

Abstract
Both wound repair and fibrosing diseases involve circulating monocytes entering a tissue and differentiating into fibroblast-like cells called fibrocytes. Fibrocyte biology has been extensively studied in both humans and mice. However, current in vitro techniques to culture murine fibrocytes can take up to two weeks and can require multiple mice to obtain enough circulating monocytes for a single experiment. An alternative source of fibrocytes is the splenic reservoir of monocytes, where one can obtain significantly more cells compared to the peripheral blood. We found that in serum free medium, fibrocytes differentiate from murine spleen cells within 5 days. To maximize fibrocyte yield, we found the optimal purification technique was to digest the spleen with a collagenase/DNase cocktail, pass the cells through a cell strainer, and lyse the red blood cells. We found that IL-13 and M-CSF significantly enhanced fibrocyte differentiation and that the optimal cell density to promote differentiation was 1.75 × 106 cells/ml. Serum amyloid P (SAP) and cross-linked IgG are two factors known to inhibit the differentiation of human monocytes into fibrocytes. We found that SAP and cross-linked IgG also inhibited the differentiation of murine spleen cells into fibrocytes. These results suggest that culturing murine spleen cells in serum free medium is a rapid and efficient system to study factors that can affect fibrocyte differentiation.
Keywords: fibrocytes, monocytes, serum-free culture, spleen
1. Introduction
To heal wounds, several cell types help form new tissue. Local fibroblasts migrate to the source of the wound and proliferate (Clark, 2001). In addition, circulating monocytes can leave the blood, enter the wound, and differentiate into spindle-shaped, fibroblast-like cells called fibrocytes (Bucala et al., 1994; Abe et al., 2001). At the site of a wound, fibrocytes secrete extracellular matrix proteins such as collagen to rebuild tissue, secrete inflammatory cytokines, stimulate angiogenesis, and promote wound closure (Chesney et al., 1998; Hartlapp et al., 2001). In addition to their beneficial effects, fibrocytes have been implicated in the formation of the scar tissue-like lesions in a variety of fibrosing diseases such as pulmonary fibrosis, congestive heart failure, cirrhosis of the liver, and nephrogenic systemic fibrosis (Cowper et al., 2003; Phillips et al., 2004; Haudek et al., 2006; Kisseleva et al., 2006; Mehrad et al., 2007). Fibrocytes have also been detected in bronchial asthma and hypertrophic scarring (Schmidt et al., 2003; Yang et al., 2005). Fibrocytes derive from a subset of CD14+ monocytes in human peripheral blood, and mature fibrocytes express markers of both hematopoietic cells (CD34, CD45, LSP-1, MHC Class II) and stromal cells (collagen I and III, fibronectin) (Bucala et al. 1994; Abe et al., 2001; Gomperts et al., 2007; Pilling et al., 2009b). Fibrocyte differentiation is inhibited by the serum protein serum amyloid P (SAP) as well as cross-linked IgG (Pilling et al., 2003, 2006). In vitro, fibrocyte differentiation is affected by cell density and media composition (Pilling et al., 2009a).
Fibrocytes have also been cultured from murine peripheral blood mononuclear cells (PBMCs), although monocytes are only 1–4% of murine peripheral blood PBMCs (Abe et al., 2001; Kile et al., 2003). However, a large reservoir of monocytes has recently been found in the subcapsular red pulp of the spleen, and these can be mobilized for wound repair (Swirski et al., 2009). Niedermeier et al. (2009) recently used this splenic reservoir to show that fibrocytes differentiate from a subpopulation of CD11b+ CD115+ Gr1+ monocytes under the control of activated CD4+ T-cells. Their protocol involved culturing the cells in the presence of 10% FBS in RPMI for 14 days.
We previously found that culturing human and murine PBMC in serum-free medium results in fibrocytes appearing within 5 days (Pilling et al., 2003, 2006, 2009a). In this study, we found that mouse spleen cells can differentiate into fibrocytes in serum free conditions within 5 days. We identified conditions where a large number of fibrocytes can be produced from a single mouse spleen, which will allow for future studies testing the effect of multiple factors on murine fibrocyte differentiation.
2. Methods
2.1 Isolation of murine SAP
Murine SAP was purified from murine serum (Gemini Bio-products, West Sacramento, CA) using calcium-dependent binding to phosphoethanolamine-conjugated agarose, as described previously (Haudek et al., 2006; Pilling et al., 2007), with the exception that Amicon Ultra-15 centrifugal filters (Millipore, Billerica, MA) were used for the 20 mM phosphate buffer exchange.
2.2 Cell fractionation
Human peripheral blood was collected into heparin vacutainer tubes (#367874; BD Bioscience, Franklin Lakes, NJ) with written consent from healthy adult volunteers and with specific approval of Rice University’s Institutional Review Board. Peripheral blood mononuclear cells (PBMC) were isolated by Ficoll-Paque Plus (GE Healthcare Biosciences, Piscataway, NJ), as previously described (Pilling et al., 2009a).
4–6 week male C57BL/6J mice (Jackson Laboratories, Bar Harbor, ME or Taconic, Hudson, NY) were used in this study. All work was done under Rice and Texas A&M IACUC approved protocols. Murine peripheral blood was drawn by cardiac puncture using heparin-coated syringes, and mononuclear cells were purified by density centrifugation using Lympholyte-Mammal (Cedarlane, Burlington, NC), following the manufacturer’s protocol. Spleens were harvested (~70–100 mg each) and cells were isolated by digesting in a cocktail of 450 U/ml collagenase I (EMD, San Diego, CA), 60 U/ml DNase I, and 60 U/ml hyaluronidase (Sigma-Aldrich, St. Louis, MO) in 1 ml RPMI (Sigma-Aldrich) for 30 minutes at 37°C. Alternatively, spleen cells were isolated by forcing diced spleen fragments through a 100 μm cell strainer (BD Biosciences, San Jose, CA) using the plunger of a 3 ml syringe (BD Medical, Franklin Lakes, NJ) and 5 ml of RPMI. Finally, some spleen cells were isolated with a combination of digest cocktail, followed by the use of a cell strainer. After digest or passage through a cell strainer, the cells were resuspended in 10 ml RPMI and collected by centrifugation at 300 × g for 10 minutes. Cells were further purified using ammonium chloride, potassium bicarbonate (ACK) lysis buffer (0.15 M NH4Cl, 10 mM KHCO3, 0.1 mM Na2EDTA; Kruisbeek, 2000), or by density centrifugation using Lympholyte-Mammal or Ficoll-Paque Plus (GE Lifesciences, Piscataway, NJ). For ACK lysis, spleen cells were resuspended in 940 μl lysis buffer for 3 minutes at room temperature, and the reaction was stopped by the addition of 14 ml PBS. The cells were collected by centrifugation at 200 × g for 10 minutes, and the PBS wash step was repeated 3 additional times. A final wash was carried out by resuspending the cells in 1.5 ml Fibrolife basal media (Lifeline Cell Technology, Walkersville, MD) with the supplements listed below, and the cells were collected by centrifugation at 300 × g for 5 minutes at 4° C. For density centrifugation, spleen cells were resuspended in 10 ml RPMI and fractionated following the manufacturer’s protocol. The mononuclear fraction was then diluted to 14 ml with PBS and cells were collected by centrifugation at 300 × g for 10 minutes. The resuspension and centrifugation was repeated three times. The cells were resuspended in 1.5 ml Fibrolife basal media with supplements and collected by centrifugation at 300 × g for 5 minutes at 4° C.
