonymous protein, dihydropyridine-sensitive calcium channel and inter-alpha-trypsin inhibitor. Biochem J. 1991;279(Pt 3):908–910. doi: 10.1042/bj2790908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Johansen J, Halpern ME, Johansen KM, Keshishian H. Stereotypic morphology of glutamatergic synapses on identified muscle cells of Drosophila larvae. J Neurosci. 1989;9:710–725. doi: 10.1523/JNEUROSCI.09-02-00710.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sink H, Whitington PM. Location and connectivity of abdominal motoneurons in the embryo and larva of Drosophila melanogaster. J Neurobiol. 1991;22:298–311. doi: 10.1002/neu.480220309. [DOI] [PubMed] [Google Scholar]
- 47.Van Vactor D, Sink H, Fambrough D, Tsoo R, Goodman CS. Genes that control neuromuscular specificity in Drosophila. Cell. 1993;73:1137–1153. doi: 10.1016/0092-8674(93)90643-5. [DOI] [PubMed] [Google Scholar]
- 48.Stowers RS, Schwarz TL. A genetic method for generating Drosophila eyes composed exclusively of mitotic clones of a single genotype. Genetics. 1999;152:1631–1639. doi: 10.1093/genetics/152.4.1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xu T, Rubin GM. Analysis of genetic mosaics in developing and adult Drosophila tissues. Development. 1993;117:1223–1237. doi: 10.1242/dev.117.4.1223. [DOI] [PubMed] [Google Scholar]
- 50.Bischof J, Maeda RK, Hediger M, Karch F, Basler K. An optimized transgenesis system for Drosophila using germ-line-specific phiC31 integrases. Proc Natl Acad Sci U S A. 2007;104:3312–3317. doi: 10.1073/pnas.0611511104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.