

Abstract
Background
Congenital heart defect (CHD) account for 25% of all human congenital abnormalities. However, very few CHD-causing genes have been identified so far. A promising approach for the identification of essential cardiac regulators whose mutations may be linked to human CHD, is the molecular and genetic analysis of heart development. With the use of a triple retinoic acid competitive antagonist (BMS189453) we previously developed a mouse model of congenital heart defects (81%), thymic abnormalities (98%) and neural tube defects (20%). D-TGA (D-transposition of great arteries) was the most prevalent cardiac defect observed (61%). Recently we were able to partially rescue this abnormal phenotype (CHD were reduced to 64.8%, p = 0.05), by oral administration of folic acid (FA). Now we have performed a microarray analysis in our mouse models to discover genes/transcripts potentially implicated in the pathogenesis of this CHD.
Results
We analysed mouse embryos (8.5 dpc) treated with BMS189453 alone and with BMS189453 plus folic acid (FA) by microarray and qRT-PCR. By selecting a fold change (FC) ≥ ± 1.5, we detected 447 genes that were differentially expressed in BMS-treated embryos vs. untreated control embryos, while 239 genes were differentially expressed in BMS-treated embryos whose mothers had also received FA supplementation vs. BMS-treated embryos. On the basis of microarray and qRT-PCR results, we further analysed the Hif1α gene. In fact Hif1α is down-regulated in BMS-treated embryos vs. untreated controls (FCmicro = -1.79; FCqRT-PCR = -1.76; p = 0.005) and its expression level is increased in BMS+FA-treated embryos compared to BMS-treated embryos (FCmicro = +1.17; FCqRT-PCR = +1.28: p = 0.005). Immunofluorescence experiments confirmed the under-expression of Hif1α protein in BMS-treated embryos compared to untreated and BMS+FA-treated embryos and, moreover, we demonstrated that at 8.5 dpc, Hif1α is mainly expressed in the embryo heart region.
Conclusions
We propose that Hif1α down-regulation in response to blocking retinoic acid binding may contribute to the development of cardiac defects in mouse newborns. In line with our hypothesis, when Hif1α expression level is restored (by supplementation of folic acid), a decrement of CHD is found. To the best of our knowledge, this is the first report that links retinoic acid metabolism to Hif1α regulation and the development of D-TGA.
Background
Congenital heart defects affect 1-2% of newborns and are the leading cause of death in infants under one year of age [1]. While the overwhelming majority of congenital heart malformations do not segregate in Mendelian ratios, they do show familial aggregation, which suggests that genetic factors play a role in their development [2,3]. Despite this, a limited number of CHD-causing genes have been identified so far [4].
Isolated D-Transposition of great arteries (D-TGA, OMIM 608808) accounts for 5% of all congenital heart diseases [5]. Its incidence is estimated at 1 in 3,500-5,000 live births [6]. Most D-TGA cases are sporadic, but familial cases have also been reported [7]. A discrete number of causing genes have been identified so far (ZIC3, CFC1, THRAP2, GDF1, NODAL), but their mutation explains only a minority of cases [8-13]. Interestingly, many of these genes participate in embryonic left-right axis patterning [14]. Moreover, D-TGA has been observed to be frequently related to laterality defects (failure to establish a normal left-right asymmetry during embryonic development), in particular, in patients with asplenia/right isomerism. Conversely, one of the most prevalent types of CHD in lateralisation defects is D-TGA [15].
Transcriptome analysis using DNA microarrays has become a standard approach for investigating the molecular basis of human disease in both clinical and experimental settings, as the pattern of transcriptional deregulation may provide insights into the cause of abnormal phenotypes, including congenital defects [16-20].
In the present study we have analysed the transcriptome of mouse embryos whose development was dramatically altered by temporarily blocking retinoic acid signalling and of embryos in which the abnormal developmental phenotype was rescued by a concomitant supplementation with folic acid [21,22].
We previously administered to pregnant mice BMS189453, a synthetic retinoic acid (RA) antagonist having good (82-98%) oral bioavailability in rats and monkeys [21]. BMS189453 binds, but does not activate, the α, β, and γ retinoid receptors [23]. Oral administration of BMS189453 to pregnant mice twice, at 7.25/7.75 dpc (days post coitum), induces cardiac defects (81%), thymic abnormalities (98%) and neural tube defects (20%) at birth [21]. Concomitant oral supplementation with FA, during pregnancy, partially rescues this abnormal phenotype [22]. In particular, FA reduces congenital heart diseases from 81.3% to 64.8%, neural tube defects from 20.3% to 3.7% and thymic abnormalities from 98.4% to 27.8%, restoring a normal number of differentiated thymic cells [22].
To better identify genes/transcripts involved in the pathogenesis of the congenital defects observed in our mouse models, we performed a global microarray analysis on embryos. To identify the best developmental stage for microarray screening, we first analysed the gene expression pattern of Rarα, a retinoic acid responsive gene in mouse embryos, at 8.5, 9.5 and 11.5 dpc. At 8.5 dpc, all embryos analysed showed down-regulation of Rarα mRNA, compared to only 70% of the embryos at 9.5 dpc and 50% of embryos at 11.5 dpc (data not shown). Thus, we thus decided to analyse the gene expression pattern in 8.5 dpc embryos.
The data presented in this paper reveal that changes in the expression level of Hif1α (hypoxia-inducible factor 1 alpha subunit) during mouse embryogenesis are associated with CHD observed in our mouse models.
Methods
BMS-189453 and folic acid treatment protocol and embryo recovery
Outbred CD1, Swiss mice (Charles River, Calco, Italy) were housed and mated under standard laboratory conditions that conform to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996); an Italian ministerial authorization (DL 116/92) was obtained to carry out the experimental treatment protocols. During the experimental period, all animals had free access to water and a conventional laboratory diet (Standard diet n.48) until sacrifice. Room temperature was kept at 21 ± 2°C and 12 h of light was automatically alternated with 12 h of darkness.
Pregnant mice were randomly divided into three experimental groups of 6 mice each (untreated controls, BMS-treated and BMS+FA-treated group). Administration of BMS189453 (Bristol-Myers-Squibb, Princeton, NJ, USA) both alone and together with folic acid to pregnant mice was performed as described [21,22].
Mouse embryos were collected at 8.5 dpc, and placentas were removed from embryonic tissue. Three to five embryos were pooled within each litter, and stored in RNAlater (Ambion) until RNA extraction.
RNA extraction, preparation and hybridisation of cDNA probe
Total RNA was extracted and purified using TriZol reagent (Invitrogen), and its quality and quantity was assessed using a Nanodrop spectrophotometer (Thermo Scientific) and agarose gel electrophoresis.
