

Abstract
BACKGROUND & AIMS
Gastric cancer evolves in the setting of a pathologic mucosal milieu characterized by both loss of acid-secreting parietal cells and mucous cell metaplasias. Indeed, mucous cell metaplasia is considered the critical preneoplastic lesion for gastric cancer. Previous investigations have shown that infection of mice with Helicobacter felis or induction of acute parietal cell loss with the drug DMP-777 leads to the emergence of a type of metaplasia designated spasmolytic polypeptide-expressing metaplasia (SPEM). We have hypothesized that SPEM arises from proliferating cells in gland bases, either from a cryptic progenitor cell or by transdifferentiation of mature chief cells.
METHODS
Taking advantage of the chief cell-restricted expression of Mist1-Cre-ERT2, we used lineage mapping to examine whether SPEM lineages were derived from chief cells in 3 independent models of induction by DMP-777 treatment, L-635 treatment, or H felis infection.
RESULTS
Treatment of mice with L-635 for 3 days led to rapid parietal cell loss, induction of a prominent inflammatory infiltrate, and emergence of SPEM. In all 3 models, SPEM developed, at least in part, from transdifferentiation of chief cells. We further found that acute parietal cell loss in the setting of inflammation (L-635 treatment) led to more rapid induction and expansion of SPEM derived from transdifferentiation of chief cells.
CONCLUSIONS
These studies provide direct evidence by lineage tracing that SPEM evolves from differentiated chief cells. Thus, mature gastric chief cells have the ability to act as cryptic progenitors and reacquire proliferative capacity within the context of mucosal injury and inflammation.
Keywords: SPEM, Chief Cell, Transdifferentiation, Metaplasia
In the normal gastric fundic mucosa, cell lineages differentiate from progenitor cells located in the neck regions of glands through the initial differentiation of 3 types of second-order progenitor cells: presurface, preparietal, and preneck cells.1 Of particular relevance to the present discussion, preneck cells differentiate into mucous neck cells as they migrate toward the base of the glands and then redifferentiate at the bottoms of glands into zymogen-secreting chief cells.2 Intestinal-type gastric cancer predominantly develops in the setting of parietal cell loss (oxyntic atrophy) and mucous cell metaplasia.3 Although loss of parietal cells from the gastric epithelium appears to lead to mucous cell metaplasia, the origin of these metaplastic lineages remains obscure. Two types of mucous cell metaplasia develop in the stomach of human beings: spasmolytic polypeptide-expressing metaplasia (SPEM), a metaplasia in the gastric fundus resembling deep antral gland cells, expresses Trefoil Factor 2 (TFF2; also known as spasmolytic polypeptide) and MUC6.4 Intestinal metaplasia develops in both the fundus and antrum and resembles intestinal goblet cells with expression of both TFF3 and MUC2.5,6 Recent investigations suggest that intestinal metaplasia in the fundus likely develops from precedent SPEM.7,8 However, in mouse models of either Helicobacter infection or acute oxyntic atrophy, only SPEM is observed.9,10 C57BL6 mice infected with Helicobacter felis for more than 9 months develop SPEM and progress to dysplasia by 1 year of infection,10 indicating a direct link between SPEM and gastric neoplasia.11
Although previous studies have indicated that SPEM in mice is the precursor for dysplasia,10,11 the origin of SPEM has remained unclear. To understand better the factors that lead to the emergence of SPEM, we have studied the induction of metaplasia after the acute destruction of parietal cells by treatment with DMP-777, a parietal cell–specific protonophore that partitions into the apical acid secretory membranes of parietal cells, leading to acute death after acid secretion.9 Importantly, because DMP-777 is also a potent neutrophil elastase inhibitor, we observed no significant inflammatory response in reaction to this acute parietal cell loss. Still, loss of parietal cells led to the emergence at the bases of fundic glands of SPEM after 10 days of DMP-777 treatment.12 Observation of SPEM was preceded by an apparent loss of normal chief cells, which express the bHLH transcription factor Mist1 and secrete pepsinogen and intrinsic factor.13 Although the normal proliferative zone for the gastric fundus is located toward the lumen in fundic gastric glands, in regions of emerging SPEM, we observed scattered proliferating mucosal cells at the bases of gastric glands.12,14 In evaluating the SPEM in gastrin-deficient mice and other models, we determined that the most reliable reflection of the emergence of SPEM was the presence at the bases of gastric glands of cells that co-expressed both TFF2 and intrinsic factor.12,15 We therefore hypothesized that SPEM cells are derived from transdifferentiation of mature chief cells.
To address this hypothesis, we performed lineage mapping studies using Mist1CreER/+/Rosa26RLacZ mice, which express bacterial β-galactosidase after tamoxifen-induced activation of Cre recombinase. The β-galactosidase is expressed exclusively in mature chief cells because tamoxifen-responsive Cre is knocked into the chief cell-specific Mist1 locus. In 3 different models of SPEM induction, SPEM cells predominantly were derived from mature (ie, Mist1-expressing) chief cells. Importantly, in models of SPEM that also induced inflammatory infiltrates, we observed a substantial expansion of the chief cell-derived, proliferative SPEM lineage. These results show that a key gastric metaplastic mucous cell lineage derives in large part from trans-differentiation of mature chief cells. Because similar scenarios for mucous cell metaplasia are linked to gastric carcinogenesis in human beings,3 our results may have major implications for our understanding of the origins of human gastric neoplasms.