2.3 Culture conditions and fibrocyte differentiation assay
PBMC and spleen cells were resuspended in 1 ml Fibrolife basal media (Lifeline Cell Technology), supplemented with 10 mM HEPES (Sigma-Aldrich), 2 x non-essential amino acids (Sigma-Aldrich), 2 mM sodium pyruvate (Sigma-Aldrich), 4 mM glutamine (Invitrogen, Carlsbad, CA), 100 U/ml penicillin, 100 g/ml streptomycin (Sigma-Aldrich), 2 x ITS-3 (Sigma-Aldrich), and 50 μm 2-mercaptoethanol (EMD). For some experiments, spleen cells were cultured in the presence or absence of 50 ng/ml murine IL-13 and 25 ng/ml murine M-CSF (Peprotech, Rocky Hill, NJ). Additionally, murine IL-4 (Peprotech), IL-13, and M-CSF were tested alone and in combination at the indicated concentrations. PBMC and spleen cells were cultured in flat-bottomed 96 well tissue culture plates (353072, BD Biosciences) at 200 μl per well at the indicated cell densities in a humidified incubator containing 5% CO2 at 37° C. On day 3 of the incubation, wells with IL-13 and M-CSF were further supplemented with 5 μl of a cocktail containing 1 μg/ml IL-13 and 0.5 μg/ml M-CSF in Fibrolife SFM. After 5 days, plates were air dried, fixed with methanol, and stained with Hema 3 stain (Fisher Scientific, Hampton, NH). Fibrocytes were counted in five different 900 μm diameter fields of view for each well of a 96 well plate, using the following criteria: an adherent cell with elongated spindle-shaped morphology and an oval nucleus (Pilling et al., 2003, 2006, and 2009a; Shao et al., 2008). The total number of fibrocytes per 105 input cells for each well was then calculated.
2.4 Immunohistochemistry
Mouse spleen cells, isolated by combination of digest cocktail, cell strainer, and ACK lysis, were cultured in 8 well glass slides (177402, Lab-tek, Nalge-Nunc, Naperville, IL) or 8 well CC2 glass slides (154941, Nalge-Nunc) at 4 × 106 cells/ml and 250 μl per well for 5 days in the presence or absence of IL-13 and M-CSF. Slides were air dried overnight, fixed in acetone, and stained as described previously (Pilling et al., 2009a) with the following modifications. Slides were stained for CD34 (clone RAM34, rat IgG2a, eBioscience, San Diego, CA), CD11c (clone 223H7, rat IgG2a, MBL Int., Woburn, MA), CD11b (clone M1/70, rat IgG2b, BioLegend, San Diego, CA), CD45 (clone 30-F11, rat IgG2b, BD Biosciences), syk (sc-1077, rabbit polyclonal, Santa Cruz Biotech., Santa Cruz, CA), or collagen I (600-401-103-01, rabbit polyclonal, Rockland, Inc., Gilbertsville, PA). Negative controls were rat IgG2a and IgG2b (BioLegend) and rabbit IgG (Jackson Immunoresearch, West Grove, PA). Primary antibodies were incubated at 5 μg/ml in PBS/BSA for 1 hour, except anti-CD34 was incubated at 10 μg/ml. Slides were then washed in 5 changes of 50 ml PBS over 15 minutes and incubated for 30 minutes in PBS/BSA with 2.5 μg/ml biotinylated mouse F(ab′)2 anti-rat IgG (Jackson Immunoresearch) or 2.5 μg/ml biotinylated goat F(ab′)2 anti-rabbit IgG (Southern Biotech, Birmingham, AL). The slides were then washed and developed as previously described (Pilling et al., 2009a).
2.5 Flow cytometry
For fibrocyte collagen analysis, spleen cells were cultured for 5 days in the presence of IL-13 and M-CSF. Adherent cells were dislodged with a 10 minute incubation in trypsin-EDTA (Sigma-Aldrich) at 37° C. Cells were washed with 14 ml PBS/10% mouse serum (Gemini) and collected by centrifugation at 300 × g for 10 minutes. The cells were resuspended in 100 μl ice-cold PBS containing 10% rat serum (Sigma-Aldrich), and incubated on ice for 15 minutes. Cells were collected by centrifugation at 300 × g for 5 minutes, and resuspended in 100 μl of PBS/4% BSA containing 2.5 μg/ml PE-conjugated anti-CD11b antibody (clone M1/70, rat IgG2b, eBioscience). After a 30 minute incubation on ice, the cells were washed twice with 1.4 ml ice-cold PBS and fixed with 100 μl of 4% paraformaldehyde in PBS for 15 minutes at room temperature. The cells were washed twice with 1.4 ml ice-cold PBS/4% BSA and permeabilized by resuspension in 100 μl PBS/4% BSA/0.1% saponin (Invitrogen). After a 15 minute incubation on ice, the cells were washed once with 1.4 ml ice-cold PBS/0.1% saponin, collected by centrifugation at 300 × g for 5 minutes, and then stained with antibodies against collagen type I (ab292-100, rabbit polyclonal, Abcam, Cambridge, MA), collagen type III (600-401-105-.1, rabbit polyclonal, Rockland), or syk (Santa Cruz) at 5 μg/ml in 100 μl PBS/4% BSA/0.1% saponin. After a 30 minute incubation on ice, cells were washed twice with 1.4 ml ice-cold PBS/0.1% saponin, and resuspend in 100 μl of PBS/4% BSA/0.1% saponin containing 2.5 μg/ml FITC-conjugated goat F(ab′)2 anti-rabbit IgG (Southern Biotech). After a 30 minute incubation on ice, cells were washed twice with 1.4 ml ice-cold PBS, resuspended in 100 μl PBS/BSA/saponin, and staining was analyzed using a C6 flow cytometer (Accuri, Ann Arbor, MI).