Synthesis of the labelled first strand cDNA was conducted according to manufacturer's instructions (Superscript Indirect cDNA labelling system, Invitrogen, USA) with starting material of 10 μg of total RNA. Briefly, the amino-allyl labelled dNTP mix was added to the reaction to generate amino-allyl labelled second strand cDNA. Following the hydrolysis reaction, single-stranded cDNA probes were purified using a Purification Module (Invitrogen). Probe mixtures where then evaporated in a vacuum centrifuge, and the cDNA pellet resuspended in 3 μL of water. The dye-coupling reactions were performed by mixing the cDNA samples with AlexaFluor Dyes 555 or 647 and were incubated for overnight in the dark. The reactions were purified with a Purification Module (Invitrogen) to remove the unincorporated/quenched dyes. After the purification, samples were combined for hybridisation. The labelled cDNAs were co-hybridised to slides in duplicate with one dye swap.
Microarray slides contained approximately 33.000 oligonucleotides corresponding to the whole mouse genome (AECOM, USA, http://microarray1k.aecom.yu.edu/).
Image Analysis and Processing
Each slide was scanned on the GenePix 4000B Microarray Scanner at the optimal wavelength for the Alexa555 (F532) and Alexa647 (F635) dyes.
The spots were automatically segmented; total intensities as well as the fluorescence ratios of the two dyes for each spot were then calculated. The spots were flagged when they exhibited poor hybridisation signals and when they were saturated (F635 or F532 median = 65535). Spots with a signal to background ratio below 1.5 were filtered together with flagged spots. We decided to subtract the local spot background signal from the foreground signal depending on the correlation of foreground to background intensity ratios, as in the method suggested by Scharpf e al. [24].
We removed systematic bias in the data by applying lowess (smoother span 2/3) [25] and dye-swap normalisations to have the least possible information loss. Dye-swap normalisation makes use of reverse labelling in the two microarray replicates directly [26]. To establish the significance of the observed regulation for each gene, we used a one sample t-test and corrected the p-value for multiple comparisons controlling the false discovery rate [27]. We transformed FC < 1 in FC* = -1/FC. Finally, only genes with a satisfactory effect (Fold Change, FC≥ ± 1.5) were considered.
Validation of relative gene expression by real-time RT-PCR
Two μg of total RNA was reverse-transcribed into cDNA according to manufacturer's instructions (High-Capacity cDNA Archive Kit, Applied Biosystems, Foster City, CA USA). The expression levels of the selected genes and an internal reference (ribosomal 18S) were measured by multiplex PCR using Assay-on-Demand™ gene expression products (Applied Biosystems, Foster City, CA USA) labelled respectively with 6 carboxyfluorescein (FAM) (selected genes) and VIC (internal reference) (Applied Biosystems). We analysed the following genes: Hif1α (Mm00468869_m1), Mospd3 (Mm00551672_g1), Mgp (Mm00485009_m1), Sat1 (Mm01198850_m1), Canx (Mm00500330_m1), Tfpi (Mm00803534_m1), Rarα (Mm00436264_m1), Cited2 (Mm00516121_m1). We performed PCRs using the TaqMan Universal PCR Master Mix and the ABI PRISM 7000 Sequence Detection System. All samples were run in triplicate and average values were calculated. Each qRT-PCR experiment was repeated at least twice.
Relative quantification of gene expression among each sample was achieved by normalisation against ribosomal 18S endogenous control using the ΔΔCt method of quantification. The relative amount of mRNA was calculated as 2-ΔΔCt. Data are mean ± standard error of the mean (SEM). A one-way analysis of variance (ANOVA) and t-test were applied to look for significant differences between experimental conditions for each candidate gene. A p value < 0.05 was considered statistically significant. Calculations were performed using the 2.9.1 version of R software (http://www.r-project.org/).
Immunofluorescence analysis
Embryos at 8.5 dpc were fixed in 4% paraformaldehyde overnight at 4°C and processed for paraffin embedding and sectioning, following standard procedures. After blocking non-specific antibody binding in 10% normal donkey serum for 1 h at room temperature, 10-μm sections were incubated with antibodies against Hif1α (10 μg/ml; Novus Biologicals) and α-actinin (2 μg/ml; Abcam) overnight at 4°C. After several washes in PBS, sections were incubated with donkey anti-rabbit AlexaFluor488 or donkey anti-rat Alexa568 (2 μg/ml; Invitrogen), depending on the primary antibody used. Nuclei were counterstained with 0.5 μg/ml Hoechst 33258 in PBS. Slides were mounted with Möwiol and fluorescent images were taken with a Zeiss Axioplan2 microscope.
In silico search for RARE elements
We searched for retinoic acid response elements (RARE) in the aligned human and murine 5-Kb region upstream of the transcription start site of Hif1α gene. RAREs consist of direct repeats (DR) of two nucleotide motifs PuG(G/T)TCA usually separated by 5, 2 or 1 intervening nucleotides. However, few other forms of RARE have been characterised, with different half-site consensus sequences or with diverse spacer length [28,29].
The 5-kb human and murine regions were obtained from the UCSC Genome Bioinformatics Site (http://genome.ucsc.edu) and potential RARE sequences were searched using in-house written codes.
Results
Transcriptome analysis of BMS189453-treated embryos
We previously reported that BMS189453 oral administration to pregnant mice was responsible for congenital defects in newborns [21], while a concomitant FA supplementation during pregnancy partially rescued the abnormal phenotype [22].
In this study, we first analysed the transcriptome dysregulation induced by altering RA binding in 8.5 dpc mouse embryos using a comparative microarray approach. The expression data of all of the experiments are available as a specific GEO Sample record with accession number GSE19012 (http://www.ncbi.nlm.nih.gov/geo/).
After data normalisation 447 genes were differentially expressed in BMS-treated embryos vs. untreated control embryos (Additional file 1: Table S1). Among these genes, 276 were down-regulated while 171 were up-regulated. According to GO classification, the down-regulated genes code for proteins involved in the cell cycle and differentiation (9.0%), signal transduction (8.3%), cellular metabolism (7.6%) and protein metabolism (7.6%), while the up-regulated genes were mainly involved in transport (11.1%), cellular metabolism (10.5%) and protein metabolism (10.5%) (Figure 1A).
Figure 1.

Graphic illustration of GO classification of up- and down-regulated genes (FC≥ ± 1.5) in BMS-treated (A) and BMS+FA-treated embryos (B).
Comparison of the gene expression profiles of BMS-treated and BMS+FA-treated embryos
Next, we analysed the gene expression pattern of embryos exposed to BMS-189453 only with that of embryos exposed to FA supplementation as well. In BMS-treated embryos whose mothers had also received FA supplementation vs. BMS-treated ones, 239 genes were differentially expressed after data normalisation. Interestingly, among these 239 genes, 235 were down-regulated, while only four were up-regulated (Additional file 2: Table S2). According to GO classification, the down-regulated genes were mainly involved in protein metabolism (14%) and cellular metabolism (8%) while the four up-regulated genes code for proteins involved in signal transduction (Figure 1B).