Materials and Methods
Mice
Eight- to 10-week-old mice were used for all studies. Generation of Mist1CreER/+ and Rosa26RLacZ mice has been described previously.16 Mist1CreER/+ mice were generated by standard embryonic stem cell targeting in which the complete Mist1 coding region was replaced with the CreERT2 coding region. Cre recombinase was activated in Mist1CreER/+/Rosa26RLacZ mice by intraperitoneal injection of tamoxifen (1 mg/0.1 mL corn oil) for 3 doses every other day. H felis infection was performed as previously described.17 During the experiments, the mice were maintained with regular mouse chow and water ad libitum in a temperature-controlled room under a 12-hour light/dark cycle. The care, maintenance, and treatment of animals in these studies followed protocols approved by the Institutional Animal Care and Use Committee of Vanderbilt University.
Drugs
Preparation and treatment of the DMP-777 have been described previously.18 L-635 was a gift of Merck & Co, Inc. Mice were administered L-635 orally as a gavage (350 mg/kg) once daily for 3 days.
H felis Challenge
H felis infection was performed as previously described.17 H felis was grown from freezer stock on trypticase soy agar plates containing 5% sheep blood (Becton Dickinson), at 37°C with 5% CO2, and inoculated into Brucella broth containing 5% heat-inactivated fetal bovine serum and 10 mg/mL vancomycin. After 24 hours, cells were harvested by centrifugation. Mice were fasted overnight and orogastrically inoculated 3 times (2-day interval) with 0.5 mL of an H felis cell suspension of 1 × 109 colony-forming units/mL using feeding needles.
β-Galactosidase Analysis
Assessment of β-galactosidase activity was used to examine the efficiency of recombination at the Rosa26RLacZ reporter allele after induction of Cre activity. Stomach whole-mounts were prepared, fixed, and exposed to X-gal substrate using a method previously reported.19 The stomachs were opened along the greater curvature and washed with ice-cold fixative containing 1% formaldehyde, 0.2% glutaraldehyde, and 0.02% NP40 in phosphate-buffered saline (PBS) (Ca+ and Mg+ free). The stomachs were immediately incubated for 2 hours in a 20-fold volume of ice-cold fixative at 4°C on a rolling platform. The fixative was removed and the tissues were washed twice in PBS for 20 minutes at room temperature (20°C) on a rolling platform. The β-galactosidase substrate (5 mmol/L K3Fe(CN)6, 5 mmol/L K4Fe(CN)6 × 3 H2O, 2 mmol/L MgCl2, 0.02% NP40, 0.1% sodium deoxycholate, and 1 mg/mL X-gal in PBS) then was added and the tissues were incubated in the dark overnight at room temperature. The substrate was removed and the tissues were washed twice in PBS for 20 minutes at room temperature on a rolling platform. The tissues then were fixed overnight in a 20-fold volume of 4% paraformaldehyde in PBS at 4°C in the dark on a rolling platform. The paraformaldehyde was removed and the tissues were washed twice in PBS for 20 minutes at room temperature on a rolling platform. The stained tissues were transferred to tissue cassettes and paraffin blocks prepared using standard methods. Tissue sections were prepared and counterstained with neutral red.
Immunohistochemistry
We performed immunohistochemistry according to the manufacturer’s recommendations, typically using a modified citric acid unmasking protocol followed by standard detection with 3,3-diaminobenzidine or alkaline phosphatase using a kit from Vector Laboratories. Samples were counterstained with hematoxylin. In some cases, secondary antibodies were conjugated to AlexaFluor 488 (Invitrogen, Camarillo, CA) or Cy-3 and nuclei were counterstained with 4,6-diamidino-2-phenylindole. We used the following primary antibodies: mouse anti-TFF2 (1:100; a gift from Dr Nicholas Wright, Cancer UK, London, England), mouse anti–H+/K+-adenosine triphosphatase (ATPase) (1:2000; a gift from of Dr Adam Smolka, Medical University of South Carolina, Charleston, SC), rabbit anti-intrinsic factor (1:1000; a gift from Dr David Alpers, Washington University, St. Louis, MO), rabbit anti–β-galactosidase (1:200; Abcam, Cambridge, MA), rat anti-CD45 (1:1000; BD Biosciences, San Jose, CA), goat anti–CD3-ε (1:1000; Santa Cruz), rat anti-CD45R (1:200; BD Biosciences), rat anti-F4/80 (1:500; Invitrogen), rat anti-MCA771G (1:500; AbD Serotec, Oxford, UK), rabbit anti–phospho-signal transducers and activators of transcription (STAT) 1 (1:50; Cell Signaling, Danvers, MA), rabbit anti–phospho-STAT 3 (1:50; Cell Signaling), rabbit anti–phospho-STAT 6 (1:1000; Abcam), and rabbit anti-MCM2 (1:100; Abcam).
Quantitation
X-gal–positive area and MCM2-positive cells were analyzed using an Ariol SL-50 automated slide scanner (Applied Imaging) as described previously.14
Statistics
The data were analyzed with the JMP software package (version 4.0; SAS Institute, Cary, NC). X-gal–positive areas and MCM2-positive cell numbers were compared with analysis of variance followed by post hoc analysis of significant means by the Dunnett test. For all comparisons, P values less than .05 were considered statistically significant.
RNA Extraction and Real-Time Reverse-Transcription Polymerase Chain Reaction
Total RNA was isolated from the gastric fundus of untreated C57BL/6 mice and mice treated with L-635 for 3 days, treated with DMP-777 for 14 days, or infected with H felis for 9 months (3 animals in each group) using TRIzol (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions. The RNA (1 μg) was treated with RQ1 RNase-free DNase (Promega, Madison, WI) and then reverse-transcribed using the Superscript III reverse transcriptase (Invitrogen, Carlsbad, CA).
