

Abstract
Objective
This study tested the hypothesis that valvular calcium deposition, pro-osteogenic signaling, and function can be altered in mice with advanced aortic valve disease.
Methods/Results
“Reversa” mice were placed on a Western-type diet for 12 months and screened for the presence of aortic valve stenosis. Mice with advanced valve disease were assigned to two groups: 1) continued progression for 2 months, and 2) “regression” for 2 months, in which lipid lowering was accomplished by a “genetic switch”. Control mice were normocholesterolemic for 14 months. Mice with advanced valve disease had massive valvular calcification that was associated with increases in bone morphogenetic protein signaling, Wnt/β-catenin signaling, and markers of osteoblast-like cell differentiation. Remarkably, reducing plasma lipids with a “genetic switch” dramatically reduced markers of pro-osteogenic signaling and significantly reduced valvular calcium deposition. Nevertheless, despite a marked reduction in valvular calcium deposition, valve function remained markedly impaired. Phospho-Smad2 levels and myofibroblast activation (indices of pro-fibrotic signaling) remained elevated.
Conclusion
Molecular processes that contribute to valvular calcification and osteogenesis remain remarkably labile during end stages of aortic valve stenosis. While reductions in valvular calcium deposition were not sufficient to improve valvular function in our animals, these findings demonstrate that aortic valve calcification is a remarkably dynamic process that can be modified therapeutically even in the presence of advanced aortic valve disease.
Background
Recently, there have been substantial advances in understanding pathobiological processes in the calcifying aortic valve. Valvular calcification, like vascular calcification, was once thought to be a passive, degenerative process. It is now clear, however, that osteoblast-like cells are present in the valves of most patients with aortic valve stenosis1-3, which suggests that calcium deposition in the valve is an active process.
Non-surgical treatments aimed at slowing or halting the progression of aortic valve stenosis have proven to be elusive. Studies in hypercholesterolemic animals have demonstrated that lipid lowering therapy can dramatically reduce osteogenic markers and calcium deposition in the valve4-6, and when initiated in early stages of valve disease, can halt the progression of aortic valve stenosis4. Three clinical trials which examined effects of lipid lowering on the progression of aortic valve stenosis have yielded negative results5-7. It is, however, difficult to interpret these findings in a biological context. Many cells in the aortic valve and vasculature express markers of terminally differentiated osteoblasts1-3, and it is unclear whether lipid lowering results in de-differentiation of these cells to a less osteogenic phenotype. Furthermore, while calcifying vascular smooth muscle cells are remarkably labile with regards to their osteogenic activity in vitro8-10, recent work in cultured valvular interstitial cells demonstrated that stiffening of the extracellular matrix associated with end stage aortic valvular stenosis may reduce the efficacy of anti-calcific treatments11.
The aim of the current study was to determine whether lipid lowering with a “genetic switch”12 would reduce pro-osteogenic signaling and calcium deposition in hypercholesterolemic mice with advanced aortic valve disease.
Methods
Animal model and protocol
At 6–8 weeks of age, “Reversa” (ldlr-/-/apoB100/100/mttpfl/fl/Mx1-Cre) littermates were assigned to either “control”, hypercholesterolemic, or regression/“reversed” groups, as described previously4. Control mice were given 4 injections of polyinosinic-polycytidylic acid (pI-pC, 225 μg, i.p.) at two-day intervals and maintained on a chow diet for 14 months. Progression mice were placed on a Western diet (Harlan Teklad #TD88137, 42% of calories from fat, 0.25% cholesterol) for 14 months. Regression/“reversed” mice were placed on a Western diet for 12 months, and then were given 4 injections of pI-pC (225 μg, i.p.), switched to a chow diet, and followed for an additional 2 months.
Evaluation of aortic valve function
Aortic valve function was evaluated as described previously. Briefly, mice were sedated with midazolam (0.15 mg subcutaneously); each mouse was cradled in the left lateral recumbent position while a 15-MHz linear-array probe was applied horizontally to the chest. The imaging probe was coupled to a Sonos 5500 imager (Philips Medical Systems, Bothell, Wash), generating 180–200 two-dimensional frames per second in both short- and long-axis left ventricular (LV) planes.Images of the aortic valve were acquired in M mode, at a nominal sampling rate of 1000 frames/second, with two-dimensional images used for guidance.