We then compared the 447 differentially expressed genes from the first experiment (BMS-treated embryos vs. untreated ones) with those (239) of the second experiment (BMS+FA-treated vs. BMS-treated-embryos). A total of 140 genes were commonly expressed in both the experiments. Furthermore, we analysed the potential involvement of these 140 genes in the development of the heart region by a combination of extensive database mining and in silico analysis of gene expression (GenePaint database). We identified 44 genes that might be potentially relevant to mouse cardiogenesis (Table 1, Table 2).
Table 1.
List of the 26 genes that have similar expression patterns in BMS- and BM+FA-treated embryos.
| Gene symbol | Gene name | FC BMS/untreated controls |
FC BMS+FA/BMS |
GO classification |
|---|---|---|---|---|
| Tmem176b | transmembrane protein 176B | -2.58 | -1.08 | cellular differentiation |
| Pcsk5 | proprotein convertase subtilisin/kexin type 5 | -2.06 | -1.08 | cardiac development |
| Ankrd13c | ankyrin repeat domain 13c | -1.58 | -1.06 | unknown biological process |
| Slc7a15 | solute carrier family 7 member 15 | -2.23 | -1.12 | amino acid transporter |
| Canx | calnexin | -1.51 | -1.07 | protein folding |
| Rab11b | RAB11B, member RAS oncogene family | -1.60 | -1.31 | signal transduction |
| Ralbp1 | ralA binding protein 1 | -1.53 | -1.12 | signal transduction |
| Sat1 | spermidine/spermine N1-acetyl transferase1 | -1.62 | -2.28 | regulation of cell proliferation |
| Fn1 | fibronectin 1 | -1.58 | -1.24 | cell-matrix adhesion |
| Pcbp1 | poly(rC) binding protein 1 | -1.57 | -1.59 | mRNA processing |
| Naca | nascent polypeptide-associated complex alpha polypeptide | -1.64 | -1.70 | regulation of transcription |
| Rpl39 | ribosomal protein L39 | 1.71 | -2.10 | translation |
| Mospd3 | motile sperm domain containing 3 | -1.92 | -1.24 | cardiac development |
| Eif4a1 | eukaryotic translation initiation factor 4A1 | -1.56 | -1.19 | translation initiation factor activity |
| Rps15a | ribosomal protein S15a | -1.77 | -2.54 | translation |
| Gpr82 | G protein-coupled receptor 82 | -1.59 | -1.21 | G-protein coupled receptor protein signalling pathway |
| Eif5a | eukaryotic translation initiation factor 5A | -1.81 | -1.36 | translation initiation factor activity |
|
EG23 4159 |
predicted gene, EG234159 | -1.50 | -1.01 | unknown |
|
LOC63 3468 |
similar to H3 histone, family 3B | -1.77 | -1.27 | unknown |
| XM_920276 | / | -1.58 | -3.10 | unknown |
| XM_921367 | / | -4.86 | -1.00 | unknown |
|
OTTMUSG 0000000 4999 |
predicted gene, OTTMUSG 00000004999 |
-1.64 | -2.14 | unknown |
| XM_980000 | / | -1.96 | -2.42 | unknown |
| XM_990335 | / | -4.16 | -1.04 | unknown |
| XR_002657 | / | -2.11 | -6.27 | unknown |
Genes in bold have been analysed by qRT-PCR
Table 2.
List of the 18 genes that have different expression patterns in BMS- and BMS+FA-treated embryos.
| Gene symbol | Gene name | FC BMS/untreated controls |
FC BMS+FA/BMS | GO classification |
|---|---|---|---|---|
| Tfpi | tissue factor pathway inhibitor | -1.79 | +1.18 | coagulation |
| Mgp | Matrix Gla protein | +1.73 | -2. 77 | cell differentiation |
| Rragb | Ras-related GTP binding B | -1.589 | +1.11 | signal transduction |
| Hs3st6 | heparan sulphate (glucosamine) 3-O-sulfotransferase 6 | -1.55 | +1.40 | heparan sulphate synthesis |
| B2m | beta-2 microglobulin | -1.71 | +1.28 | immune response |
| Hif1α | hypoxia inducible factor 1, alpha subunit | -1.79 | +1.17 | cardiac looping |
| Rpn2 | ribophorin II | +1.78 | -1.59 | protein glycosylation |
| Rhoj | ras homolog gene family, member J | -1.67 | +1.30 | signal transduction |
| Spcs2 | signal peptidase complex subunit 2 homolog (S. cerevisiae) | +1.70 | 1.38 | signal peptide processing |
| Atp6v1b1 | ATPase, H+ transporting, lysosomal V1 subunit B1 | +2.08 | 1.01 | proton transport |
| Lmbrd2 | LMBR1 domain containing 2 | -1.88 | +1.16 | unknown biological process |
| Rnf217 | ring finger protein 217 | +2.05 | -1.26 | unknown biological process |
| Vmn2r52 | vomeronasal 2, receptor 52 | -1.67 | +1.26 | transmembrane receptor |
| D14Abb1e | DNA segment, Chr 14, Abbott 1 expressed | -1.70 | +1.38 | unknown |
| XM_981869 | +1.78 | -1.75 | unknown | |
| XM_991521 | +1.55 | -1.24 | unknown | |
| XM_994830 | -2.37 | +1.13 | unknown | |
| NM_133970 | -1.51 | +1.40 | unknown |
Genes in bold have been analysed by qRT-PCR.
qRT-PCR assays
Additionally, to confirm the expression pattern revealed by our microarray assays, we analysed six differentially expressed genes (Mgp, Tfpi, Hif1α, Mospd3, Canx, Sat1) by qRT-PCR (Table 1 and Table 2, in bold). We selected genes that had a similar expression pattern (up-regulated or down-regulated) in both embryos groups (Canx, Sat1, Mospd3; Table 1) and genes that showed a different expression pattern in both embryos groups (i.e. if down-regulated in the BMS-treated group, they were up-regulated in the BMS+FA-treated group, and vice-versa) (Mgp, Tfpi, Hif1α; Table 2). The housekeeping gene 18SRNA was used as an internal control.
The mRNA expression values of these selected genes in untreated controls, and BMS- and BMS+FA-treated embryos are represented in Figure 2A. We also evaluated the expression level of each gene in BMS+FA- vs. BMS-treated embryos (Figure 2B). As indicated in Figure 2, the qRT-PCR results are in very good agreement with the expression patterns of the microarray experiments (Table 1 and Table 2).
Figure 2.