Equal amounts of each complementary DNA were analyzed for the expression of tumor necrosis factor-α, interleukin (IL)-1β, IL-4, IL-10, and interferon-γ by real-time polymerase chain reaction (PCR) using specific primers (200 nmol/L) and the EXPRESS SYBR GreenER quantitative PCR SuperMix (Invitrogen, Carlsbad, CA) in an ABI StepOnePlus Real-Time PCR System (Applied Biosystems, Foster City, CA). The cycling conditions were as indicated by the SYBR Green supermix manufacturer’s protocol. Each sample was measured in triplicate. The primer sequences were as follows: tumor necrosis factor-α (forward: 5′-CTGTGAAGGGAATGGGTGTT-3′ and reverse: 5′-GGTCACTGTCCCAGCATCTT-3′); IL-1β (forward: 5′-CGTGGACCTTCCAGGATGAG-3′ and reverse: 5′-ATGGGAACGTCACACACCAG-3′), IL-4 (forward: 5′-TCACAGCAACGAAGAACACC-3′ and reverse: 5′-CTGCAGCTCCATGAGAACAC-3′); IL-10 (forward: 5′-CAAAGGACCAGCTGGACAAC-3′ and reverse: 5′-TCATTTCCGATAAGGCTTGG-3′); interferon-γ (forward: 5′-GCCACGGCACAGTCATTGAA-3′ and reverse: 5′-CGCCTTGCTGTTGCTGAAGA-3′). Cycle threshold was converted to relative expression according to the 2−ΔΔ cycle threshold method, using TATA– box-binding protein as an endogenous control. For each relative expression analysis, the mean value of the normalized cycle thresholds of all normal mouse samples was taken as reference.
Statistical significance (P < .05) of the differences in the expression levels was determined using an unpaired t test with Welch’s correction.
Results
Lineage Mapping of SPEM in DMP-777–Treated Mice, a Model of Parietal Cell Loss Without Inflammation
We performed lineage tracing of chief cells using Mist1CreER/+/Rosa26RLacZ mice, which express Cre-ERT2 from the Mist1 locus in mature chief cells.13 The mice (n = 8) were first treated with tamoxifen for 3 days to induce Rosa26RLacZ recombination and β-galactosidase expression in mature chief cells, and then recovered drug-free for 10 days before administration of DMP-777 for 14 days to elicit SPEM. In mice that received tamoxifen, we observed strong β-galactosidase expression in chief cells of the gastric fundus (Figure 1A), Brunner’s gland cells (Figure 1B), and pancreatic acinar cells (Figure 1C), but no staining was observed in the gastric antrum (Figure 1D). In animals that were not treated with DMP-777 (n = 4), we observed complete separation of TFF2 immunostaining mucous neck cells and X-gal staining Mist1-expressing chief cells (Figure 1E). Although a previous study has indicated that some prezymogenic cells in the transition zone between mucous neck cells and chief cells transiently express markers of both lineages,20 we did not observe any cells that stained for both TFF2 and X-gal. These results suggest that, after the 10-day period after tamoxifen treatment, all of the β-galactosidase– expressing cells in the fundus were mature chief cells. As previously reported,18 DMP-777 induced both a marked loss of parietal cells and prominent emergence of SPEM stained with antibodies against TFF2 (Figure 1F). We observed basal glandular cells labeling with antibodies against TFF2, which concomitantly showed β-galactosidase enzymatic activity (Figure 1F), indicating that these SPEM cells were derived from mature, Mist1-expressing chief cells. These cells also showed immunoreactivity for β-galactosidase protein using both immunohistochemistry and immunofluorescence detection (Supplementary Figures 1 and 2). No β-galactosidase staining was detected in Rosa26RLacZmice (n = 8) treated with DMP-777 (n = 2), in Mist1CreER/+/Rosa26RLacZ mice without tamoxifen induction treated with DMP-777 (n = 2) or in wild-type mice treated with DMP-777 (n = 2) (Supplementary Figures 3–5). We also observed an expansion of TFF2-expressing cells in the midgland region in mice treated with DMP-777. Those cells did not show expression of β-galactosidase, suggesting that they may arise from mucous neck cells.20 Therefore, the phenotype of SPEM glands in DMP-777–treated mice was composed of 2 groups of TFF2-expressing mucous cells: one unequivocally derived from transdifferentiation of chief cells and another likely derived from mucous neck cells, which normally express TFF2, likely through arrest of forward differentiation into chief cells.
Figure 1.

Mist1CreER/+/Rosa26RLacZ mice and DMP-777 treatment. (A–D) β-galactosidase expression in Mist1CreER/+/Rosa26RLacZ mice was induced by tamoxifen treatment. β-galactosidase activity was detected with incubation of tissue with X-gal. In mice receiving tamoxifen, strong X-gal staining (blue) was observed in chief cells of the (A) gastric fundus, (B) Brunner’s gland cells, and (C) pancreatic acinar cells, but no staining was observed in the (D) gastric antrum. (E) Stomach of a Mist1CreER/+/Rosa26RLacZ mouse treated with tamoxifen and then killed 10 days after the last tamoxifen injection stained with X-gal and antibodies against TFF2 (brown). Note the complete separation of TFF2 immunostaining mucous neck cells and X-gal–staining Mist1-expressing chief cells. (F) Tamoxifen-treated mice receiving further DMP-777 treatment. DMP-777 induced a marked loss of parietal cells and prominent SPEM, which co-stained for X-gal and TFF2. Right panel: higher magnification view. Note the co-staining of β-galactosidase–expressing, X-gal–stained cells at the bases of glands with antibodies against TFF2. Bar, 50 μm.