Blood lipids, oxidative stress, histology, and immunohistochemistry were examined as described previously11. Methods for image quantitation and immunofluorescent image background correction are described in the online supplement.
Results
Plasma lipid levels
Total plasma cholesterol was markedly elevated in hypercholesterolemic mice (571 ± 54 mg/dl) versus control mice (150 ± 10 mg/dl, p < 0.01). Lowering cholesterol levels with a genetic switch at 12 months significantly lowered cholesterol levels in the “reversed” group (200 ± 9, p< 0.01 versus hypercholesterolemic group).
Histological changes in aortic valve
In control mice, there were small amounts of calcium deposition in the aortic valve at 14 months (Figure 1). In hypercholesterolemic mice, calcium deposition in the valve cusps was dramatically increased (Figure 1). Remarkably, calcium deposition in the valve cusps was significantly reduced by lipid lowering for 2 months (Figure 1).
Figure 1.

Effects of reducing plasma lipids on valvular calcium deposition (Alizarin Red staining) in A) control mice (CTRL), B) hypercholesterolemic mice (HCHOL), and C) “reversed” mice (REV). Images show valve cusps extending into the lumen of the aorta—arrows highlight corresponding/similar anatomical regions of the valve cusp in each image. Note that there is massive calcification of the aortic valve in HCHOL mice, which can be markedly reduced in a relatively short period of time by reducing blood lipids. D) Quantitation of histological data. * = p < 0.05 versus control mice, † = p < 0.05 versus hypercholesterolemic mice.
Pro-calcific signaling in aortic valve
Levels of phospho-smad1/5/8 were relatively low in control mice, but were significantly elevated in hypercholesterolemic mice following 14 months of severe hypercholesterolemia (Figure 2A). Reducing plasma lipids for 2 months significantly reduced phospho-smad1/5/8 immunofluorescence in “reversed” mice (Figure 2A).
Figure 2.

Effects of reducing plasma lipids on pro-osteogenic signaling. Phospho-Smad1/5/8 immunofluorescence (indicative of bone morphogenetic protein signaling) in A) control mice (CTRL), B) hypercholesterolemic mice (HCHOL), and C) “reversed” mice (REV). D) Quantitation of phospho-Smad 1/5/8 immunofluorescent data. Beta-catenin immunofluorescence (indicative of Wnt signaling) in E) control mice, F) hypercholesterolemic mice, and G) “reversed” mice. H) Quantitation of beta-catenin immunofluorescent data. * = p < 0.05 versus control mice, † = p < 0.05 versus hypercholesterolemic mice. Images were acquired using a 60x objective and include only valvular tissue near the base/attachment of the valve.
Immunofluorescence of β-catenin was virtually undetectable in control mice (Figure 2B). β-catenin immunofluorescence was significantly increased in mice subjected to 14 months of severe hypercholesterolemia, and was significantly attenuated by reducing plasma lipids for 2 months (Figure 2B).
Immunofluorescence of the smad1/5/8 target gene CBFA1/Runx2 was very low in control mice, but was significantly increased in hypercholesterolemic mice at 14 months (Figure 3). Reducing plasma lipid levels for 2 months significantly attenuated immunofluorescence of CBFA1/Runx2 in the aortic valve cusps (Figure 3).
Figure 3.

Effects of reducing plasma lipids on Runx2, a key regulator of osteoblast differentiation in A) control mice, B) hypercholesterolemic mice, and C) “reversed” mice. D) Quantitation of histological data showing that changes in Runx2 levels parallel P-Smad1/5/8 and β-catenin (both of which can independently drive expression of Runx2). * = p < 0.05 versus control mice, † = p < 0.05 versus hypercholesterolemic mice. Images were acquired using a 60x objective and include only valvular tissue near the base/attachment of the valve.
Pro-fibrotic signaling in aortic valve
Myofibroblast activation (assessed by immunofluorescence of alpha smooth muscle actin (α-SMA)) was very low in control mice, but robustly increased in hypercholesterolemic mice at 14 months. Reducing plasma lipids for 2 months significantly reduced myofibroblast activation in “reversed” mice (Figure 4).
Figure 4.