Quantitative RT-PCR gene expression analysis in BMS treated and BMS+FA-treated embryos. Fold change in gene expression was analysed by the 2-ΔΔCt (see methods for details) of six selected genes in untreated controls, BMS- and BMS+FA-treated embryos (A) and in BMS+FA vs. BMS-treated embryos (B). Data are mean ± S.E.M., n = 6; * p < 0.05, ** p < 0.01, *** p < 0.0001.
Hif1α mRNA and protein are down-regulated in BMS-treated embryos, but their expression is recovered by supplementation with folic acid
Among the six genes analysed by qRT-PCR, Hif1α expression pattern was noteworthy; in fact, Hif1α was down-regulated in BMS-treated embryos (FCmicro = -1.79; FCqRT-PCR = -1.76, p = 0.005 Table 1, Figure 2A and 2B) but its mRNA level increased by about 73% in BMS+FA-treated embryos (FCmicro = +1.17; FCqRT-PCR = +1.28, p = 0.005; Table 1; Figure 2A and 2B).
To investigate whether changes in Hif1α mRNA expression are accompanied by variations at the protein level and to analyse Hif1α localisation in 8.5 dpc mouse embryos, an immunofluorescence analysis was performed on paraffin sections.
Hif1α is particularly expressed in the myocardium of untreated controls (Figure 3A') compared to the control gene α-actinin (Figure 3A''), while it is clearly under-expressed in BMS-treated embryos (Figure 3B') compared to the control gene (Figure 3B'') and compared to untreated controls (Figure 3A') and BMS+FA-treated animals (Figure 3C').
Figure 3.

Immunofluorescence results of Hif1α expression in untreated controls (A'), BMS- (B') and BMS+FA-treated (C') embryos at 8.5dpc. Transverse sections of untreated controls (A), BMS-treated (B) and BMS+FA-treated (C) embryos at 8.5 dpc were counterstained with Hoechst to show nuclei (blue). Positive Hif1α staining is evident in the myocardium of untreated controls (A') and BMS+FA-treated embryos (C'). Bc = bulbus cordis; Da = dorsal aorta; Ec = endocardium or endocardial tissue; Fd = foregut diverticulum; Lhsv = wall of the left horn of sinus venosus; Mc = myocardium; Nt = neural tube; Pv = primitive ventricle
In silico-identification of RARE elements in the promoter region of Hif1α
There is no evidence in the literature of a specific regulation of the expression of Hif1α by retinoic acid. Luo et al. (1997), who cloned and characterised the 2,000 bp region upstream of the ATG of mouse Hif1α, described its GC-rich promoter as typical of the so-called housekeeping genes [30].
To verify the presence of putative RAREs far from the ATG, we analysed the aligned mouse and human 5,000 bp sequences. Interestingly, we found numerously sequences with high homology to RAREs. Within a highly conserved region of 476 nt (Figure 4), we found a putative RARE at -2,177nt in the mouse promoter region (Figure 4, bottom sequence). This putative RARE is comprised of a direct repeat (DR) separated by a 2 bp (DR2) element (Figure 4, underlined). In the same highly conserved region of 476 nt, we found a motif resembling a DR3 in the human genomic region (Figure 4, top sequence, in grey).
Figure 4.

Alignment of a common 476 bp sequence in the human HIF1α (top sequence) and mouse Hif1α (bottom sequence) promoter region. The putative DR2 RARE element in the mouse promoter region is underlined, while the putative DR3 RARE element in the human promoter region is grey coloured.
Gene expression analysis of Hif1α downstream target genes
Hif1α gene encodes the alpha subunit of the heterodimeric transcription factor HIF-1 (Hypoxia Inducible Factor-1), which can promote or repress the transcription of a broad range of genes that are involved in maintaining biological homeostasis.
To test whether the observed expression pattern of Hif1α in our embryos is correlated with that of a HIF-1 target gene, Cited2, we performed qRT-PCR assays. We choose to analyse Cited2 for three reasons: 1. it was not present in our microarray slides; 2. it is an important negative regulator of Hif1α; 3. several papers demonstrated its role in cardiac looping and TGA [31]. As expected, Cited2 is down-regulated in BMS-treated embryos (FC = -1.94, p < 0.001), but it also remains down-regulated in BMS+FA-treated embryos (FC = -2.36, p < 0.001) (Figure 5).
Figure 5.
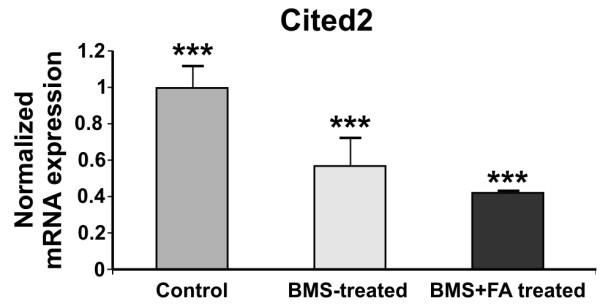
Cited2 expression in treated embryos. Fold change in gene expression analysed by the 2-ΔΔCt (see methods for details) of Cited2 in untreated controls, and BMS- and BMS+FA-treated embryos (8.5 dpc). Data are mean ± S.E.M., n = 6; *p < 0.05, **p < 0.01, ***p < 0.0001.
To assess the expression level of Hif1α and Cited2 during normal mouse development we analysed total RNA isolated from 6.5 to 18.5 dpc mouse embryos by qRT-PCR (Seegene Inc, Korea). Both these genes are widely expressed throughout embryonic development, with a higher level of Hif1α compared to Cited2 (Figure 6). Hif1α shows its highest expression between 7.5 and 8.5 dpc, while for Cited2, a peak is observed between 8.5 and 9.5 dpc (Figure 6). Because it is known that the stages from 7.5 to 11.5 dpc are relevant for normal cardiac development, these data further suggest the essential role of a correct dosage of Hif1α and its downstream targets, such as Cited2, for a normal morphogenesis of the heart.
Figure 6.

Hif1α and Cited2 mRNA expression pattern during normal mouse embryogenesis. Data are mean ± S.E.M., n = 3.
Discussion
Hif1α down-regulation might be responsible for the cardiac defects of mouse embryos
Heart defects are a common feature of congenital human syndromes, as they are present in 25% of all congenital abnormalities, suggesting that genes important in patterning the heart may also have a role in the development of other embryonic structures [1,3,4].
It is well known that a precisely regulated supply of RA is essential for normal cardiogenesis, because both an excess and a deficiency of RA (or vitamin A) have been found to cause teratogenic effects during early heart development [21,32-35].
