

Abstract
AIM
To propose a relevant grading scale for clinical adverse events or laboratory results, electrocardiogram (ECG) and vital sign findings supporting both dose escalation and stopping decisions in first-entry-into-man (FIM) studies conducted in young healthy subjects.
METHODS
A three-level scale was used for the proposed grading system. The grading is directly derived from the observed severity of discontinuous variables, as are most of clinical adverse events. A ‘combined method’ based on normal ranges and spontaneous variation is suggested for grading the findings which are continuous variables mainly numerical in nature. One grade, at the subject level, and one algorithm, at the cohort level, support the proposed decision rules. This work was managed by a Club Phase I working group.
RESULTS
Examples of grade 1, 2 and 3 limits are given for the most frequent clinical adverse events and laboratory tests, ECG and vital sign findings. When available, the proposed NIH and FDA limits are also provided. The safety recommendation is to use the grade 2 at least as an alert for caution and the grade 3 as a maximum for stopping, applying the algorithm at the cohort level.
CONCLUSIONS
This paper proposes a safety grading system based on relevant criteria which might be used by investigators and sponsors to support and rationalize dose escalation decisions in healthy young subject FIM studies. These proposals are designed not to be a guideline but some ‘points to consider’ helping the dose escalation process. This paper supports the recent reinforcement of the safety requirements for FIM studies by European authorities.
Keywords: dose escalation, grading, healthy subjects, phase 1, stopping rules
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
After the TGN1412 incident in London, the European authorities reinforced the safety requirements for first-entry-into-man (FIM) studies. Their recommendations on risk management included a risk minimization strategy based on clearly defined stopping rules. However to date, there are no approved grading scales for clinical adverse events or safety findings to support dose escalation and to define stopping rules in order to mitigate the risks for healthy subjects participating in FIM trials.
WHAT THIS STUDY ADDS
This paper proposes standardized methods for the grading of safety data (adverse events, laboratory tests, electrocardiogram and vital sign findings) based on relevant criteria. The proposed grading scale provides support both for dose escalation and the design of stopping rules. The derived safety thresholds are applicable at either an individual subject level or at a cohort level, and are specifically adapted to young male healthy subjects, who represent the majority of the subjects participating in FIM trials.
Introduction
After the TGN1412 tragedy [1] the authorities issued several recommendations and guidance with regards to improving safety in first-entry-into-man studies (FIM) using risk assessment and risk minimization strategies. One of the proposed risk minimization strategies was to clarify the decision rules for dose escalation and dose stopping during a FIM study. Application of these recommendations and guidelines firstly requires a homogeneity in the grading of adverse events (AEs) and non clinically significant findings (laboratory tests, ECG and vital signs) and secondly a relevant method to define the stopping rules, which at the same time adequately protects the healthy subjects and permits an appropriate safety evaluation of a new molecule.
To date, there are no approved grading systems for clinical adverse events or safety findings or methods to define the stopping rules in FIM studies conducted in healthy young subjects. There are however two grading systems that are published for use in oncology studies ‘Common Terminology Criteria for Adverse Events (CTCAE)’ by the National Cancer Institute (NCI) and the National Institute of Health (NIH [2, 3]) and ‘WHO toxicity criteria by grade’ by the World Health Organization (WHO [4]). These scales are well suited for cytotoxic compounds, being assessed in cancer patients, where they are regularly and successfully used. However, they have limited validity and usefulness in the assessment of non-cytotoxic drugs in healthy subjects. More recently, two additional guidelines have been issued: ‘Table for grading the severity of adult and paediatric adverse events – December 2004’ by the NIH to cover AIDS clinical trials [5] and ‘Toxicity Grading Scale for Healthy Adult and Adolescent Volunteers enrolled in Preventive Vaccine Clinical trials – Draft April 2005/Final September 2007’ by the FDA to cover vaccine clinical trials [6]. Even if the two proposed grading systems are similar on several points, a difference of opinion is observed: the NIH argued against a ‘separate toxicity scale’ for vaccines and asked for a more rigorous definition of a ‘healthy’ subject, along with a clear distinction between normal ranges and abnormalities. Comments made by the NIH emphasize the difficulty of defining ‘healthy’ or establishing a grade I threshold [7]. Even if these two guidelines offer several meaningful and realistic solutions, they are not supported by any relevant or accurate methodology for the development of decision criteria and they are not specifically tailored to healthy young subjects. Thus the specific needs for healthy subjects in FIM are not covered by the existing guidance documents.