Effects of reducing plasma lipids on pro-fibrotic/TGF-β signaling and myofibroblast activation. Phospho-Smad2 immunofluorescence (indicative of TGF-β signaling) in A) control mice, B) hypercholesterolemic mice, and C) “reversed” mice. D) Quantitation of phospho-Smad2 immunofluorescent data. Alpha smooth muscle actin levels (indicative of myofibroblast activation) in E) control mice (CTRL), F) hypercholesterolemic mice (HCHOL), and G) “reversed” mice (REV). H) Quantitation of α-SMA immunofluorescent data. * = p < 0.05 versus control mice, † = p < 0.05 versus hypercholesterolemic mice. Images were acquired using a 60x objective and include only valvular tissue near the base/attachment of the valve.
Levels of phospho-smad2/3 (an indicator of TGF-β signaling) were low in control mice, but significantly elevated in mice following 14 months of severe hypercholesterolemia. Reducing plasma lipids for 2 months, however, did not significantly alter phospho-Smad2/3 immunofluorescence (Figure 4).
Oxidative stress in the aortic valve
Superoxide levels were low in control mice at 14 months, and were significantly increased in hypercholesterolemic mice (Figure S1). Reducing plasma lipids for 2 months, however, did not significantly reduce superoxide levels in the aortic valve cusps (Figure S1).
Aortic valve function
Cusp separation distance in control mice was 0.84 ± 0.03 mm at 12 months of treatment, and did not change significantly from 12 to 14 months (Figure 5). Cusp separation distance was significantly reduced in mice after 12 months of hypercholesterolemia (0.67 ±0.02 mm), but did not decrease significantly following an additional two months of hypercholesterolemia (Figure 5). Cusp separation distance was not significantly altered by reducing blood lipids for two months (Figure 5).
Figure 5.
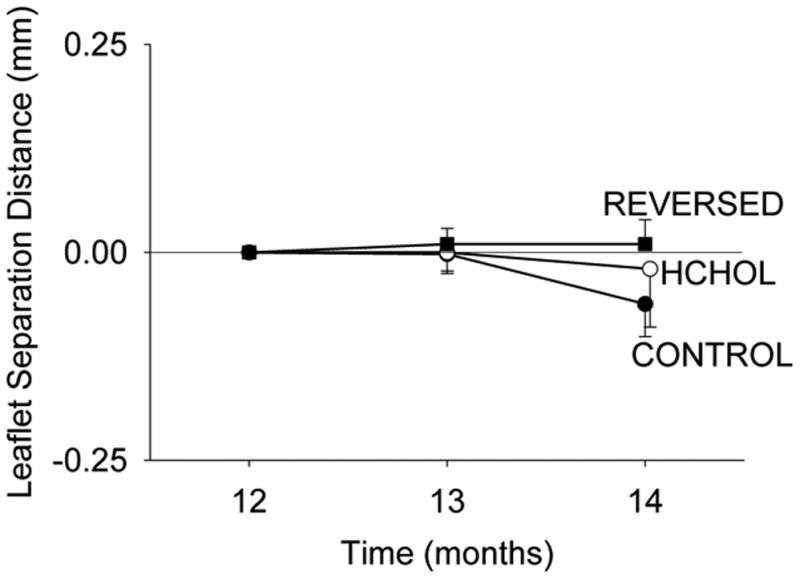
Effects of reducing plasma lipids on changes in aortic valve function. Aortic valve function is not improved by reduction of blood lipids, despite remarkable reduction of calcification of the valve. Absolute values of leaflet separation distance at 12 months are provided in the text.
Heart and lung weights
Compared to control mice, both heart weight and heart weight/body weight ratios were significantly increased after prolonged hypercholesterolemia (see Table 1). Reducing plasma lipids for two months did not significantly reduce heart or lung wet weight in the “reversed” group (see Table 1).