A Novel Model of Acute Parietal Cell Loss With Inflammation
As noted earlier, DMP-777 abrogates development of inflammation, presumably by its inhibition of neutrophil elastase, blocking the inflammatory response early in the innate immune phases. However, in human beings, metaplasia usually develops in the setting of severe inflammation. We therefore examined the effects of a structurally related β-lactam compound, Merck L-635 (L-635), which retains potent parietal cell protonophore activity, but does not have any significant activity against neutrophil elastase.9 We treated C57BL/6 mice with 3 daily doses of L-635 (n = 4) and examined the stomachs. Compared with the normal gastric mucosa (Figure 2A), and as seen with DMP-777 treatment, L-635 caused prominent parietal cell loss (Figure 2B and C). However, in contrast with DMP-777 treatment, we also observed a prominent submucosal and intramucosal inflammatory infiltrate (Figure 2B). Although DMP-777 treatment for only 3 days does not elicit any metaplasia,18 L-635 over the same interval caused a marked mucous cell metaplasia that dominated the fundic mucosa and was strongly positive for TFF2 (Figure 2D). As previously reported for SPEM induced by DMP-777, the L-635–induced metaplasia showed dual expression of both TFF2 and intrinsic factor (Figure 2E). L-635–induced metaplastic cells also stained for Ki-67, indicating the adoption of proliferative capacity in the metaplastic cells (Figure 2F). It is important to note that although 3 doses of DMP-777 does elicit parietal cell loss in the fundic mucosa, we do not observe induction of SPEM until 10 –14 days of drug treatment.18 These results indicated that a combination of parietal cell loss and inflammation could potentiate the development of SPEM.
Figure 2.

Characteristics of stomachs from C57BL/6 mice treated with L-635. (A) Cell lineage markers in normal mouse fundus. First panel: H&E staining; second panel: periodic acid–Schiff (PAS) staining; third panel: H+/K+-ATPase staining for parietal cells; and fourth panel: dual-immunofluorescence staining for both intrinsic factor and TFF2. A few dual-stained cells were located at the junction between the mucous neck cell and chief cell zones. Green, intrinsic factor for chief cell; red, TFF2; blue, 4,6-diamidino-2-phenylindole (DAPI). (B–F) Extensive SPEM accompanying inflammation induced by L-635. (B) H&E staining of gastric mucosa from a mouse treated with L-635 showing metaplasia with parietal cell loss and a prominent submucosal and intramucosal inflammatory infiltrate. Right panel: higher magnification view. (D) PAS staining showing prominent mucous cell metaplasia. (C) H+/K+-ATPase staining showing marked loss of parietal cells. (D) Immunostaining for TFF2 (red) in L-635–treated mice showing a marked TFF2-staining mucous cell metaplasia that dominated the fundic mucosa. (E) Dual-immunofluorescence staining for both intrinsic factor and TFF2 showing cells with dual expression of both TFF2 and intrinsic factor at the bases of fundic glands. Green, intrinsic factor; red, TFF2; blue, DAPI. (F) L-635–induced metaplastic cells also were strongly stained for Ki-67, indicating the adoption of proliferative capacity in the metaplastic cells. Green, intrinsic factor; red, Ki-67. Bar, 50 μm.
Given the apparent effects of inflammation on the development of SPEM, we compared the inflammatory infiltrates observed in L-635–treated mice with H felis–infected mice. Both models showed prominent intramucosal and submucosal lymphocytic infiltrates comprising both B and T cells (Supplementary Figure 6). Infiltrates in both models also contained both neutrophils and macrophages (Supplementary Figure 7). To evaluate the functional impact of these mixed immune cell infiltrates, we studied the expression of cytokines by quantitative PCR. Supplementary Figure 8 shows that, similar to previous reports, chronic H felis infection elicited significant increases in the expression of IL-1β, tumor necrosis factor-α, and IL-4. In contrast, acute L-635 treatment elicited significant increases in IL-1β as well as IL-10. DMP-777 treatment does not cause a significant infiltrate, and we did not observe any significant increases in any of the cytokines tested. In concert with these quantitative PCR studies, we also stained sections of L-635–treated and H felis–infected mouse stomachs for activated phosphorylated-STAT proteins as downstream indicators of cytokine activity (Supplementary Figure 9). In H felis–infected mice, the nuclei of epithelial cells were stained extensively at the bases of fundic glands with antibodies against phospho-STAT3, and phospho-STAT1 staining was observed in the nuclei of scattered cells in the upper portions of the glands. Nevertheless, no phospho-STAT staining was observed in L-635–treated mice, perhaps reflecting the acute nature of the inflammation.
Lineage Mapping of SPEM in Mouse Models of Parietal Cell Loss and Inflammation
To evaluate the origin of metaplasia in L-635–treated mice, we treated the Mist1CreER/+/Rosa26RLacZ mice with tamoxifen to induce β-galactosidase activity in all mature chief cells. Ten days later, we treated those mice with 3 doses of L-635 (n = 6) and evaluated X-gal staining (Figure 3). Compared with both untreated animals (n = 4) and DMP-777–treated mice (n = 8), L-635 treatment caused a significant expansion of the number of cells showing β-galactosidase enzymatic activity (as assessed by X-gal staining; Figure 3C). These cells also showed β-galactosidase protein immunoreactivity (Supplementary Figures 1C and D and 2). TFF2 expression was observed in all of the X-gal–stained cells in mice treated with L-635 (Figure 3E and F). These results suggested that L-635 induced transdifferentiation of chief cells into SPEM and that the attendant inflammation induced rapid expansion of that metaplasia.
Figure 3.

Mist1CreER/+/Rosa26RLacZ mice treated with DMP-777 or L-635. (A and B) Mist1 cell lineage analysis (X-gal staining) after treatment with either (A) DMP-777 or (B) L-635 for inducing SPEM. (C) β-galactosidase–expressing cells were quantitated as an X-gal–stained area per 1.5 mm2 of total mucosal tissue (±standard error of the mean). Compared with both untreated animals (n = 4) and DMP-777–treated mice (n = 8), L-635 treatment (n = 6) caused a significant expansion of the number of X-gal–stained cells. (D–F) Immunostaining for TFF2 in X-gal–stained sections from DMP-777 and L-635–treated mice. Brown (3,3-diaminobenzidine) = TFF2. (D) DMP-777 induced a marked loss of parietal cells and prominent SPEM, with co-staining for X-gal and TFF2 at the bases of glands (brackets). (E) TFF2 expression was observed throughout the X-gal–stained cells in mice treated with L-635. (F) Higher magnification view of panel E. Bar, 100 μm.