Healthy subject protection vs. a thorough clinical evaluation of drug properties (safety, pharmacokinetics and pharmacodynamic activity) undoubtedly generates a conflict-of-interest. On the one hand, the possible risk of discomfort, adverse consequence on subject activity or person, argue for stopping the study drug administration at the individual level. On the other hand, the data may not be considered as relevant enough to justify stopping dose escalation or qualifying the maximal tolerated dose or the maximum achievable exposure, the primary objective at this stage of drug development. Therefore, insufficient data would argue for continuing study progression and dose escalation. A certain number of rules are therefore required to support this decision process in routine practice and these common processes ‘stopping rules’ have to be supported by an accurate algorithm.
Club Phase I (CPI), a French non-profitable association of clinical pharmacologists from pharmaceutical companies, contract research organizations and academia, decided in 2006 to set-up a working group whose objectives were to provide useful and relevant points to consider (not a guideline) specifically focused on healthy young subjects and FIM dose escalation studies, to grade clinical AEs and other significant findings. This grading will thereby permit a rationalized approach defining stopping rules. This approach is illustrated with a few common examples of clinical AEs, laboratory tests, ECG and vital sign findings.
Methods
This work was managed by a working group of CPI, including senior clinical pharmacologists. The proposals were sent by email for comment to CPI members and discussed within two internal meetings. Lastly, a preliminary version was presented to the international meeting sponsored by CPI and AGAH (3rd Joint Annual Meeting: Managing challenges in early drug development: Biologicals and small molecules, April 28 and 29, 2009. Espace Tête d'Or, Lyon, France).
Scope
This work focuses specifically on FIM single or multiple dose escalation studies and is only appropriate for healthy young subjects.
Definition of adverse events and safety findings
The adverse event (AE) definition from the International Conference of Harmonization [8]: ‘Any untoward medical occurrence in a patient or clinical investigation subject administered any pharmaceutical product, which does not necessarily have to have a causal relationship with this treatment’ applies here.
Under FIM study conditions, safety data are obtained through spontaneous reporting of AEs by the subject or obtained in answer to the investigator's customary question: ‘Have you anything to report from the time of drug administration?’ or from any observation related to safety procedures (e.g. vital signs, ECG, routine laboratory tests, etc.). In this paper, the term ‘event’ will be used only for a clinical AE and the word ‘finding’ will be the preferred term for any significant result from routine laboratory tests, ECGs or vital sign evaluation.
Grading system
A three-level scale was used as the basis of the proposed grading system.
Scaling to grade 1
There are two approaches for grading adverse events and safety findings to level 1.
1) One for which the grading is directly derived from the ‘observed intensity/severity’, the ‘daily life consequences’ and the need for concomitant rescue medication. This is generally applicable for discontinuous variables and most of the clinical AEs belong to this category.
Grade 1: does not interfere with daily activity,
Grade 2: interferes with daily activity, no treatment required, except paracetamol,
Grade 3: prevents daily activity or requires treatment.
A Grade 4, which corresponds to an event which is life-threatening, is usually proposed. Actually, it is exceptional in phase 1 clinical trials and it obviously supports a stopping decision. Therefore, it is not developed in this paper.
2) For findings, which are continuous variables mainly numerical in nature, the grading is based on the ‘likelihood of risk or consequence’ but no longer on an observed severity.
If some thresholds have already been established by the medical community to define diseases, they must be used and applied to healthy phase I subjects. Example includes definitions of anaemia, diabetes, hypertension or liver injury as described in Hy's law [9]. The relevance of such a threshold is not questionable since per se they support a disease definition. In the other situations, a specific method is required to determine a relevant threshold for the continuous variables. The CPI recommendation is that a ‘combined method’[10] be used based on the normal range and spontaneous variability in young healthy subjects: ‘A relevant threshold for adverse change during a phase I study is required to discriminate between ‘normal’ and ‘abnormal’ values under study conditions, within or exceeding spontaneous normal variations. If a value is ‘abnormal’ under phase I study conditions, it must be considered as significant, and as a potential ‘finding’ whatever the cause. Thus an accurate method should be based both on the limits of normal range and on the range of variation. In phase I studies this means that the following must be taken into consideration: (i) upper or lower limit of normal ranges, as appropriate and (ii) spontaneous variation from baseline in phase I under identical conditions’[10]. Such a method determines a threshold of ‘abnormality’ from a statistical point of view.
The normal ranges and the calculation of the normal spontaneous variation reported here as an example are derived from data collected in one clinical pharmacology unit (Association de Recherche Thérapeutique, Lyon, France), and from young male healthy subjects (YMHS) only, under phase I study conditions [11]. Such parameter variations are then applied to upper or lower limits of normal ranges (ULNR or LLNR), as appropriate.
Then, the normal ranges published by the New England Journal of Medicine (NEJM) [12] are also provided and may be used more easily as a reference as they are widely accepted worldwide.