Table 1. Changes in body weight and cardiac weight.
| Control | Hypercholesterolemic | Reversed | |
|---|---|---|---|
| Body Weight (g) | 18.3 ± 0.4 | 22.0 ± 1.3 | 18.4 ± 2.0 |
| Heart Wet Weight (μg) | 106 ± 5 | 156 ± 15* | 193 ± 19* |
| Heart Wet Weight/Body Weight | 5.6 ± 0.2 | 7.2 ± 1 | 11.1 ± 1.8* |
Body weight, heart wet weight, and heart wet weight/body weight ratios in normocholesterolemic control mice, hypercholesterolemic mice, and “reversed” mice that had reduced blood lipids for 2 months (mean ± SE).
p < 0.05 versus normocholesterolemic control mice
Discussion
The major findings of this study are: 1) lipid lowering dramatically reduces levels of several pro-osteogenic proteins in hypercholesterolemic mice with advanced aortic valve disease, 2) lipid lowering significantly reduces calcium deposition in aortic valve of mice with advanced disease, and 3) despite biological and histological improvements in the aortic valve, lipid lowering did not improve aortic valve function or cardiac hypertrophy in hypercholesterolemic mice.
Pro-osteogenic signaling and valvular calcium deposition
After 14 months of hypercholesterolemia, we observed large increases in pro-osteogenic proteins in hypercholesterolemic mice. Importantly, we observed marked activation of both canonical bone morphogenetic protein signaling and Wnt/β-catenin signaling in hypercholesterolemic mice—two pathways that are consistently and robustly activated in human aortic valve stenosis12-14.
Increases in pro-calcific signaling were associated with massive calcification of the aortic valve cusps. Although we did not conduct a detailed analysis of the composition and ultrastructure of these calcium deposits, previous work examining tissue from humans with severe aortic valve stenosis identified matrix components that are virtually indistinguishable from bone, and often resemble endochondral ossification 13-15. Similar observations have been made in calcified arterial plaques of humans and mice16-21, suggesting that osteoblast-like cells in the cardiovascular system can drive the formation of bone-like tissue.
After activating a “genetic switch” and normalizing cholesterol levels from 12 to 14 months, pro-osteogenic signaling was markedly reduced in the aortic valve cusps. Remarkably, reductions in pro-osteogenic protein levels also were associated with significant reductions in valvular calcium deposition. These findings contrast with observations from humans with severe calcification of the coronary arteries22 or aortic valve7, 23, where lipid lowering therapy has a negligible impact on calcium burden. These findings are consistent, however, with previous observations in animals with early aortic valve disease4 and in primary prevention studies. It is not clear how much the aortic valve and atherosclerotic plaque differ with regards to resorption of calcium and regression of advanced lesions, or whether mice have a greater ability to resorb/regress ectopic calcium deposition compared to primates24.
Oxidative stress
After 14 months of hypercholesterolemia, we observed significant increases in superoxide levels in the aortic valve. This finding is consistent with previous observations from our group and others that superoxide levels are significantly increased during all stages of aortic valve stenosis2, 25, and there is no “burn-out” during terminal stages of the disease.
A surprising finding was that reduction of blood lipids for two months did not reduce superoxide levels in the aortic valve. This finding contrasts with our previous study, where we demonstrated that superoxide levels could be significantly reduced if lipid lowering was initiated during early stages of aortic valve disease4. This finding has two important implications. First, increased oxidative stress is not likely to be a primary stimulus for increased pro-calcific signaling in end stage stenosis, but may instead play an important role in amplification of pro-calcific signaling and matrix remodeling. This concept is supported in part from observations in calcifying vascular smooth muscle cells, where addition of exogenous oxidative stress accelerates calcium deposition and nodule formation only in the presence of calcifying media9, 26. Second, it is possible that sustained increases in valvular oxidative stress following normalization of plasma lipids in advanced aortic valve disease may be secondary to increased mechanical stresses/pressures placed on the valve27.
Aortic valve function
Despite its dramatic effects on pro-calcific signaling and calcium deposition in the valve, reducing cholesterol levels failed to improve aortic valve function in the present study. This observation is relevant to the principal findings of three recent clinical trials7-9, which demonstrated no benefit of statin therapy in patients with advanced aortic valve disease. There are several potential explanations for this observation. First, reductions in valvular calcium may not have passed a “threshold” for improving valve function, as calcium deposition in the “regression” mice remained higher than what we had previously observed4. Second, it is possible that valvular fibrosis plays an important role in determining valve function in advanced stages of disease. Our data demonstrating that TGF-β signaling and myofibroblast activation do not decrease following lipid lowering is consistent with the persistence of a pro-fibrotic environment in the aortic valve28. Although we did not assess changes in valvular fibrosis in the present study, we previously reported that valvular fibrosis does not improve following six months of lipid lowering; it is unlikely that fibrosis improves with this shorter period of time4. Finally, it is possible that stenotic aortic valves may reach a “point of no return”, at least in relation to lipid lowering, at which impairment in valvular function is sustained indefinitely, even after reversal of its initiating stimulus and amelioration of pro-osteogenic signaling.