We previously developed a mouse model for congenital defects, in particular cardiac and thymic abnormalities, by administration to pregnant mice of BMS-189453, an antagonist of retinoic acid that selectively binds to RAR receptors (α, β and γ), and blocks the intake of RA inside the cells. Mice born from pregnant females treated with BMS-189453 showed thymic abnormalities (98%), cardiac defects (81%, 61% of which were D-TGA) and neural tube defects (20%) [21]. These abnormal phenotypes could be partly rescued by oral administration of FA to the pregnant females exposed to BMS-189453 [22].
A comparative analysis of microarray gene expression patterns, identified Hif1α as a gene that is down-regulated in BMS-treated embryos and whose expression level is restored (73% vs untreated control) by FA administration. Microarray data were confirmed by qRT-PCR. In parallel, to further support the mRNA results, we used specific antibodies and immunofluorescence analysis, to evaluate Hif1α expression and localisation in mouse embryos at 8.5 dpc. We found that at this embryonic stage Hif1α is expressed in the myocardium (Figure 3A') and that, in line with our microarray and qRT-PCR observations, Hif1α was not detected in sections of BMS-exposed embryos (Figure 3B'), while untreated control (Figure 3A') and BMS+FA-treated embryos (Figure 3C') displayed comparable levels of expression.
Hif1α has an essential role in cardiovascular development, as mice lacking this transcription factor show cardia bifida and cardiac looping defects [36]. Moreover, it has been recently demonstrated that cardiac development in the mouse is characterised by a hypoxic environment with high levels of Hif1α protein [37]. In particular Hif1α is expressed in wild type hearts between E8.0 to E11.5; the highest levels were found at 8.5 and 9.5 dpc, with a decrement at 10.5 that became undetectable by 11.5dpc. In particular, the hypoxic environment in the developing heart initiates a HIF-1 mediated transcriptional program that facilitates the development of a functional heart [37]. These data confirm that Hif1α is not only expressed, but it is functional in the 8.5 dpc developing heart, which is the developmental stage we analysed in our mouse models.
Interestingly, Cited2, which is a negative Hif1α regulator and also a HIF-1 inducible gene [38], has an important role in heart development and morphogenesis, in particular, in controlling left-right patterning through a Nodal-Pitx2c pathway [39]. Intriguingly, the human homologue was found to be mutated in patients with D-TGA and heterotaxia [40]. Because Cited2 was not present in our microarray, we analysed its mRNA by qRT-PCR. As expected, Cited2 is down-regulated in BMS-treated embryos, but it is further down-regulated in BMS+FA-treated embryos (Figure 5). This last result might indicate that when Hif1α expression is raised by supplementation of FA, there is a time window necessary to recover Cited2 expression. This is reasonable because it is known that Cited2 and Hif1α participate to a unique regulatory feedback mechanism to limit excess HIF-1 activation and to maintain normal tissue homeostasis; in this regulatory mechanism, Cited2 dissociates p300 from Hif1α and represses Hif1α activity [41,42]. In line with this hypothesis, we found that the Hif1α expression pattern during mouse embryogenesis follows a trend sequentially to Cited2 (Figure 6). In fact, Hif1α highest expression levels are found at 7.5 and 8.5 dpc, while the highest Cited2 levels are at 8.5 and 9.5 dpc (Figure 6).
Finally, while an important role has been well established for HIFα in many human cancers, especially for those that are highly hypoxic [43], and in the pathophysiological responses to hypoxia in pulmonary hypertension and myocardial ischemia [44], the present are the first data linking Hif1α to human CHDs to the best of our knowledge.
What is the link between Hif1α and retinoic acid?
Both excess RA administration and vitamin A deficiency can disturb cardiac looping in embryos [32,33,45,46]. Moreover, it was demonstrated that treatment with retinoic acid induced D-TGA in mice [21,46]. Because it is also well known that Hif1α deficiency causes cardiac looping disturbance [36,37,47], it is reasonable to think that a alterations in the metabolism of retinoic acid might influence a correct dosage of Hif1α.
However, there is no evidence that RA directly regulates the expression of Hif1α, and in fact it was elegantly demonstrated that at E8.5, RA activity is present only in the posterior portion of the heart (atria and inflow tract), whereas Hif1α is expressed throughout the heart [48]. Moreover, in a different system, it was demonstrated that ATRA (all-trans retinoic acid) increases HIF1α protein levels [49]. By microarray and qRT-PCR assays, we found that Hif1α is down-regulated when RA signalling is inhibited; moreover, an in silico search performed on the 5,000 bp genomic region upstream the ATG revealed a conserved region of approximately 500 nt in both human and mouse Hif1α, containing a putative RARE element resembling a DR2 in the mouse promoter (top sequence, Figure 4). Because it is known that the heterodimer RXR/RAR interacts preferentially with DR2 and DR5 [50]; it would be interesting to verify whether this RARE sequence is functional.
What is the link between Hif1α and folic acid?
Folic acid is an essential vitamin for a wide spectrum of biochemical reactions involved in DNA and RNA metabolism. Impaired folate-dependent metabolism can lead to several pathologies including megaloblastic anaemia, cardiovascular diseases and neural tube and congenital heart defects [51]. While it is well known that maternal supplementation with folic acid during pregnancy lowers the risk of congenital birth defects, the molecular mechanism of action for this phenomenon is still unclear [51,52]. It has recently been shown that in the mouse, maternal folate deficiency significantly affects myocardial cell proliferation with no change in apoptosis levels [53].
Considering that Hif1α does not belong to the category of genes involved in FA metabolism, it is difficult to establish what relationship exists with FA. Due to the well-known ability of FA to affect DNA methylation [54], it may be speculated that FA might epigenetically regulate Hif1α expression.
Conclusions
A finely tuned regulation of gene expression during embryogenesis and development is crucial for a normal anatomy and physiology. Though very little is currently known regarding factors that influence and regulate developmental gene expression of the cardiovascular system, emerging large-scale technologies should be of great help to unravel the complex and highly regulated interplay of genes and cell-cell interactions in the developing heart by permitting a complete analysis of embryos transcriptomes [18,20,55].
In this paper, we have identified a discrete number of altered genes that might be involved in congenital defects in the mouse. These defects, in particular CHD and D-TGA, caused by a dysregulation of RA metabolism, were consistently rescued by exogenous administration of folic acid in vivo.
Among the altered genes, we have more extensively analysed the expression pattern of Hif1α. The down-regulation of Hif1α (both at the mRNA and protein levels) following blocking of retinoic acid intake in the developing mouse, its recovery after oral supplementation with folic acid and its localisation in the cardiac primordia suggest that the observed congenital heart malformation might be due to a de-regulation of Hif1α and its downstream targets (e.g. Cited2). Because alteration of both of these genes causes defects in left-right patterning of developing embryos, and the major cardiac defect in our mouse model was D-TGA, the present data support the suggestion to include D-TGA in the group of heterotaxy, a disorder characterised by abnormal lateralisation of normally asymmetric thoracic and abdominal organs.