For monocyte analysis, mouse spleen cells were subjected to flow analysis as described previously (Pilling et al., 2009a) with the following modifications. Cells were stained for Ly6G (clone 1A8, rat IgG2a, BioLegend). The secondary antibody was a FITC-conjugated mouse F(ab′)2 anti-rat IgG (Jackson Immunoresearch), used at 2.5 μg/ml. After incubating the cells with the secondary antibody and washing, the cells were resuspended in 100 μl ice-cold PBS containing 10% rat serum (Sigma-Aldrich), and incubated on ice for 30 minutes. Cells were collected by centrifugation at 300 × g for 5 minutes, and resuspended in 100 μl of PBS/4% BSA containing 2.5 μg/ml PE-conjugated anti-CD11b antibody (clone M1/70, rat IgG2b, eBioscience). After a 30 minute incubation on ice, cells were washed twice with 1.4 ml ice-cold PBS, resuspended in 100 μl ice-cold PBS/BSA, and then analyzed using a C6 flow cytometer (Accuri).
2.6 Monocyte enrichment and CD11b depletion of spleen cells
Mouse spleen cells were isolated by the combination of digest cocktail, cell strainer, and ACK lysis. Monocytes were enriched from spleen cells using an EasySep mouse monocyte negative selection kit (Stemcell Technologies, Vancouver, BC), following the manufacturer’s protocol. Alternatively, CD11b+ cells were depleted from spleen cells using the EasySep Mouse CD11b positive selection kit (Stemcell Technologies), following the manufacturer’s protocol. The CD11b-depleted cells were then subjected to ACK lysis. The purity of the cells was checked by morphology, with a monocyte defined as a large cell (10–15 μm) with a kidney-shaped nucleus. Cytospins were prepared by centrifugation at 400 rpm for 5 minutes in a Shandon Cytospin 2 (Thermo Scientific, Waltham, MA) and stained with a Hema 3 kit (Fisher). The purity was also checked by staining for CD11b and Ly6G using flow cytometry, as described above. The cells were cultured for 5 days as described above, except the monocyte-enriched cells were plated at 5 × 105 cells/ml while the unfractionated spleen cells and CD11b-depleted cells were plated at 3 × 106 cells/ml.
2.7 Determining the effect of SAP and IgG on fibrocyte differentiation
Lympholyte-Mammal isolated murine PBMCs were cultured at 2.5 × 106 cells/ml. Spleen cells, isolated by the combination of digest cocktail, cell strainer, and ACK lysis, were cultured at 3.5 × 106 cells/ml. Murine and human SAP (EMD-Calbiochem) in 20 mM sodium phosphate buffer, pH 7.4, was added to the cells at the indicated concentrations. The cells were then cultured for 5 days, and fibrocytes were counted as described above.
Spleen cells were also tested for sensitivity to IgG. Chromopure murine IgG (Jackson Immunoresearch) was clarified by centrifugation at 14,000 × g for 15 minutes at 4° C to remove aggregates. Mouse spleen cells at 5 × 106 cells/ml in 500 μl were incubated with 100, 10, 1, or 0 μg/ml IgG for 30 minutes at 4° C. Cells were washed with 1.4 ml ice-cold Fibrolife medium and collected by centrifugation at 300 × g for 5 minutes at 4° C. Cells were resuspended in 500 μl Fibrolife medium and incubated with 500 ng/ml goat F(ab′)2 anti-mouse IgG (Southern Biotech) for 30 minutes at 4° C.
Cells were washed with 1.4 ml Fibrolife, collected by centrifugation at 300 × g for 5 minutes, and resuspended to 500 μl. Cells were counted and diluted to 3.5 × 106 cells/ml. The cells were then cultured for 5 days, and fibrocytes were counted.
2.8 Statistical analysis
Statistical analysis was performed using GraphPad Prism software (Graphpad Software, San Diego, CA). Significance was defined as p < 0.05.
3. Results
To study factors that regulate the differentiation of monocytes to fibrocytes, peripheral blood mononuclear cells can be isolated from blood and cultured in vitro. When murine PBMCs are utilized, the cells must be cultured at higher densities due to the low percentage of monocytes found in mouse blood (Wirth et al., 1982; Kile et al., 2003); therefore, the blood from multiple mice must be pooled together to obtain enough cells for a single experiment (Pilling et al, 2009a). Murine spleens have recently been shown to contain a monocyte population that can be mobilized for wound repair (Swirski et al., 2009), and spleen cells have been shown to differentiate into fibrocytes, although this process requires serum and an incubation period of 14 days (Niedermeier et al., 2009). Therefore, we tested if cultured mouse spleen cells can differentiate into fibrocytes in serum free conditions.
3.1 Spindle-shaped cells that appear in cultures of spleen cells express fibrocyte markers
We previously observed the appearance of spindle-shaped cells in 5 day serum-free cultures of human and murine PBMCs (Pilling et al., 2003, 2006, 2009a; Shao et al., 2008). These cells showed positive staining for markers that have been used to classify cells as fibrocytes, including the general leukocyte marker CD45, the stem cell marker CD34, the monocyte marker CD11b, and the matrix biosynthesis marker collagen I (Bucala et al., 1994; Abe et al., 2001, Pilling et al., 2003). To assess the viability of culturing mouse spleen cells to study fibrocyte differentiation, we first confirmed the presence of fibrocytes in mouse spleen cell cultures. Mouse spleen cells were initially isolated by passage through a cell strainer and further purified by density centrifugation. After a 5 day incubation in serum-free media, we observed spindle-shaped cells in the culture. To confirm the identity of the spindle-shaped cells, we stained the cells for a variety of markers. The spindle-shaped cells were positive for well-known markers of fibrocytes, including CD11b (Figure 1D), CD45 (Figure 1E), and weakly positive for collagen I (Figure 1H). The cells also stained positive for CD34 (Figure 1A) with variable expression levels, similar to the variability found in human fibrocytes (Pilling et al., 2003 and 2009b; Phillips et al., 2004). Fibrocytes are known to produce low levels of collagen (Wang et al., 2007; Pilling et al., 2009b), but to further confirm the presence of collagen, we measured the intracellular levels of collagen on 5 day cultured spleen cells by flow cytometry. The overall cell population (Figure 2A) showed little to no staining for collagen (Figure 2C), but CD11b+ cells (Figure 2B) stained positive for both collagen I and III (Figures 2D and 2E). Similar to the immunohistochemistry results, the CD11b+ cells only stained weakly positive for collagen I with a median fluorescent intensity of 3.4 × 103 ± 0.1 × 103 compared to 5.8 × 104 ± 0.6 × 104 for the positive control Syk (Figure 2D). To confirm that the spindle-shaped cells were not dendritic cells, we stained by immunohistochemistry for CD11c, a marker for dendritic cells (Shortman et al., 2002). We observed that cells in culture with a dendritic shape stained strongly for CD11c, while the spindle-shaped cells were either negative or very weakly positive (Figure 1B). This suggests that the spindle-shaped cells are fibrocytes rather than dendritic cells. Together, these observations suggest that murine spleen cells cultured for 5 days in serum-free media can differentiate into fibrocytes. We can therefore use murine spleen cells to further study conditions and factors that affect fibrocyte differentiation.
Figure 1. Cultured mouse spleen cells express markers of fibrocytes.