Lineage Mapping of SPEM in 6-Month-Old H felis–Infected Mice
Previous studies have shown that infection of C57BL6 mice with H felis leads to development of SPEM in the presence of a prominent inflammatory infiltrate.10,12,21,22 We therefore sought to determine whether SPEM elicited by H felis infection also was derived from chief cell transdifferentiation. Mist1CreER/+/Rosa26RLacZ mice were treated with tamoxifen and then 10 days later were infected with H felis (n = 6). Mice were killed 6 months after challenge and evaluated for expression of β-galactosidase and TFF2. The H felis–infected mice showed prominent X-gal staining lineages in the fundic mucosa, comprising more than half the height of the glands (Figure 4A–C). The X-gal–positive cells were stained prominently with TFF2 antibodies throughout the length of the glands (Figure 4D and E and Supplementary Figure 2), indicating that SPEM elicited by H felis infection was derived from mature chief cells. Dual immunostaining for TFF2 and β-galactosidase protein also was observed throughout the SPEM cells (Supplementary Figure 2). These data indicate that SPEM induced and sustained in the setting of chronic inflammation is derived predominantly from chief cells.
Figure 4.
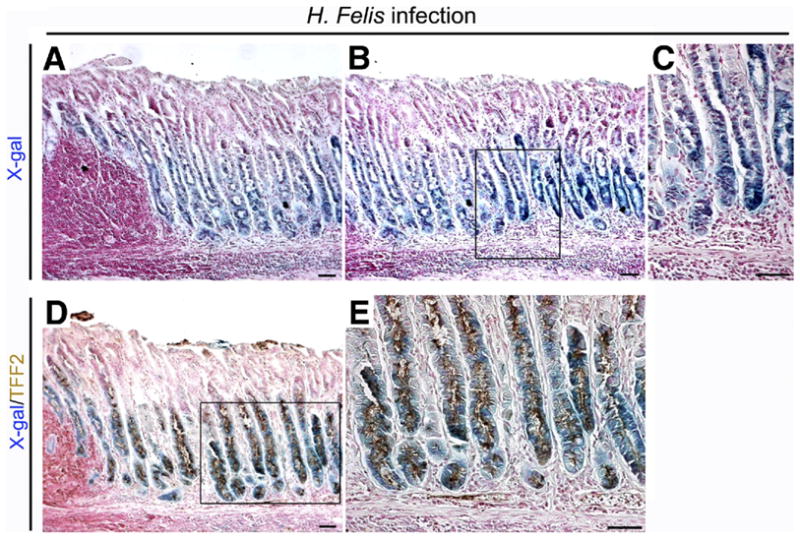
Lineage mapping of SPEM in 6-month-old H felis–infected Mist1CreER/+/Rosa26RLacZ mice. Sections of fundic mucosa stained with (A–C) X-gal or dual stained for (D and E) X-gal and TFF2 (brown). Chronic H felis infection caused an expansion of the number of X-gal–stained cells. (A) Note the lymphoid follicle and inflammatory cell infiltration in the mucosa. (B) Mucosa adjacent to section in panel A. (C) Higher magnification view of panel B. (D) Dual immunostaining for TFF2 in X-gal–stained sections. Brown (3,3-diaminobenzidine) = TFF2. TFF2 expression was observed throughout the X-gal–stained cells in H felis–infected mice. (E) Higher magnification view of panel D. Bar, 50 μm.
Proliferative Cell Lineage Analysis in SPEM Induced by DMP-777 and Acute and Chronic Inflammation
To confirm that the attendant inflammation induced rapid expansion of metaplasia, we analyzed the proliferative cell lineages in the 3 different SPEM models. We evaluated MCM2 staining of proliferative cells23 in SPEM induced by DMP-777 (no inflammation), L-635 (acute inflammation) and H felis infection (chronic inflammation) (Figure 5). In untreated mice (n = 4), MCM2 stained only proliferating normal progenitor cells in the upper gland neck, although no proliferating cells were observed among the X-gal–staining chief cells (Figure 5A and E). In DMP-777–treated mice (n = 8), MCM2-expressing proliferative cells were also X-gal positive at the bases of fundic glands (Figure 5B and E). In mice treated with L-635 (n = 6) and in H felis–infected mice (n = 6), MCM2 staining was observed in X-gal–staining cells along the entire gland length (Figure 5C and D). The number of MCM2-labeled proliferative cells in L-635–treated animals and H felis–infected mice was significantly greater than in DMP-777–treated mice (Figure 5E). Similar results were observed for assessment of proliferating cells with either bromodeoxyuridine or Ki-67 staining (data not shown). Thus, metaplastic cells derived from transdifferentiating chief cells in L-635–treated mice and H felis–infected mice acquire substantial proliferative capacity.
Figure 5.

Proliferative cell lineage analysis in SPEM induced by DMP-777, L-635, and H felis infection in Mist1CreER/+/Rosa26RLacZ mice. Proliferating cells in mice were visualized with antibodies against MCM2. (A) MCM2 stained only proliferating normal progenitor cells in the upper fundic gland neck, whereas no proliferating cells were observed in the X-gal–staining chief cells (n = 4). (B) In DMP-777–treated mice (n = 8), we observed MCM2 expression in X-gal–staining cells at the bases of fundic glands. Right panel: higher magnification view. (C) In mice treated with L-635 (n = 6), MCM2 staining was observed in X-gal–staining cells along the entire gland length. Right 2 panels: higher magnification views. (D) In H felis–infected mice (n = 6), MCM2-staining nuclei were present in X-gal–staining cells throughout the length of the glands. SM, submucosa; LF, lymphoid follicle. Right panel: higher magnification view. (E) MCM2-positive proliferative cells were quantitated (±standard error of the mean). The number of proliferating chief cell-derived, X-gal–positive cells in L-635–treated animals and H felis–infected mice was significantly greater than in DMP-777–treated mice. **P < .01. Bar, 50 μm.