Abbreviations
D-TGA: D-transposition of great arteries; FA: folic acid; dpc: days post coitum; FC: fold change; Hif1α: hypoxia inducible factor 1 alpha subunit; BMS-treated: BMS189453 treatment; BMS+FA-treated: BMS189453 treatment plus folic acid supplementation; RARE: retinoic acid responsive elements.
Authors' contributions
FA designed most of the experiments, coordinated the study and wrote the manuscript. LD performed microarray experiments and participated in the interpretation of the results and in writing part of the manuscript. LC carried out immunofluorescence experiments and participated in the interpretation of the results and in writing part of the manuscript. LV carried out qRT-PCRs and participated to the interpretation of the results. DC performed mouse treatments. SB, GP, GP, AD analysed microarray raw data, interpreted the results and wrote part of the manuscript. GS participated in the interpretation of immunofluorescence results and revised the manuscript. BM and GN together contributed to coordinate the study and edited the manuscript. All authors read and approved the final manuscript.
Supplementary Material
Table S1: List of the 477 differentially expressed genes (FC≥ ± 1.5) in BMS189453-treated embryos
Table S2: List of the 239 differentially expressed genes (FC≥ ± 1.5) in BMS+FA-treated embryos
Contributor Information
Francesca Amati, Email: amati@med.uniroma2.it.
Laura Diano, Email: laura.diano@gmail.com.
Luisa Campagnolo, Email: campagno@med.uniroma2.it.
Lucia Vecchione, Email: luciavecchione@yahoo.it.
Daria Cipollone, Email: idaria@libero.it.
Susana Bueno, Email: Susana.Bueno@caspur.it.
Gianluca Prosperini, Email: g.prosperini@caspur.it.
Alessandro Desideri, Email: desideri@bio.uniroma2.it.
Gregorio Siracusa, Email: g.siracusa@uniroma2.it.
Giovanni Chillemi, Email: chillemi@caspur.it.
Bruno Marino, Email: bruno.marino@uniroma1.it.
Giuseppe Novelli, Email: novelli@med.uniroma2.it.
Acknowledgements
We thank Graziano Bonelli for expert graphic assistance and Gabriele Rossi for technical assistance in immunofluorescence.
References
- Garg V. Insights into the genetic basis of congenital heart disease. Cell Mol Life Sci. 2006;63:1141–1148. doi: 10.1007/s00018-005-5532-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weismann CG, Gelb BD. The genetics of congenital heart disease: a review of recent developments. Curr Opin Cardiol. 2007;22:200–206. doi: 10.1097/HCO.0b013e3280f629c7. [DOI] [PubMed] [Google Scholar]
- Jing-Bin H Ying-Long L Pei-Wu S Xiao-Dong L Ming D Xiang-Ming F Molecular mechanisms of congenital heart disease Cardiovasc Pathol in press Corrected Proof, Available online 10 September 200919747853
- Nemer M. Genetic insights into normal and abnormal heart development. Cardiovasc Pathol. 2008;17:48–54. doi: 10.1016/j.carpath.2007.06.005. [DOI] [PubMed] [Google Scholar]
- Samánek M, Slavík Z, Zborilová B, Hrobonová V, Vorísková M, Skovránek J. Prevalence, treatment, and outcome of heart disease in live-born children: a prospective analysis of 91,823 live-born children. Pediatr Cardiol. 1989;10:205–211. doi: 10.1007/BF02083294. [DOI] [PubMed] [Google Scholar]
- Martins P, Cautela P. Transposition of the great arteries. Orphanet J Rare Dis. 2008;3:27. doi: 10.1186/1750-1172-3-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Digilio MC, Casey B, Toscano A, Calabrò R, Pacileo G, Marasini M, Banaudi E, Giannotti A, Dallapiccola B, Marino B. Complete transposition of the great arteries: patterns of congenital heart disease in familial precurrence. Circulation. 2001;104:2809–2814. doi: 10.1161/hc4701.099786. [DOI] [PubMed] [Google Scholar]
- Mégarbané A, Salem N, Stephan E, Ashoush R, Lenoir D, Delague V, Kassab R, Loiselet J, Bouvagnet P. X-linked transposition of the great arteries and incomplete penetrance among males with a nonsense mutation in ZIC3. Eur J Hum Genet. 2000;8:704–708. doi: 10.1038/sj.ejhg.5200526. [DOI] [PubMed] [Google Scholar]
- Bamford RN, Roessler E, Burdine RD, Saplakoğlu U, dela Cruz J, Splitt M, Goodship JA, Towbin J, Bowers P, Ferrero GB, Marino B, Schier AF, Shen MM, Muenke M, Casey B. Loss-of-function mutations in the EGF-CFC gene CFC1 are associated with human left-right laterality defects. Nat Genet. 2000;26:365–369. doi: 10.1038/81695. [DOI] [PubMed] [Google Scholar]
- Muncke N, Jung C, Rüdiger H, Ulmer H, Roeth R, Hubert A, Goldmuntz E, Driscoll D, Goodship J, Schön K, Rappold G. Missense mutations and gene interruption in PROSIT240, a novel TRAP240-like gene, in patients with congenital heart defect (transposition of the great arteries) Circulation. 2003;108:2843–2850. doi: 10.1161/01.CIR.0000103684.77636.CD. [DOI] [PubMed] [Google Scholar]
- Karkera JD, Lee JS, Roessler E, Banerjee-Basu S, Ouspenskaia MV, Mez J, Goldmuntz E, Bowers P, Towbin J, Belmont JW, Baxevanis AD, Schier AF, Muenke M. Loss-of-function mutations in growth differentiation factor-1 (GDF1) are associated with congenital heart defects in humans. Am J Hum Genet. 2007;81:987–994. doi: 10.1086/522890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roessler E, Ouspenskaia MV, Karkera JD, Vélez JI, Kantipong A, Lacbawan F, Bowers P, Belmont JW, Towbin JA, Goldmuntz E, Feldman B, Muenke M. Reduced NODAL signaling strength via mutation of several pathway members including FOXH1 is linked to human heart defects and holoprosencephaly. Am J Hum Genet. 2008;83:18–29. doi: 10.1016/j.ajhg.2008.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohapatra B, Casey B, Li H, Ho-Dawson T, Smith L, Fernbach SD, Molinari L, Niesh SR, Jefferies JL, Craigen WJ, Towbin JA, Belmont JW, Ware SM. Identification and functional characterization of NODAL rare variants in heterotaxy and isolated cardiovascular malformations. Hum Mol Genet. 2009;18:861–871. doi: 10.1093/hmg/ddn411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Luca A, Sarkozy A, Consoli F, Ferese R, Guida V, Dentici ML, Mingarelli R, Bellacchio E, Tuo G, Limongelli G, Digilio MC, Marino B, Dallapiccola B. Familial transposition of great arteries caused by multiple mutations in laterality genes. Heart. 2009;96:673–677. doi: 10.1136/hrt.2009.181685. [DOI] [PubMed] [Google Scholar]