Limitations
Although hypercholesterolemia is a risk factor for the development of aortic valve stenosis, there are other risk factors (including age) that have stronger associations with development of aortic valve stenosis. The experimental model is unique, however, because it captures two key aspects of human valve disease: 1) hemodynamically significant impairments in aortic valve function, and 2) activation of pro-osteogenic signaling cascades that is similar to what is observed in human disease. While there is likely to be context-dependent signaling for a number of pro-osteogenic signaling cascades (i.e., modulation by the presence/absence of hypercholesterolemia), this nevertheless serves as a powerful model to facilitate studies of development and treatment of aortic valve stenosis.
There are methodological limitations associated with the present investigation, many of which are inherent to working with mice. Because only a small amount of tissue is available from mouse aortic valves, we are unable to perform Western blotting to assess protein levels of pro-osteogenic signaling molecules. Also, the number of sections available for staining is small because the number of animals per group is limited by the extended duration of time needed to generate animals. Consequently, we were not able to determine whether osteoclasts were present in aortic valves during progression or regression of calcification. Nevertheless, the data are consistent with the hypothesis that calcium deposition in the aortic valve is an active and labile process.
Conclusions
The major findings of this study are that profound reductions in pro-osteogenic signaling and valvular calcium deposition can occur in mice with advanced aortic valve disease. The data suggest that aortic valve calcification in end-stage disease remains an active process that can be modified therapeutically. Thus, although lipid lowering per se does not improve aortic valve function, the finding that pro-osteogenic signaling and calcium deposition are labile even in advanced aortic valve stenosis implies that it may be feasible to improve valve function despite advanced aortic valve disease.
Supplementary Material
Figure I. Effects of reducing plasma lipids on superoxide levels in the aortic valve. Note that valvular superoxide is not altered by reduction of blood lipids. CTRL = control, HCHOL = high fat/hypercholesterolemic group, REV = “reversed” group.
Acknowledgments
These studies were supported by National Institutes of Health grants HL-092235, HL-62984, NS-24621, M058939, RR-017369, and by a Carver Research Program of Excellence.
References
- 1.Otto CM, Kuusisto J, Reichenbach DD, Gown AM, O'Brien KD. Characterization of the early lesion of ‘degenerative’ valvular aortic stenosis. Histological and immunohistochemical studies. Circulation. 1994;90:844–853. doi: 10.1161/01.cir.90.2.844. [DOI] [PubMed] [Google Scholar]
- 2.Miller JD, Chu Y, Brooks RM, Richenbacher WE, Pena-Silva R, Heistad DD. Dysregulation of antioxidant mechanisms contributes to increased oxidative stress in calcific aortic valvular stenosis in humans. Journal of the American College of Cardiology. 2008;52:843–850. doi: 10.1016/j.jacc.2008.05.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rajamannan NM, Subramaniam M, Rickard D, Stock SR, Donovan J, Springett M, Orszulak T, Fullerton DA, Tajik AJ, Bonow RO, Spelsberg T. Human aortic valve calcification is associated with an osteoblast phenotype. Circulation. 2003;107:2181–2184. doi: 10.1161/01.CIR.0000070591.21548.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Miller JD, Weiss RM, Serrano KM, Brooks RM, 2nd, Berry CJ, Zimmerman K, Young SG, Heistad DD. Lowering plasma cholesterol levels halts progression of aortic valve disease in mice. Circulation. 2009;119:2693–2701. doi: 10.1161/CIRCULATIONAHA.108.834614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chan KL, Teo K, Dumesnil JG, Ni A, Tam J. Effect of Lipid lowering with rosuvastatin on progression of aortic stenosis: results of the aortic stenosis progression observation: measuring effects of rosuvastatin (ASTRONOMER) trial. Circulation. 121:306–314. doi: 10.1161/CIRCULATIONAHA.109.900027. [DOI] [PubMed] [Google Scholar]