Isolated spleen cells were cultured for 5 days at 4 × 106 cells/ml on 8 well CC2 slides, then air dried, fixed, and stained with antibodies against (A) CD34, (B) CD11c, (C) rat IgG 2a control for A and B, (D) CD11b, (E) CD45, (F) rat IgG 2b control for D and E, (G) syk, (H) collagen I, and (I) rabbit IgG control for G and H. Cells were counterstained with hematoxylin to identify nuclei. Bar is 50 μm, and arrow in A indicates a fibrocyte. Images are representative of four independent experiments.
Figure 2. Expression of collagen by 5 day cultured spleen cells.

ACK-treated spleen cells were cultured for 5 days at 3.5 × 105 cells per well in the presence of IL-13 and M-CSF. Adherent cells were removed by trypsin-EDTA treatment and stained for the presence of CD11b. The cells were then fixed, permeabilized, stained with rabbit polyclonal antibodies, and analyzed by flow cytometry. A) Forward and side scatter characteristics of 5 day cultured spleen cells. (B) Forward and side scatter analysis of CD11b+ cells. C and D) Histograms show fluorescence intensity of FITC-conjugated goat F(ab′)2 anti-rabbit 2° (black line) compared to collagen I (red line), collagen III (blue line), and as a positive control syk (yellow line). Flow cytometry plots are representative of 3 independent experiments. C) When gating the entire live cell population R1 (from A), collagen was nearly undetectable, but when gating (D) for CD11b+ cells and region P1 (from A and B) there was an increase in the levels of both collagen I and III. E) Compared to FITC control, CD11b+ cells had a significant increase in median fluorescent intensity for collagen I and III staining (1-way ANOVA, Dunnett’s test). **, p < 0.01; ***, p < 0.001.
3.2 Effect of purification method on fibrocyte differentiation
Murine splenocytes are typically isolated either by passage through a cell strainer or by digestion with enzymes (Swirski et al., 2009). Mononuclear cells are typically further purified by density centrifugation, and leukocytes are purified by lysis of red blood cells (Kruisbeek et al., 2000). We tested a variety of cell purification methods to optimize spleen cell recovery and fibrocyte differentiation. After removal of the spleen, we isolated cells by passage through a cell strainer, digestion with collagenase and DNase, or a combination of the two. We further purified the isolated spleen cells by density centrifugation with Lympholyte-Mammal or Ficoll, or by ACK lysis to remove red blood cells. For the initial isolation, the digest cocktail or the combination of cell strainer and digest resulted in the highest recovery of spleen cells (Figures 3A, 3C, and 3E). For the additional purification step, ACK lysis resulted in a significantly higher recovery of spleen cells compared to density centrifugation by Lympholyte-Mammal or Ficoll (Figures 3A, 3C, and 3E). For fibrocyte yield, there was no significant difference between the initial isolation techniques (Figures 3B, 3D, and 3F). However, for the additional purification step, ACK lysis showed a trend of increased fibrocyte differentiation compared to density centrifugation (Figures 3B, 3D, and 3F). Additionally, the cultures prepared by ACK lysis and density centrifugation were checked for the presence of any contaminating fibroblasts by morphology on day 1 and day 5 of the culture. The characteristic morphology of fibroblasts was never observed in the cultures, nor was there strong collagen staining of any cell in the culture, which would be indicative of a fibroblast. Due to high spleen cell recovery and yielding the highest number of fibrocytes, the remaining experiments were performed using the digest cocktail plus cell strainer technique, followed by ACK lysis.
Figure 3. Effect of spleen cell isolation and purification techniques on fibrocyte differentiation.

A and B) After removal, the spleen was treated with collagenase/DNase, and the cells were further purified through ACK lysis or density centrifugation. C and D) After removal, the spleen was passed through a 100 μm cell strainer, and the cells were further purified by ACK lysis or density centrifugation. E and F) After removal, the spleen was treated with collagenase/DNase and then passed through a 100 μm cell strainer. The cells were further purified by ACK lysis or Lympholyte-Mammal density centrifugation. For A, C, and E, the total number of cells recovered by the indicated purification technique was measured. For B, D, and F, the purified spleen cells were cultured in SFM in the presence or absence of IL-13 and M-CSF for 5 days. Cells were air dried, fixed, stained, and the number of fibrocytes was counted. Results are mean ± SEM (n=3).
3.3 Effect of culture conditions on fibrocyte differentiation
We previously found that human fibrocyte differentiation was increased when PBMC were cultured with the pro-fibrotic cytokines IL-4 and IL-13 (Shao et al., 2008). Spleen monocytes have also been successfully cultured in the presence of GM-CSF and M-CSF (Swirski et al., 2009), and fibrocyte differentiation from murine spleen cells was markedly enhanced using conditioned media from CD4+ T cells (Niedermeier et al., 2009). Therefore, we tested the effect of cytokines on spleen cell differentiation in our serum-free medium culture conditions. We cultured ACK-treated spleen cells in the presence of murine M-CSF to promote monocyte survival, along with IL-4 and IL-13 as pro-fibrotic cytokines, both individually and in combination. Both M-CSF alone and the combination of IL-13 and M-CSF increased the number of fibrocytes in culture, but M-CSF alone tended to promote macrophage and dendritic-like cell differentiation as well as spindle-shaped, pseudo-fibrocyte cells that did not meet the criteria of a fibrocyte, i.e., an elongated spindle-shaped cell with an oval nucleus (Figure 4A). These pseudo-fibrocytes tended to have round nuclei, shorter processes with increased width, and were not counted as fibrocytes. The addition of IL-13 and M-CSF resulted in significantly more elongated spindle-shaped cells with oval nuclei (Figures 4B), which also express the same fibrocyte markers found in Figure 1 (data not shown). When added individually, IL-4, at concentrations above 12.5 ng/ml, significantly decreased the number of fibrocytes, IL-13 had no effect on fibrocyte differentiation, and 25 ng/ml M-CSF significantly increased the number of fibrocytes (Figure 4C). When used in combination, IL-13 and M-CSF showed a significant increase in fibrocyte number compared to M-CSF alone (Figure 4D) and compared to cells not treated with cytokines (Figures 3B, 3D, 3F; Figure 4D; Figure 5). When IL-4 was cultured with M-CSF or in combination with M-CSF and IL-13, the number of fibrocytes was reduced to control levels, suggesting that the inhibitory effect of IL-4 counteracts the potentiating effects of M-CSF and IL-13 on fibrocyte differentiation (Figure 4D). Together, these results suggest that IL-13 and M-CSF can be used to promote the differentiation of fibrocytes from ACK-treated spleen cells in serum-free culture.
Figure 4. The effect of cytokines on spleen fibrocyte differentiation.