- Marino B, Digilio MC, Versacci P, Anaclerio S, Dallapiccola B. Transposition of great arteries. Understanding its pathogenesis. Ital Heart J Suppl. 2002;3:154–160. [PubMed] [Google Scholar]
- Brown V, Jin P, Ceman S, Darnell JC, O'Donnell WT, Tenenbaum SA, Jin X, Feng Y, Wilkinson KD, Keene JD, Darnell RB, Warren ST. Microarray identification of FMRP-associated brain mRNAs and altered mRNA translational profiles in fragile X syndrome. Cell. 2001;107:477–487. doi: 10.1016/S0092-8674(01)00568-2. [DOI] [PubMed] [Google Scholar]
- Amati F, Biancolella M, D'Apice MR, Gambardella S, Mango R, Sbraccia P, D'Adamo M, Margiotti K, Nardone A, Lewis M, Novelli G. Gene expression profiling of fibroblasts from a human progeroid disease (mandibuloacral dysplasia, MAD #248370) through cDNA microarrays. Gene Expr. 2004;12:39–47. doi: 10.3727/000000004783992189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma HS, Peters TH, Moorhouse MJ, van der Spek PJ, Bogers AJ. DNA microarray analysis for human congenital heart disease. Cell Biochem Biophys. 2006;44:1–9. doi: 10.1385/CBB:44:1:001. [DOI] [PubMed] [Google Scholar]
- Nishimura Y, Martin CL, Vazquez-Lopez A, Spence SJ, Alvarez-Retuerto AI, Sigman M, Steindler C, Pellegrini S, Schanen NC, Warren ST, Geschwind DH. Genome-wide expression profiling of lymphoblastoid cell lines distinguishes different forms of autism and reveals shared pathways. Hum Mol Genet. 2007;16:1682–1698. doi: 10.1093/hmg/ddm116. [DOI] [PubMed] [Google Scholar]
- Amati F, Chillemi G, Novelli G. Developmental gene expression regulation. Nova Science Publishers; 2009. Gene Expression Analysis during Development by High-Throughput Methods; pp. 189–214. [Google Scholar]
- Cipollone D, Amati F, Carsetti R, Placidi S, Biancolella M, D'Amati G, Novelli G, Siracusa G, Marino B. A multiple retinoic acid antagonist induces conotruncal anomalies, including transposition of the great arteries, in mice. Cardiovasc Pathol. 2006;15:194–202. doi: 10.1016/j.carpath.2006.04.004. [DOI] [PubMed] [Google Scholar]
- Cipollone D, Carsetti R, Tagliani A, Rosado MM, Borgiani P, Novelli G, D'Amati G, Fumagalli L, Marino B, Businaro R. Folic acid and methionine in the prevention of teratogen-induced congenital defects in mice. Cardiovasc Pathol. 2009;18:100–109. doi: 10.1016/j.carpath.2008.02.007. [DOI] [PubMed] [Google Scholar]
- Géhin M, Vivat V, Wurtz JM, Losson R, Chambon P, Moras D, Gronemeyer H. Structural basis for engineering of retinoic acid receptor isotype-selective agonists and antagonists. Chem Biol. 1999;6:519–529. doi: 10.1016/S1074-5521(99)80084-2. [DOI] [PubMed] [Google Scholar]
- Scharpf RB, Iacobuzio-Donahue CA, Sneddon JB, Parmigiani G. When should one subtract background fluorescence in 2-color microarrays? Biostatistics. 2007;8:695–707. doi: 10.1093/biostatistics/kxl041. [DOI] [PubMed] [Google Scholar]
- Yang YH, Dudoit S, Luu P, Lin DM, Peng V, Ngai J, Speed TP. Normalization for cDNA microarray data: a robust composite method addressing single and multiple slide systematic variation. Nucleic Acids Res. 2002;30:e15. doi: 10.1093/nar/30.4.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang Y, Brass A, Hoyle DC, Hayes A, Bashein A, Oliver SG, Waddington D, Rattray M. A model-based analysis of microarray experimental error and normalisation. Nucleic Acids Res. 2003;31:e96. doi: 10.1093/nar/gng097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J Roy Statist Soc Ser B. 1995;57:289–300. [Google Scholar]
- Zechel C, Shen XQ, Chambon P, Gronemeyer H. Dimerization interfaces formed between the DNA binding domains determine the cooperative binding of RXR/RAR and RXR/TR heterodimers to DR5 and DR4 elements. EMBO J. 1994;13:1414–1424. doi: 10.1002/j.1460-2075.1994.tb06395.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zechel C, Shen XQ, Chen JY, Chen ZP, Chambon P, Gronemeyer H. The dimerization interfaces formed between the DNA binding domains of RXR, RAR and TR determine the binding specificity and polarity of the full-length receptors to direct repeats. EMBO J. 1994;13:1425–1433. doi: 10.1002/j.1460-2075.1994.tb06396.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo G, Gu YZ, Jain S, Chan WK, Carr KM, Hogenesch JB, Bradfield CA. Molecular characterization of the murine Hif-1 alpha locus. Gene Expr. 1997;6:287–299. [PMC free article] [PubMed] [Google Scholar]
- Weninger WJ, Lopes Floro K, Bennett MB, Withington SL, Preis JI, Barbera JP, Mohun TJ, Dunwoodie SL. Cited2 is required both for heart morphogenesis and establishment of the left-right axis in mouse development. Development. 2005;132:1337–1348. doi: 10.1242/dev.01696. [DOI] [PubMed] [Google Scholar]
- Bouman HG, Broekhuizen ML, Baasten AM, Gittenberger-De Groot AC, Wenink AC. Spectrum of looping disturbances in stage 34 chicken hearts after retinoic acid treatment. Anat Rec. 1995;243:101–108. doi: 10.1002/ar.1092430112. [DOI] [PubMed] [Google Scholar]
- Chazaud C, Chambon P, Dollé P. Retinoic acid is required in the mouse embryo for left-right asymmetry determination and heart morphogenesis. Development. 1999;126:2589–2596. doi: 10.1242/dev.126.12.2589. [DOI] [PubMed] [Google Scholar]