Discussion
In human beings, metaplastic cell lineages are acknowledged as critical precursors for the development of intestinal-type gastric cancer.3 Although human beings show both intestinal metaplasia and SPEM as preneoplastic lesions, in mice infected with Helicobacter species only SPEM develops and progresses toward dysplasia. In both human beings and mice, the cellular origin of metaplastic lineages has remained elusive. Previous concepts of preneoplasia have focused on derivation of metaplasia from aberrant differentiation of professional progenitor cells. The current results have defined a new paradigm for the induction of preneoplastic metaplasia in the stomach. An initial loss of parietal cells is required for induction of metaplasia. Although parietal cells are best known for their secretion of acid into the gastric lumen, they also secrete a number of important mucosal growth factors including transforming growth factor-α, amphiregulin, HB-EGF, and Shh.24–27 The loss of these important factors may induce the transdifferentiation of mature chief cells into SPEM. Our results suggest that chief cells represent a repository of potential progenitor cells at the bases of fundic glands. We show here that modification of the gland milieu with loss of parietal cells alone (as in the case of DMP-777 treatment) is sufficient to activate transdifferentiation of chief cells. Nevertheless, the additional presence of inflammation in L-635–treated and H felis–infected mice leads to expansion of SPEM and conversion from a relatively indolent metaplasia into an expanded proliferative metaplasia. These effects appear to be further amplified and sustained in SPEM observed in the setting of chronic inflammation after 6 months of H felis infection. The relationship of chief cells to intestinal metaplasia cannot be addressed in mice. However, recent investigations in both H pylori–infected Mongolian gerbils8 and in human beings28 indicates that intestinal metaplasia may arise from pre-existing SPEM. Thus, the transdifferentiation of chief cells into SPEM represents the critical proximate step in the development of metaplasia and gastric carcinogenesis in the setting of oxyntic atrophy.
Previous studies have shown an extremely rapid induction of SPEM in gastrin-deficient mice treated with DMP-777.15,18 Microdissection of the SPEM lineages forming in gastrin-deficient mice suggested that parietal cell loss leads to both alteration in differentiation transcripts and re-entry into proliferation.15 In the present investigations, a key hallmark of the transdifferentiation process for chief cells appears to be the recrudescence of proliferative capacity that is augmented in the presence of inflammation. The present studies as well as previous investigations have not noted any patterns of apoptosis in chief cell populations after loss of parietal cells.9 These results along with the rapid progression of SPEM in gastrin-deficient mice treated with DMP-777 and L-635–treated mice suggests that transdifferentiation represents a pervasive phenomenon affecting a broad population of chief cells. Recent studies in the intestine have highlighted a role for discrete stem cell populations expressing Lgr5 in renewal of the normal mucosa and promotion of carcinogenesis.29,30 Further investigations have documented the presence of progenitor cells expressing Lgr5 at the bases of antral glands, but few Lgr5-positive cells were present in the fundic mucosa, and those were located only along the lesser curvature.31 In addition, we have performed a number of studies with laser capture microdissection of SPEM from both mouse models and human tissue and have not observed Lgr5 expression in metaplasia.15,32 In contrast, our results reported here indicate a novel mechanism for the generation of preneoplastic metaplasia in stomach. Although recent studies have suggested that neoplasias are fueled from rare abnormal progenitors (cancer stem cells), presumably derived from normal progenitor cells, the present investigations show that mature, terminally differentiated cells, can transdifferentiate into proliferating metaplastic mucous cells. Whether specific subpopulations of transdifferentiating chief cells respond to inflammatory influences remains to be determined.
Although the present studies do not address the further question of how metaplasia progresses to dysplasia, these findings on the origin of metaplastic lineages have important implications for a general understanding of gastric carcinogenesis. Importantly, mature chief cells gave rise to metaplasia induced by infection with Helicobacter, which is considered the major etiologic factor for preneoplastic metaplasia and a recognized carcinogen in human beings.33 Indeed, the persistence of metaplastic lineages derived from mature chief cells 6 months after Cre-mediated induction of β-galactosidase expression suggests that H felis promotes the persistence of self-renewing metaplastic cells. Because SPEM is associated directly with the development of dysplasia in mice and human beings,4,10,22 these findings suggest that gastric neoplasias in human beings ultimately may develop from metaplasias that arise from transdifferentiation of chief cells. Recent investigations also have indicated that trans-differentiation of pancreatic acinar cells may lead to ductular adenocarcinoma.34–36 Thus, in contrast with the concept of a cancer stem cell derived from normal progenitor cells, we now implicate here an alternative origin of neoplastic cells: an array of mature cells may have the potential to transdifferentiate, adopting metaplastic characteristics and becoming proliferative in the presence of inflammatory mediators. Because these metaplastic cells do not arise from professional progenitors, but rather have re-entered the cell cycle from a formerly postmitotic state, they may be predisposed to aberrant growth and acquisition of mutations. In any case, our current studies suggest that metaplasia itself is not pre-neoplastic without inflammatory influences. Further study will be required to better understand the nature of the metaplastic cells and to define the particular inflammatory regulators that promote neoplastic transformation of metaplastic cells.