ACK-treated spleen cells were cultured for 5 days at 3.5 × 105 cells per well in the presence of the indicated cytokines. Cells were air dried, fixed, stained, and the number of fibrocytes was counted. Wells treated with (A) M-CSF alone resulted in an increase of macrophage and dendritic-like cells (*) and pseudo-fibrocytes (white arrow), with round nuclei and shorter processes with increased width. Wells treated with (B) M-CSF and IL-13 in combination had more elongated, spindle-shaped cells with oval nuclei (black arrow). Bar is 100 μm, pictures are representative of 3 independent experiments. (C) IL-4, IL-13, and M-CSF were added to cultures individually to the indicated concentrations, or (D) in combination at 50 ng/ml for IL-4 and IL-13 and 25 ng/ml for M-CSF. Compared to SFM, ≥ 25 ng/ml IL-4 significantly reduced fibrocyte differentiation, while 25 ng/ml M-CSF significantly increased fibrocyte differentiation (1-way ANOVA, Dunnett’s test). The combination of IL-13 and M-CSF signficanty increased fibrocyte differentiation compared to M-CSF alone (1-way ANOVA, Dunnett’s test). *, p < 0.05; **, p < 0.01. Results are mean ± SEM (n=3).
Figure 5. The effect of cell density on spleen fibrocyte differentiation.
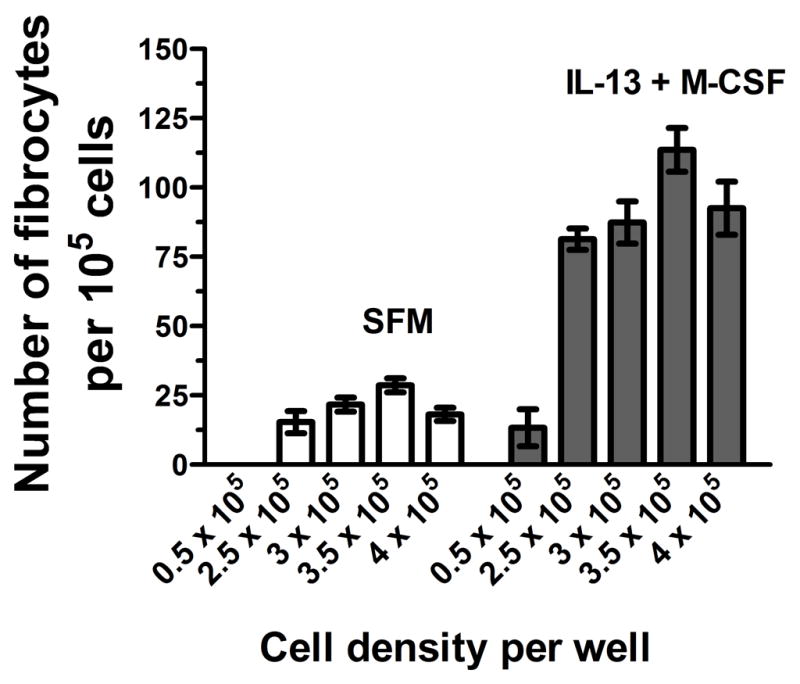
ACK-treated spleen cells were cultured in SFM in the presence or absence of IL-13 and M-CSF for 5 days at a range of cell densities. Cells were air dried, fixed, stained, and fibrocytes were counted. Results are mean ± SEM (n=3).
In vitro, human fibrocyte differentiation is dependent on cell density (Pilling et al., 2009a). We therefore cultured ACK-treated spleen cells at different cell densities to determine the optimal culture condition. The highest yield of fibrocytes was observed at 3.5×105 cells per well (Figure 5). For the remaining assays, cells were cultured at 3.5 × 105 cells per well unless otherwise noted.
3.4 Monocyte origin of spleen-derived fibrocytes
Fibrocyte precursors appear to differentiate from human CD14+ peripheral blood monocytes (Abe et al., 2001 Yang et al., 2002; Pilling et al., 2003 & 2006). Niedermeier et al. (2009) found that murine fibrocytes derive from a GR-1+, Ly6G- monocyte subpopulation using splenocytes cultured for 14 days in serum. To confirm the origin of the fibrocytes in our serum-free 5 day culture system, we isolated monocytes from ACK-treated spleen cells. When the spleen cells were enriched for monocytes using an EasySep kit, the population of CD11b+, Ly6G- cells increased from less than 2.7% of total cells to more than 60% (Figures 6A, 6B, 6D, and 6E). Additionally, the monocyte-enriched population had an increase in the number of cells with kidney-shaped nuclei (Figures 6C and 6F) compared to spleen cell controls. The morphology of the cells along with the flow cytometry data suggests that the population was predominantly composed of monocytes. When cultured for 5 days, the monocyte-enriched population had a significant increase in fibrocyte differentiation (Figure 6G) compared to ACK-treated spleen cultures.
Figure 6. Enrichment of monocytes from spleen cells significantly enhances fibrocyte differentiation.

Spleen cells were isolated through enzymatic digest, passed through a 100 μm cell strainer, and further purified by ACK lysis. The ACK-treated spleen cells were then enriched for monocytes through negative selection. ACK-treated spleen and monocyte-enriched cells were stained with rat monoclonal antibodies and analyzed by flow cytometry. Histograms show fluorescence intensity of isotype control antibody (black line) compared to the indicated antibody (red line). Compared to ACK-treated spleen cells (A and B), monocyte-enriched cells (D and E) had an increased number of CD11b+, Ly6G- cells. Flow cytometry plots are representative of 3 independent experiments. ACK-treated spleen and monocyte-enriched cells were analyzed for morphology. Monocyte-enriched cultures (F) showed an increased number of cells with kidney shaped nuclei (stained dark purple) compared to ACK-treated spleen cells (C). Bar is 20 μm. Pictures are representative of 3 independent experiments. G) ACK-treated spleen and monocyte-enriched cells were cultured for 5 days at 3.5 × 105 cells per well and 0.5 × 105 cells per well, respectively, in the presence or absence of IL-13 and M-CSF. Cells were air dried, fixed, stained, and the number of fibrocytes was counted. Results are mean ± SEM (n=3). Monocyte-enrichment significantly increased the number of fibrocytes (t-test). *, p < 0.05; **, p <0.01.
Niedermeier et al. (2009) found that the appearance of fibrocytes was dependent on CD11b+ monocytes. When we depleted monocytes and other CD11b+ cells from ACK-treated spleen cells using an EasySep CD11b kit, fibrocytes did not appear in our serum-free culture (data not shown). Together, the data suggest that the fibrocytes from our serum free 5 day culture are of monocyte origin, comparable to what has been observed in other systems (Abe et al., 2001; Pilling et al., 2003; Niedermeier et al., 2009).