- Rosenthal N, Xavier-Neto J. From the bottom of the heart: anteroposterior decisions in cardiac muscle differentiation. Curr Opin Cell Biol. 2000;12:742–746. doi: 10.1016/S0955-0674(00)00162-9. [DOI] [PubMed] [Google Scholar]
- Pan J, Baker KM. Retinoic acid and the heart. Vitam Horm. 2007;75:257–283. doi: 10.1016/S0083-6729(06)75010-5. full_text. [DOI] [PubMed] [Google Scholar]
- Compernolle V, Brusselmans K, Franco D, Moorman A, Dewerchin M, Collen D, Carmeliet P. Cardia bifida, defective heart development and abnormal neural crest migration in embryos lacking hypoxia-inducible factor-1alpha. Cardiovasc Res. 2003;60:569–579. doi: 10.1016/j.cardiores.2003.07.003. [DOI] [PubMed] [Google Scholar]
- Krishnan J, Ahuja P, Bodenmann S, Knapik D, Perriard E, Krek W, Perriard JC. Essential role of developmentally activated hypoxia-Inducible factor 1α for cardiac morphogenesis and function. Circ Res. 2008;103:1139–1146. doi: 10.1161/01.RES.0000338613.89841.c1. [DOI] [PubMed] [Google Scholar]
- Bhattacharya S, Michels CL, Leung MK, Arany ZP, Kung AL, Livingston DM. Functional role of p35srj, a novel p300/CBP binding protein, during transactivation by HIF-1. Genes Dev. 1999;13:64–75. doi: 10.1101/gad.13.1.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamforth SD, Bragança J, Farthing CR, Schneider JE, Broadbent C, Michell AC, Clarke K, Neubauer S, Norris D, Brown NA, Anderson RH, Bhattacharya S. Cited2 controls left-right patterning and heart development through a Nodal-Pitx2c pathway. Nat Genet. 2004;36:1189–1196. doi: 10.1038/ng1446. [DOI] [PubMed] [Google Scholar]
- Sperling S, Grimm CH, Dunkel I, Mebus S, Sperling HP, Ebner A, Galli R, Lehrach H, Fusch C, Berger F, Hammer S. Identification and functional analysis of CITED2 mutations in patients with congenital heart defects. Hum Mutat. 2005;26:575–582. doi: 10.1002/humu.20262. [DOI] [PubMed] [Google Scholar]
- Shin DH, Li SH, Chun YS, Huang LE, Kim MS, Park JW. CITED2 mediates the paradoxical responses of HIF-1a to proteasome inhibition. Oncogene. 2008;27:1939–1944. doi: 10.1038/sj.onc.1210826. [DOI] [PubMed] [Google Scholar]
- Freedman SJ, Sun ZY, Kung AL, France DS, Wagner G, Eck MJ. Structural basis for negative regulation of hypoxia-inducible factor-1alpha by CITED2. Nat Struct Biol. 2003;10:504–12. doi: 10.1038/nsb936. [DOI] [PubMed] [Google Scholar]
- Poon E, Harris AL, Ashcroft M. Targeting the hypoxia-inducible factor (HIF) pathway in cancer. Expert Rev Mol Med. 2009;11:e26. doi: 10.1017/S1462399409001173. [DOI] [PubMed] [Google Scholar]
- Semenza GL, Agani F, Feldser D, Iyer N, Kotch L, Laughner E, Yu A. Hypoxia, HIF-1, and the pathophysiology of common human diseases. Adv Exp Med Biol. 2000;475:123–130. doi: 10.1007/0-306-46825-5_12. full_text. [DOI] [PubMed] [Google Scholar]
- Yasui H, Morishima M, Nakazawa M, Aikawa E. Anomalous looping, atrioventricular cushion dysplasia, and unilateral ventricular hypoplasia in the mouse embryos with right isomerism induced by retinoic acid. Anat Rec. 1998;250:210–219. doi: 10.1002/(SICI)1097-0185(199802)250:2<210::AID-AR11>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- Yasui H, Nakazawa M, Morishima M, Miyagawa-Tomita S, Momma K. Morphological observations on the pathogenetic process of transposition of the great arteries induced by retinoic acid in mice. Circulation. 1995;91:2478–2486. doi: 10.1161/01.cir.91.9.2478. [DOI] [PubMed] [Google Scholar]
- Licht AH, Müller-Holtkamp F, Flamme I, Breier G. Inhibition of hypoxia-inducible factor activity in endothelial cells disrupts embryonic cardiovascular development. Blood. 2006;107:584–590. doi: 10.1182/blood-2005-07-3033. [DOI] [PubMed] [Google Scholar]
- Moss JB, Xavier-Neto J, Shapiro MD, Nayeem SM, McCaffery P, Dräger UC, Rosenthal N. Dynamic patterns of retinoic acid synthesis and response in the developing mammalian heart. Dev Biol. 1998;199:55–71. doi: 10.1006/dbio.1998.8911. [DOI] [PubMed] [Google Scholar]
- Zhang J, Song LP, Huang Y, Zhao Q, Zhao KW, Chen GQ. Accumulation of hypoxia inducible factor-1α protein and its role in the differentiation of myeloid leukemic cells induced by all-trans retinoic acid. Haematologica. 2008;93:1480–1487. doi: 10.3324/haematol.13096. [DOI] [PubMed] [Google Scholar]
- Mader S, Chen JY, Chen Z, White J, Chambon P, Gronemeyer H. The patterns of binding of RAR, RXR and TR homo- and heterodimers to direct repeats are dictated by the binding specificites of the DNA binding domains. EMBO J. 1993;12:5029–5041. doi: 10.1002/j.1460-2075.1993.tb06196.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ifergan I, Assaraf YG. Molecular mechanisms of adaptation to folate deficiency. Vitam Horm. 2008;79:99–143. doi: 10.1016/S0083-6729(08)00404-4. [DOI] [PubMed] [Google Scholar]
- Botto LD, Mulinare J, Erickson JD. Do multivitamin or folic acid supplements reduce the risk for congenital heart defects? Evidence and gaps. Am J Med Genet A. 2003;121A:95–101. doi: 10.1002/ajmg.a.20132. [DOI] [PubMed] [Google Scholar]
- Li D, Rozen R. Maternal folate deficiency affects proliferation, but not apoptosis, in embryonic mouse heart. J Nutr. 2006;136:1774–1778. doi: 10.1093/jn/136.7.1774. [DOI] [PubMed] [Google Scholar]
- Nafee T, Farrell W, Carroll W, Fryer A, Ismail K. Epigenetic control of fetal gene expression. BJOG. 2008;115:158–168. doi: 10.1111/j.1471-0528.2007.01528.x. [DOI] [PubMed] [Google Scholar]
- Sperling S. Array analysis applied to malformed hearts: Molecular dissection of tetralogy of Fallot. Methods Mol Med. 2006;126:233–246. doi: 10.1385/1-59745-088-X:233. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1: List of the 477 differentially expressed genes (FC≥ ± 1.5) in BMS189453-treated embryos
Table S2: List of the 239 differentially expressed genes (FC≥ ± 1.5) in BMS+FA-treated embryos