In summary, using lineage mapping with Mist1CreER/+ mice, we have shown in 3 separate mouse models of oxyntic atrophy that resulting SPEM originates in part or predominantly from transdifferentiation of chief cells. The results further suggest that metaplasias derived from chief cells undergo expansion in the presence of inflammatory infiltrates. These findings indicate that preneoplastic metaplastic lineages in the gastric fundic mucosa can arise from differentiated cell lineages, rather than professional progenitor populations.
Supplementary Material
Acknowledgments
The authors thank Drs Adam Smolka, Nicholas Wright, and David Alpers for the gifts of antibodies and James West, Rupesh Chaturvedi, and Keith Wilson for quantitative polymerase chain reaction primers.
Funding
These studies were supported by grants from a Department of Veterans Affairs Merit Review Award (RO1 DK071590), and the AGA Funderburg Award in Gastric Biology Related to Cancer (J.R.G.); by National Institutes of Health grant RO1 DK079798 (J.C.M.), by National Institutes of Health grant RO1 DK55489 and RO1 CA124586 (S.F.K.), and by R01 DK58587, R01 CA77955, and P01 CA116087 (R.M.P.). This work was supported by core resources of the Vanderbilt Digestive Disease Center (P30 DK058404).
Abbreviations used in this paper
- PCR
polymerase chain reaction
- SPEM
spasmolytic polypeptide expressing metaplasia
- STAT
signal transducers and activators of transcription
- TFF2
trefoil factor family 2
Footnotes
Conflicts of interest
The authors disclose no conflicts.
Note: To access the supplementary material accompanying this article, visit the online version of Gastroenterology at www.gastrojournal.org, and at doi: 10.1053/j.gastro.2010.09.005.
References
- 1.Karam SM, Leblond CP. Dynamics of epithelial cells in the corpus of the mouse stomach. I. Identification of proliferative cell types and pinpointing of the stem cell. Anat Rec. 1993;236:259–279. doi: 10.1002/ar.1092360202. [DOI] [PubMed] [Google Scholar]
- 2.Karam SM, Leblond CP. Dynamics of epithelial cells in the corpus of the mouse stomach. III. Inward migration of neck cells followed by progressive transformation into zymogenic cells. Anat Rec. 1993;236:297–313. doi: 10.1002/ar.1092360204. [DOI] [PubMed] [Google Scholar]
- 3.Correa P. A human model of gastric carcinogenesis. Cancer Res. 1988;48:3554–3560. [PubMed] [Google Scholar]
- 4.Schmidt PH, Lee JR, Joshi V, et al. Identification of a metaplastic cell lineage associated with human gastric adenocarcinoma. Lab Invest. 1999;79:639–646. [PMC free article] [PubMed] [Google Scholar]
- 5.El-Zimaity HM, Ramchatesingh J, Saeed MA, et al. Gastric intestinal metaplasia: subtypes and natural history. J Clin Pathol. 2001;54:679–683. doi: 10.1136/jcp.54.9.679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Filipe MI, Munoz N, Matko I, et al. Intestinal metaplasia types and the risk of gastric cancer: a cohort study in Slovenia. Int J Cancer. 1994;57:324–329. doi: 10.1002/ijc.2910570306. [DOI] [PubMed] [Google Scholar]
- 7.Nam KT, Lee HJ, Mok H, et al. Amphiregulin-deficient mice develop spasmolytic polypeptide expressing metaplasia and intestinal metaplasia. Gastroenterology. 2009;136:1288–1296. doi: 10.1053/j.gastro.2008.12.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yoshizawa N, Takenaka Y, Yamaguchi H, et al. Emergence of spasmolytic polypeptide-expressing metaplasia in Mongolian gerbils infected with Helicobacter pylori. Lab Invest. 2007;87:1265–1276. doi: 10.1038/labinvest.3700682. [DOI] [PubMed] [Google Scholar]
- 9.Goldenring JR, Ray GS, Coffey RJ, et al. Reversible drug-induced oxyntic atrophy in rats. Gastroenterology. 2000;118:1080–1093. doi: 10.1016/s0016-5085(00)70361-1. [DOI] [PubMed] [Google Scholar]
- 10.Wang TC, Dangler CA, Chen D, et al. Synergistic interaction between hypergastrinemia and Helicobacter infection in a mouse model of gastric cancer. Gastroenterology. 2000;118:36–47. doi: 10.1016/s0016-5085(00)70412-4. [DOI] [PubMed] [Google Scholar]
- 11.Houghton J, Stoicov C, Nomura S, et al. Gastric cancer originating from bone marrow-derived cells. Science. 2004;306:1568–1571. doi: 10.1126/science.1099513. [DOI] [PubMed] [Google Scholar]
- 12.Nomura S, Baxter T, Yamaguchi H, et al. Spasmolytic polypeptide expressing metaplasia to preneoplasia in H. felis-infected mice. Gastroenterology. 2004;127:582–594. doi: 10.1053/j.gastro.2004.05.029. [DOI] [PubMed] [Google Scholar]
- 13.Ramsey VG, Doherty JM, Chen CC, et al. The maturation of mucus-secreting gastric epithelial progenitors into digestive-enzyme secreting zymogenic cells requires Mist1. Development. 2007;134:211–222. doi: 10.1242/dev.02700. [DOI] [PubMed] [Google Scholar]
- 14.Nam KT, Varro A, Coffey RJ, et al. Potentiation of oxyntic atrophy-induced gastric metaplasia in amphiregulin-deficient mice. Gastroenterology. 2007;132:1804–1819. doi: 10.1053/j.gastro.2007.03.040. [DOI] [PubMed] [Google Scholar]