- 6.Rossebo AB, Pedersen TR, Boman K, Brudi P, Chambers JB, Egstrup K, Gerdts E, Gohlke-Barwolf C, Holme I, Kesaniemi YA, Malbecq W, Nienaber CA, Ray S, Skjaerpe T, Wachtell K, Willenheimer R. Intensive lipid lowering with simvastatin and ezetimibe in aortic stenosis. The New England journal of medicine. 2008;359:1343–1356. doi: 10.1056/NEJMoa0804602. [DOI] [PubMed] [Google Scholar]
- 7.Cowell SJ, Newby DE, Prescott RJ, Bloomfield P, Reid J, Northridge DB, Boon NA. A randomized trial of intensive lipid-lowering therapy in calcific aortic stenosis. The New England journal of medicine. 2005;352:2389–2397. doi: 10.1056/NEJMoa043876. [DOI] [PubMed] [Google Scholar]
- 8.Abedin M, Lim J, Tang TB, Park D, Demer LL, Tintut Y. N-3 fatty acids inhibit vascular calcification via the p38-mitogen-activated protein kinase and peroxisome proliferator-activated receptor-gamma pathways. Circulation research. 2006;98:727–729. doi: 10.1161/01.RES.0000216009.68958.e6. [DOI] [PubMed] [Google Scholar]
- 9.Byon CH, Javed A, Dai Q, Kappes JC, Clemens TL, Darley-Usmar VM, McDonald JM, Chen Y. Oxidative stress induces vascular calcification through modulation of the osteogenic transcription factor Runx2 by AKT signaling. The Journal of biological chemistry. 2008;283:15319–15327. doi: 10.1074/jbc.M800021200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kizu A, Shioi A, Jono S, Koyama H, Okuno Y, Nishizawa Y. Statins inhibit in vitro calcification of human vascular smooth muscle cells induced by inflammatory mediators. Journal of cellular biochemistry. 2004;93:1011–1019. doi: 10.1002/jcb.20207. [DOI] [PubMed] [Google Scholar]
- 11.Monzack EL, Gu X, Masters KS. Efficacy of simvastatin treatment of valvular interstitial cells varies with the extracellular environment. Arteriosclerosis, thrombosis, and vascular biology. 2009;29:246–253. doi: 10.1161/ATVBAHA.108.179218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lieu HD, Withycombe SK, Walker Q, Rong JX, Walzem RL, Wong JS, Hamilton RL, Fisher EA, Young SG. Eliminating atherogenesis in mice by switching off hepatic lipoprotein secretion. Circulation. 2003;107:1315–1321. doi: 10.1161/01.cir.0000054781.50889.0c. [DOI] [PubMed] [Google Scholar]
- 13.Ing SW, Mohler ER, Iii, Putt ME, Torigian D, Leonard MB. Correlates of valvular ossification in patients with aortic valve stenosis. Clinical and translational science. 2009;2:431–435. doi: 10.1111/j.1752-8062.2009.00168.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mohler ER, 3rd, Gannon F, Reynolds C, Zimmerman R, Keane MG, Kaplan FS. Bone formation and inflammation in cardiac valves. Circulation. 2001;103:1522–1528. doi: 10.1161/01.cir.103.11.1522. [DOI] [PubMed] [Google Scholar]
- 15.Steiner I, Kasparova P, Kohout A, Dominik J. Bone formation in cardiac valves: a histopathological study of 128 cases. Virchows Arch. 2007;450:653–657. doi: 10.1007/s00428-007-0430-7. [DOI] [PubMed] [Google Scholar]
- 16.Hunt JL, Fairman R, Mitchell ME, Carpenter JP, Golden M, Khalapyan T, Wolfe M, Neschis D, Milner R, Scoll B, Cusack A, Mohler ER., 3rd Bone formation in carotid plaques: a clinicopathological study. Stroke; a journal of cerebral circulation. 2002;33:1214–1219. doi: 10.1161/01.str.0000013741.41309.67. [DOI] [PubMed] [Google Scholar]
- 17.Pauli S, Lauwers P, Van Schil P, Hendriks J, Van den Brande F, Bogers JP, Van Marck E, Eyskens E. Lamellar bone formation in an atherosclerotic plaque of the carotid artery, with a review of histogenesis--a case report. Angiology. 2000;51:77–81. doi: 10.1177/000331970005100112. [DOI] [PubMed] [Google Scholar]