3.5 SAP and cross-linked IgG inhibit the differentiation of spleen-derived fibrocytes
We previously found that a number of factors can regulate the differentiation of human PBMCs into fibrocytes (Pilling et al., 2003, 2006; Shao et al., 2008). For instance, SAP and aggregated IgG inhibit fibrocyte differentiation (Pilling et al., 2003, 2006). To determine if purified murine spleen cells can also be used to test the bioactivity of such factors, we cultured ACK-treated murine spleen cells with different concentrations of SAP. At concentrations above 10 μg/ml, SAP significantly reduced the number of fibrocytes compared to serum-free medium and buffer controls (Figure 7). The IC50 values for SAP inhibition of murine spleen cells are comparable to those of murine PBMCs treated with SAP (Table 1). For both murine PBMCs and spleen cells, human SAP was significantly more effective at inhibiting fibrocyte differentiation than murine SAP. Together, these results suggest that cultured spleen cells can give similar biological responses as peripheral blood mononuclear cells. Interestingly, in the presence of IL-13 and M-CSF, the addition of 60 μg/ml murine SAP did not fully inhibit fibrocyte differentiation (Figure 5B); however, the IC50 of murine SAP was not significantly altered (1-way ANOVA, Tukey’s test, Table 1).
Figure 7. Murine and human SAP inhibit fibrocyte differentiation of cultured spleen cells.

Murine, ACK-treated spleen cells were cultured for 5 days at 3.5 × 105 cells per well in the (A) absence or (B) presence of IL-13 and M-CSF, and the indicated mouse and human SAP concentrations. Cells were air dried, fixed, stained, and the number of fibrocytes was counted. Results are mean ± SEM (n=3). In the absence or presence of IL-13 and M-CSF, ≥ 10 μg/ml mouse SAP or ≥ 1 μg/ml human SAP significantly inhibited fibrocyte differentiation compared to buffer control (t-test). *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Table 1.
SAP IC50 values for the inhibition of fibrocyte differentiation in human and murine cell cultures.
| Cells | Mouse SAP IC50 μg/ml | Human SAP IC50 μg/ml |
|---|---|---|
| Human PBMC | 0.78 ± 0.15 | 0.41 ± 0.05* |
| Murine PBMC | 7.1 ± 1.0 | 1.6 ± 0.1** |
| Murine Spleen | 9.0 ± 2.1 | 1.3 ± 0.3** |
| Murine Spleen + Cytokines | 15 ± 3.7 | 1.9 ± 0.3** |
Human PBMCs, murine PBMCs, and murine spleen cells were cultured for 5 days at 5 × 104 cells per well, 2.5 × 105 cells per well, and 3.5 × 105 cells per well, respectively, in the presence of human and murine SAP. Murine spleen cells were also cultured in the presence or absence of the cytokines IL-13 and M-CSF. Cells were air dried, fixed, stained, and enumerated by morphology. Using fibrocyte counts normalized to SFM controls, IC50 levels were calculated by fitting SAP bioactivity to a sigmoidal dose response curve with variable slope. Results are expressed as mean ± SEM (n=5 for spleens, n=3 for mouse PBMCs, n=6 for human PBMCs). For the inhibition of fibrocyte differentiation by murine SAP, no significant difference was observed between the IC50 values for murine PBMCs and murine spleen cells (1-way ANOVA, Tukey’s test). Human SAP was significantly more effective than murine SAP on both human cells (p<0.05) and mouse cells (p<0.01; t-test).
p < 0.05;
p < 0.01.
The differentiation of human PBMCs into fibrocytes is inhibited by cross-linked IgG (Pilling et al., 2006). To determine if murine spleen cells have a similar biological response, we cultured spleen cells in the presence or absence of monomeric and cross-linked IgG. Monomeric murine IgG at concentrations up to 100 μg/ml and goat-anti mouse IgG F(ab′)2 crosslinker alone had no effect on fibrocyte differentiation (Figure 8). The addition of the goat F(ab′)2 anti-mouse IgG to monomeric IgG at concentrations as low as 10 μg/ml led to a significant reduction in fibrocyte differentiation (Figure 8). Similar to the ability of SAP and cross-linked IgG to inhibit the differentiation of human PBMCs into fibrocytes, the differentiation of mouse spleen cells into fibrocytes is inhibited by both SAP and crosslinked IgG. Since the biological response is consistent with those observed from human PBMC cultures, mouse spleen cultures appear to be a viable alternative to peripheral blood for studying factors that affect fibrocyte differentiation.
Figure 8. Cross-linked but not monomeric IgG inhibit fibrocyte differentiation of spleen cells.

Murine, ACK-treated spleen cells were incubated with the indicated concentrations of murine IgG for 30 minutes, followed by incubation in the presence or absence of 500 ng/ml goat F(ab′)2 anti-mouse IgG cross-linker for 30 minutes. After washing the spleen cells, the cells were cultured for 5 days at 3.5 × 105 cells per well in the presence or absence of IL-13 and M-CSF. Cells were air dried, fixed, stained, and the number of fibrocytes was counted. Results are mean ± SEM (n=5). Compared to controls, cross-linked IgG at 10 and 100 μg/ml significantly reduced fibrocyte differentiation (1-way ANOVA, Dunnett’s test). *, p < 0.05; **, p < 0.01; ***, p < 0.001.
4. Discussion
We previously found that we could culture both human and murine PBMCs in serum-free culture for 5 days to obtain fibrocytes and use this system to test for factors that affect fibrocyte differentiation (Pilling et al., 2003, 2006, 2009a; Shao et al., 2008). However, to obtain enough mouse PBMCs, we had to pool blood from multiple mice. We therefore explored alternative sources for mononuclear cells, including the spleen. The spleen has been shown to contain monocytes that can participate in wound repair, and spleen cells have been shown to differentiate into fibrocytes in a 14 day serum culture (Swirski et al., 2009; Niedermeier et al., 2009). In this report, we found that murine spleen cells can also be cultured in our serum-free system to obtain fibrocytes and test for factors that can affect fibrocyte differentiation. Our study focused on the common C57BL/6 strain. For other strains, the fibrocyte yield from spleen and blood cells, and response to cytokines, could be different, although results similar to C57BL/6 mice are expected.
We found that for optimal fibrocyte differentiation, leukocytes should be isolated from the spleen using a collagenase/DNase digest cocktail, followed by passage through a cell strainer and lysis of the red blood cells with ACK lysing buffer. The use of the digest cocktail led to an increase in the number of fibrocytes, which may be due to the release of monocytes attached to the matrix of the spleen. The number of non-red blood cells isolated from a spleen is ~ 40 times higher than the number of non-red blood cells isolated from approximately 1 ml of the peripheral blood of a mouse. However, the percentage of monocytes in the spleen cell preparation is 0.5% compared to 1–2% for the peripheral blood of C57BL/6 mice (Kile et al., 2003; Swirski et al., 2009). Thus, there are ~ 10 times more monocytes obtained from a spleen than peripheral blood. We observed that ~ 6 % of the input monocytes from the spleen differentiated into fibrocytes and this percentage increased 4-fold with the addition of IL-13 and M-CSF. For peripheral blood, ~ 8 % of the input monocytes differentiated into fibrocytes. These results are comparable to the differentiation in human PBMC, where ~ 5% of the input monocytes differentiate into fibrocytes, and this percentage is increased 4-fold with the addition of IL-13 (Pilling et al., 2003 and 2009a; Shao et al., 2008).