- 15.Nozaki K, Ogawa M, Williams JA, et al. A molecular signature of gastric metaplasia arising in response to acute parietal cell loss. Gastroenterology. 2008;134:511–522. doi: 10.1053/j.gastro.2007.11.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shi G, Zhu L, Sun Y, et al. Loss of the acinar-restricted transcription factor Mist1 accelerates Kras-induced pancreatic intraepithelial neoplasia. Gastroenterology. 2009;136:1368–1378. doi: 10.1053/j.gastro.2008.12.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fox JG, Wang TC, Rogers AB, et al. Host and microbial constituents influence Helicobacter pylori-induced cancer in a murine model of hypergastrinemia. Gastroenterology. 2003;124:1879–1890. doi: 10.1016/s0016-5085(03)00406-2. [DOI] [PubMed] [Google Scholar]
- 18.Nomura S, Yamaguchi H, Ogawa M, et al. Alterations in gastric mucosal lineages induced by acute oxyntic atrophy in wild-type and gastrin-deficient mice. Am J Physiol Gastrointest Liver Physiol. 2005;288:G362–G375. doi: 10.1152/ajpgi.00160.2004. [DOI] [PubMed] [Google Scholar]
- 19.Barker N, van Es JH, Kuipers J, et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature. 2007;449:1003–1007. doi: 10.1038/nature06196. [DOI] [PubMed] [Google Scholar]
- 20.Bredemeyer AJ, Geahlen JH, Weis VG, et al. The gastric epithelial progenitor cell niche and differentiation of the zymogenic (chief) cell lineage. Dev Biol. 2009;325:211–224. doi: 10.1016/j.ydbio.2008.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fox JG, Li X, Cahill RJ, et al. Hypertrophic gastropathy in Helicobacter felis-infected wild-type C57BL/6 mice and p53 hemizygous transgenic mice. Gastroenterology. 1996;110:155–166. doi: 10.1053/gast.1996.v110.pm8536852. [DOI] [PubMed] [Google Scholar]
- 22.Wang TC, Goldenring JR, Dangler C, et al. Mice lacking secretory phospholipase A2 show altered apoptosis and differentiation with Helicobacter felis infection. Gastroenterology. 1998;114:675–689. doi: 10.1016/s0016-5085(98)70581-5. [DOI] [PubMed] [Google Scholar]
- 23.Guzinska-Ustymowicz K, Pryczynicz A, Kemona A, et al. Correlation between proliferation markers: PCNA, Ki-67, MCM-2 and antiapoptotic protein Bcl-2 in colorectal cancer. Anticancer Res. 2009;29:3049–3052. [PubMed] [Google Scholar]
- 24.Abe S, Sasano H, Katoh K, et al. Immunohistochemical studies on EGF family growth factors in normal and ulcerated human gastric mucosa. Dig Dis Sci. 1997;42:1199–1209. doi: 10.1023/a:1018897922644. [DOI] [PubMed] [Google Scholar]
- 25.Beauchamp RD, Barnard JA, McCutchen CM, et al. Localization of transforming growth factor alpha and its receptor in gastric mucosal cells. Implications for a regulatory role in acid secretion and mucosal renewal. J Clin Invest. 1989;84:1017–1023. doi: 10.1172/JCI114223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Murayama Y, Miyagawa J, Higashiyama S, et al. Localization of heparin-binding epidermal growth factor-like growth factor in human gastric mucosa. Gastroenterology. 1995;109:1051–1059. doi: 10.1016/0016-5085(95)90562-6. [DOI] [PubMed] [Google Scholar]
- 27.Stepan V, Ramamoorthy S, Nitsche H, et al. Regulation and function of the sonic hedgehog signal transduction pathway in isolated gastric parietal cells. J Biol Chem. 2005;280:15700–15708. doi: 10.1074/jbc.M413037200. [DOI] [PubMed] [Google Scholar]
- 28.Goldenring JR, Nam KT, Wang TC, et al. Spasmolytic polypeptide-expressing metaplasia and intestinal metaplasia: time for reevaluation of metaplasias and the origins of gastric cancer. Gastroenterology. 2010;138:2207–2210.e1. doi: 10.1053/j.gastro.2010.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Barker N, Ridgway RA, van Es JH, et al. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature. 2009;457:608–611. doi: 10.1038/nature07602. [DOI] [PubMed] [Google Scholar]
- 30.Sato T, Vries RG, Snippert HJ, et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature. 2009;459:262–265. doi: 10.1038/nature07935. [DOI] [PubMed] [Google Scholar]
- 31.Barker N, Huch M, Kujala P, et al. Lgr5(+ve) stem cells drive self-renewal in the stomach and build long-lived gastric units in vitro. Cell Stem Cell. 2010;6:25–36. doi: 10.1016/j.stem.2009.11.013. [DOI] [PubMed] [Google Scholar]
- 32.Lee HJ, Nam KT, Park HS, et al. Gene expression profiling of metaplastic lineages identifies CDH17 as a prognostic marker in early stage gastric cancer. Gastroenterology. 2010;139:213–225 e3. doi: 10.1053/j.gastro.2010.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cover TL, Blaser MJ. Helicobacter pylori in health and disease. Gastroenterology. 2009;136:1863–1873. doi: 10.1053/j.gastro.2009.01.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Habbe N, Shi G, Meguid RA, et al. Spontaneous induction of murine pancreatic intraepithelial neoplasia (mPanIN) by acinar cell targeting of oncogenic Kras in adult mice. Proc Natl Acad Sci U S A. 2008;105:18913–18918. doi: 10.1073/pnas.0810097105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Means AL, Meszoely IM, Suzuki K, et al. Pancreatic epithelial plasticity mediated by acinar cell transdifferentiation and generation of nestin-positive intermediates. Development. 2005;132:3767–3776. doi: 10.1242/dev.01925. [DOI] [PubMed] [Google Scholar]
- 36.Zhu L, Shi G, Schmidt CM, et al. Acinar cells contribute to the molecular heterogeneity of pancreatic intraepithelial neoplasia. Am J Pathol. 2007;171:263–273. doi: 10.2353/ajpath.2007.061176. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.