- 18.Bostrom K, Watson KE, Stanford WP, Demer LL. Atherosclerotic calcification: relation to developmental osteogenesis. The American journal of cardiology. 1995;75:88B–91B. doi: 10.1016/0002-9149(95)80020-s. [DOI] [PubMed] [Google Scholar]
- 19.Bostrom K, Watson KE, Horn S, Wortham C, Herman IM, Demer LL. Bone morphogenetic protein expression in human atherosclerotic lesions. The Journal of clinical investigation. 1993;91:1800–1809. doi: 10.1172/JCI116391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Duer MJ, Friscic T, Proudfoot D, Reid DG, Schoppet M, Shanahan CM, Skepper JN, Wise ER. Mineral surface in calcified plaque is like that of bone: further evidence for regulated mineralization. Arteriosclerosis, thrombosis, and vascular biology. 2008;28:2030–2034. doi: 10.1161/ATVBAHA.108.172387. [DOI] [PubMed] [Google Scholar]
- 21.Rattazzi M, Bennett BJ, Bea F, Kirk EA, Ricks JL, Speer M, Schwartz SM, Giachelli CM, Rosenfeld ME. Calcification of advanced atherosclerotic lesions in the innominate arteries of ApoE-deficient mice: potential role of chondrocyte-like cells. Arteriosclerosis, thrombosis, and vascular biology. 2005;25:1420–1425. doi: 10.1161/01.ATV.0000166600.58468.1b. [DOI] [PubMed] [Google Scholar]
- 22.Houslay ES, Cowell SJ, Prescott RJ, Reid J, Burton J, Northridge DB, Boon NA, Newby DE. Progressive coronary calcification despite intensive lipid-lowering treatment: a randomised controlled trial. Heart (British Cardiac Society) 2006;92:1207–1212. doi: 10.1136/hrt.2005.080929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dichtl W, Alber HF, Feuchtner GM, Hintringer F, Reinthaler M, Bartel T, Sussenbacher A, Grander W, Ulmer H, Pachinger O, Muller S. Prognosis and risk factors in patients with asymptomatic aortic stenosis and their modulation by atorvastatin (20 mg) The American journal of cardiology. 2008;102:743–748. doi: 10.1016/j.amjcard.2008.04.060. [DOI] [PubMed] [Google Scholar]
- 24.Stary HC. The development of calcium deposits in atherosclerotic lesions and their persistence after lipid regression. The American journal of cardiology. 2001;88:16E–19E. doi: 10.1016/s0002-9149(01)01713-1. [DOI] [PubMed] [Google Scholar]
- 25.Liberman M, Bassi E, Martinatti MK, Lario FC, Wosniak J, Jr, Pomerantzeff PM, Laurindo FR. Oxidant generation predominates around calcifying foci and enhances progression of aortic valve calcification. Arteriosclerosis, thrombosis, and vascular biology. 2008;28:463–470. doi: 10.1161/ATVBAHA.107.156745. [DOI] [PubMed] [Google Scholar]
- 26.Mody N, Parhami F, Sarafian TA, Demer LL. Oxidative stress modulates osteoblastic differentiation of vascular and bone cells. Free radical biology & medicine. 2001;31:509–519. doi: 10.1016/s0891-5849(01)00610-4. [DOI] [PubMed] [Google Scholar]
- 27.Vecchione C, Carnevale D, Di Pardo A, Gentile MT, Damato A, Cocozza G, Antenucci G, Mascio G, Bettarini U, Landolfi A, Iorio L, Maffei A, Lembo G. Pressure-induced vascular oxidative stress is mediated through activation of integrin-linked kinase 1/betaPIX/Rac-1 pathway. Hypertension. 2009;54:1028–1034. doi: 10.1161/HYPERTENSIONAHA.109.136572. [DOI] [PubMed] [Google Scholar]
- 28.Cushing MC, Mariner PD, Liao JT, Sims EA, Anseth KS. Fibroblast growth factor represses Smad-mediated myofibroblast activation in aortic valvular interstitial cells. Faseb J. 2008;22:1769–1777. doi: 10.1096/fj.07-087627. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure I. Effects of reducing plasma lipids on superoxide levels in the aortic valve. Note that valvular superoxide is not altered by reduction of blood lipids. CTRL = control, HCHOL = high fat/hypercholesterolemic group, REV = “reversed” group.