We further optimized fibrocyte differentiation by plating the cells at high cell density. The optimal cell density of 1.75 × 106 cells/ml is significantly higher than the optimal cell density of 2.5 × 105 cells/ml we observed for our serum free culture of human PBMCs (Pilling et al., 2009a). One possible explanation for the higher optimal cell density for spleen cells is that the percentages of monocytes in murine blood and spleen are significantly lower than that of circulating monocytes in humans (Kile et al., 2003; Swirski et al., 2009; Robbins et al., 2010). In addition, the promotion of fibrocyte differentiation by high cell density is may be due to both cell-to-cell contacts and soluble factors. In fact, T-cells have been shown to be required for the differentiation of spleen monocytes into fibrocytes (Niedermeier et al., 2009), and Th2 cells can secrete pro-fibrotic cytokines such as IL-13, a factor known to enhance fibrocyte differentiation (Shao et al, 2008).
One final optimization was to repeat all assays in the presence of IL-13 and M-CSF. IL-13 was selected for its ability to enhance fibrocyte differentiation of human PBMCs (Shao et al. 2008) and M-CSF to promote monocyte survival (Becker et al., 1987). Utilizing a combination of factors to promote leukocyte differentiation is a standard practice; for instance, GM-CSF and IL-4 are used to promote dendritic cells (Inaba et al., 1992; Sallusto et al., 1994), and M-CSF and serum are used to promote macrophages (Tushinski et al., 1982). The use of IL-13 and M-CSF more than tripled the number of fibrocytes, while not significantly altering the biological response of the spleen cells to differing culture conditions and factors such as SAP and cross-linked IgG. In contrast, IL-4 inhibited fibrocyte differentiation and negated the potentiating effects of IL-13 and M-CSF to control levels. We previously found that IL-4 promoted fibrocyte differentiation in human PBMCs (Shao et al., 2008), but murine IL-4 has the opposite effect when incubated with murine spleen cells, which was also observed by Niedermeier et al. (2009). The ability of IL-4 to counteract IL-13 and M-CSF can partially be explained by the fact that mouse monocytes are significantly more sensitive to levels of IL-4 than IL-13 (Junttila et al., 2008). Additionally, there is biological divergence between humans and mice for cytokine activity, such as the differential effects on B cells by IL-4 and IL-13 (Mestas et al., 2004), but a possible divergence has not been fully explored for fibrocyte differentiation. It is known that IL-4 is non-essential in the bleomycin-induced model of murine lung fibrosis (Izbicki, et al., 2002), while IL-13 deficient animals were protected from FITC-induced lung fibrosis (Kolodsick et al., 2004). However, both cytokines and their receptors are induced in bleomycin-induced lung fibrosis (Jakubzick et al., 2003).
We were able to culture over 3 × 104 fibrocytes from a single spleen, and this number increased to 1.1 × 105 fibrocytes in the presence of IL-13 and M-CSF. This is considerably higher than the 4 × 103 fibrocytes we could obtain from the peripheral blood of a single mouse (~ 1 ml). After 14 days in cultures containing serum, Abe et al. (2001) observed 8 – 40 × 103 fibrocytes from 1 ml of peripheral blood. The higher number of fibrocytes observed by Abe et al. (2001) could be due to the culturing of fibrocytes for 14 days in serum compared to our culturing of fibrocytes in serum-free medium for 5 days. Another possibility is that Abe et al. (2001) identified fibrocytes using flow cytometry and staining for CD11b and collagen I, while we identified fibrocytes both morphologically and by marker expression.
The serum-free culture of mouse spleen cells resulted in the differentiation of fibrocytes over a short period of time compared to the typical 14 day period required in serum-based cultures. The delay in serum-based cultures is likely due to components in serum such as IgG and SAP that can inhibit fibrocyte differentiation. We were able to identify conditions to produce a high yield of fibrocytes from a single spleen, and the cells were sensitive to factors known to inhibit fibrocyte differentiation. This will allow for efficient testing of factors that can affect fibrocyte differentiation in multiple murine backgrounds.
Acknowledgments
This work was supported by NIH grant HL083029. We would like to thank Promedior for the generous donation of human SAP. We would also like to thank Kelly Campbell for excellent technical assistance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abe R, Donnelly SC, Peng T, Bucala R, Metz CN. Peripheral blood fibrocytes: differentiation pathway and migration to wound sites. J Immunol. 2001;166:7556–7562. doi: 10.4049/jimmunol.166.12.7556. [DOI] [PubMed] [Google Scholar]
- Becker S, Warren MK, Haskill S. Colony-stimulating factor-induced monocyte survival and differentiation into macrophages in serum-free cultures. J Immunol. 1987;139:3703–9. [PubMed] [Google Scholar]
- Bucala R, Spiegel LA, Chesney J, Hogan M, Cerami A. Circulating fibrocytes define a new leukocyte subpopulation that mediates tissue repair. Mol Med. 1994;1:71–81. [PMC free article] [PubMed] [Google Scholar]
- Chesney J, Metz C, Stavitsky AB, Bacher M, Bucala R. Regulated production of type I collagen and inflammatory cytokines by peripheral blood fibrocytes. J Immunol. 1998;160:419–425. [PubMed] [Google Scholar]
- Cowper SE, Bucala R. Nephrogenic fibrosing dermopathy: suspect identified, motive unclear. Am J Dermatopathol. 2003;25:358. doi: 10.1097/00000372-200308000-00017. [DOI] [PubMed] [Google Scholar]
- Gomperts BN, Strieter RM. Fibrocytes in lung disease. J Leukoc Biol. 2007;82:449–56. doi: 10.1189/jlb.0906587. [DOI] [PubMed] [Google Scholar]
- Hartlapp I, Abe R, Saeed RW, Peng T, Voelter W, Bucala R, Metz CN. Fibrocytes induce an angiogenic phenotype in cultured endothelial cells and promote angiogenesis in vivo. The FASEB Journal. 2001;15:2215–2224. doi: 10.1096/fj.01-0049com. [DOI] [PubMed] [Google Scholar]
- Haudek SB, Xia Y, Huebener P, Lee JM, Carlson S, Crawford JR, Pilling D, Gomer RH, Trial J, Frangogiannis NG, Entman ML. Bone marrow-derived fibroblast precursors mediate ischemic cardiomyopathy in mice. Proc Natl Acad Sci USA. 2006;103:18284–9. doi: 10.1073/pnas.0608799103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inaba K, Steinman RM, Pack MW, Aya H, Inaba M, Sudo T, Wolpe S, Schuler G. Identification of proliferating dendritic cell precursors in mouse blood. J Exp Med. 1992;175:1157–67. doi: 10.1084/jem.175.5.1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junttila IS, Mizukami K, Dickensheets H, Meier-Schellersheim M, Yamane H, Donnelly RP, Paul WE. Tuning sensitivity to IL-4 and IL-13: differential expression of IL-4Ralpha, IL-13Ralpha1, and gammac regulates relative cytokine sensitivity. J Exp Med. 2008;205:2595–608. doi: 10.1084/jem.20080452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kile BT, Mason-Garrison CL, Justice MJ. Sex and strain-related differences in the peripheral blood cell values of inbred mouse strains. Mamm Genome. 2003;14:81–5. doi: 10.1007/s00335-002-2160-0. [DOI] [PubMed] [Google Scholar]
- Kisseleva T, Uchinami H, Feirt N, Quintana-Bustamante O, Segovia JC, Schwabe RF, Brenner DA. Bone marrow-derived fibrocytes participate in pathogenesis of liver fibrosis. J Hepatol. 2006;45:429–438. doi: 10.1016/j.jhep.2006.04.014. [DOI] [PubMed] [Google Scholar]
- Kruisbeek AM. Isolation and fractionation of mononuclear cell populations. Curr Protoc Immunol. 2000;39:3.1.3–3.1.5. [Google Scholar]
- Mehrad B, Burdick MD, Zisman DA, Keane MP, Belperio JA, Strieter RM. Circulating peripheral blood fibrocytes in human fibrotic interstitial lung disease. Biochem Biophys Res Commun. 2007;353:104–8. doi: 10.1016/j.bbrc.2006.11.149. [DOI] [PubMed] [Google Scholar]
- Mestas J, Hughes CC. Of mice and not men: differences between mouse and human immunology. J Immunol. 2004;172:2731–8. doi: 10.4049/jimmunol.172.5.2731. [DOI] [PubMed] [Google Scholar]
- Niedermeier M, Reich B, Rodriguez Gomez M, Denzel A, Schmidbauer K, Gobel N, Talke Y, Schweda F, Mack M. CD4+ T cells control the differentiation of Gr1+ monocytes into fibrocytes. Proc Natl Acad Sci USA. 2009;106:17892–7. doi: 10.1073/pnas.0906070106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips RJ, Burdick MD, Hong K, Lutz MA, Murray LA, Xue YY, Belperio JA, Keane MP, Strieter RM. Circulating fibrocytes traffic to the lungs in response to CXCL12 and mediate fibrosis. J Clin Invest. 2004;114:438–446. doi: 10.1172/JCI20997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilling D, Buckley CD, Salmon M, Gomer RH. Inhibition of fibrocyte differentiation by serum amyloid P. J Immunol. 2003;17:5537–5546. doi: 10.4049/jimmunol.171.10.5537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilling D, Tucker NM, Gomer RH. Aggregated IgG inhibits the differentiation of human fibrocytes. J Leukoc Biol. 2006;79:1242–51. doi: 10.1189/jlb.0805456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilling D, Roife D, Wang M, Ronkainen SD, Crawford JR, Travis EL, Gomer RH. Reduction of bleomycin-induced pulmonary fibrosis by serum amyloid P. J Immunol. 2007;179:4035–44. doi: 10.4049/jimmunol.179.6.4035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilling D, Vakil V, Gomer RH. Improved serum-free culture conditions for the differentiation of human and murine fibrocytes. J Immunol Methods. 2009a;351:62–70. doi: 10.1016/j.jim.2009.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilling D, Fan T, Huang D, Kaul B, Gomer RH. Identification of markers that distinguish monocyte-derived fibrocytes from monocytes, macrophages, and fibroblasts. PLoS One. 2009b;4:e7475. doi: 10.1371/journal.pone.0007475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins CS, Swirski FK. The multiple roles of monocyte subsets in steady state and inflammation. Cell Mol Life Sci. 2010 May 1; doi: 10.1007/s00018-010-0375-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sallusto F, Lanzavecchia A. Efficient presentation of soluble antigen by cultured human dendritic cells is maintained by granulocyte/macrophage colony-stimulating factor plus interleukin 4 and downregulated by tumor necrosis factor alpha. J Exp Med. 1994;179:1109–1118. doi: 10.1084/jem.179.4.1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt M, Sun G, Stacey MA, Mori L, Mattoli S. Identification of Circulating Fibrocytes as Precursors of Bronchial Myofibroblasts in Asthma. J Immunol. 2003;171:380–389. doi: 10.4049/jimmunol.171.1.380. [DOI] [PubMed] [Google Scholar]
- Shao DD, Suresh R, Vakil V, Gomer RH, Pilling D. Pivotal Advance: Th-1 cytokines inhibit, and Th-2 cytokines promote fibrocyte differentiation. J Leukoc Biol. 2008;83:1323–33. doi: 10.1189/jlb.1107782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shortman K, Liu YJ. Mouse and human dendritic cell subtypes. Nat Rev Immunol. 2002;2:151–161. doi: 10.1038/nri746. [DOI] [PubMed] [Google Scholar]
- Swirski F, Nahrendorf M, Etzrodt M, Wildgruber M, Cortez-Retamozo V, Panizzi P, Figueiredo J, Kohler R, Chudnovskiy A, Waterman P, Aikawa E, Mempel T, Libby P, Weissleder R, Pittet M. Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science. 2009;325:612–6. doi: 10.1126/science.1175202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tushinski RJ, Oliver IT, Guilbert LJ, Tynan PW, Warner JR, Stanley ER. Survival of mononuclear phagocytes depends on a lineage-specific growth factor that the differentiated cells selectively destroy. Cell. 1982;28:71–81. doi: 10.1016/0092-8674(82)90376-2. [DOI] [PubMed] [Google Scholar]
- Wang JF, Jiao H, Stewart TL, Shankowsky HA, Scott PG, Tredget EE. Fibrocytes from burn patients regulate the activities of fibroblasts. Wound Repair Regen. 2007;15:113–21. doi: 10.1111/j.1524-475X.2006.00192.x. [DOI] [PubMed] [Google Scholar]
- Yang L, Scott PG, Giuffre J, Shankowsky HA, Ghahary A, Tredget EE. Peripheral Blood Fibrocytes from Burn Patients: Identification and Quantification of Fibrocytes in Adherent Cells Cultured from Peripheral Blood Mononuclear Cells. Lab Invest. 2002;82:1183–1192. doi: 10.1097/01.lab.0000027841.50269.61. [DOI] [PubMed] [Google Scholar]
- Yang L, Scott PG, Dodd C, Medina A, Jiao H, Shankowsky HA, Ghahary A, Tredget EE. Identification of fibrocytes in postburn hypertrophic scar. Wound Repair Regen. 2005;13:398–404. doi: 10.1111/j.1067-1927.2005.130407.x. [DOI] [PubMed] [Google Scholar]
- Wirth JJ, Theisen MA, Crowle AJ. Culture conditions required for primary isolation and study of mouse blood monocytes. J Reticuloendothel Soc. 1982;31:325–37. [PubMed] [Google Scholar]