However, the most relevant thresholds have to be derived from local normal ranges and variations of each clinical pharmacology unit. Our unit had determined these values from its local population.
The following are required conditions before the application of grades: (i) accurate measurements, sampling and assay conditions and (ii) control of abnormality before validation into ‘finding’.
Note 1: This method is not relevant to define a subject's eligibility to participate in FIM study i.e. these thresholds are not to be used to define inclusion/exclusion screening criteria.
Note 2: The grading for findings has not been primarily designed to qualify a finding as an AE although it may be acceptable to use grade 2 or 3 depending on the finding to harmonize data collection from various studies, various investigators and various countries.
Scaling to grades 2 and 3
There is no accepted consensus or method to support grade 2 or 3 limits in healthy population. Grade 2 and 3 limits can no longer be based solely on statistical grounds, but should also integrate the possible consequences or an anticipated risk based on medical knowledge and clinical pharmacology experience. In this paper, the grade 2 and 3 assignments are selected through an anticipation of the possible risks or consequences, made by consensus within the CPI working group. As the subject's safety is paramount, these values were chosen as they are adapted specifically to the protection of young healthy subjects, who represent the main population involved in FIM trials. These grades were finalized after taking into consideration both the NIH [5] and FDA [6] proposals.
Grading application
A limited number of examples of clinical AEs and findings have been selected to illustrate these proposals. These examples were chosen as they were the most frequently reported AEs and findings in FIM studies, based on published papers [13–16] and the CPI taskforce's experience. The results of the grade limits, as proposed by the CPI taskforce, are presented in separate tables. The NIH [5] or FDA [6] proposals, if available, are shown in parallel. The results for ‘findings’ are expressed relative to the upper (U) or lower (L) limit of the normal range (NR), as appropriate, and/or as the absolute values, depending on the feasibility and on the relevance for the parameter. When needed, such a value is associated with the variation from baseline. In a few exceptions, the variation is the only parameter taken into consideration for establishing a pertinent threshold.
Note: The reported limits are exclusively defined for safety purposes and they are not intended for defining pharmacologic/pharmacodynamic acceptable limits. Specific and different thresholds are required when a defined pharmacologic activity is explored, for instance, limits of activated partial thromboplastin time (aPTT) used as a pharmacological biomarker for an anticoagulant.
Stopping rules
At the individual level, a generalized rule based on a risk estimate is applicable i.e. stop dose escalation if any event is of grade equal or superior to a certain level, usually 3. Moreover and in addition to the observed AE, this process should also take into consideration all the available pertinent information, i.e. time relative to drug administration and association with other safety signals, determining an upgrading. Any association of a finding to clinical symptom(s) or sign(s), or a rapid worsening, or a concomitant modification of any other relevant parameter(s), for example, ALT and bilirubin, CPK and AST, creatinine and hyperkalaemia would also result in an upgrading of the finding. This cautious approach is already recommended by the FDA for liver injuries [17] and by Hy's law [9].
At the dose cohort level, a more complex process is required. In fact, the decision depends on the type of event, its intensity, its monitorability, its reversibility and possible outcome (complications), the number of subjects experiencing the event, and on the type of drug administered i.e. placebo or active drug. Thus, an algorithm is needed. The proposed algorithm is based on the small number of subjects administered in a FIM study, the rarity of events, and the possible high risk potential, for example, a malaise with loss of consciousness, justifying limited un-blinding of subjects experiencing risky events.
Note: The reported rules are exclusively determined for adverse events and findings not related to the study procedures.
Results
Grading
The results are presented in the tables.
Table 1 presents the grades 1 to 3 concerning the clinical adverse events. Table 2 reports, as an example, the reference values and the reference changes in a healthy young subject population of one clinical pharmacology unit, Association de Recherche Thérapeutique, Lyon, France, and then the grade 1 limit/threshold determined by the combined method use. An application to the corresponding normal range (NR) of the NEJM [12] is also presented as a widely accepted reference. The grades 1 to 3 concerning laboratory findings are presented in the Tables 3, 4, 5 and 6, ECG findings in Table 7 and vital sign findings in Table 8.
Table 1.
Club Phase I proposed grading application to frequent clinical adverse events, including comparable FDA and NIH grading (if available)
| Intensity | Grade 1* | Grade 2* | Grade 3* |
|---|---|---|---|
| General definition | Does not interfere with activity | Interferes with activity, no treatment except paracetamol | Prevents daily activity or requires treatment |
| Headache | Transient | Several hours but less <12 h. Interferes with activity, no treatment except paracetamol | >12 h, presence during the night Prevents daily activity or requires treatment |
| FDA headache | No interference with activity | Repeated use of non-narcotic pain reliever >24 h or some interference with activity | Significant; any use of narcotic pain reliever or prevents daily activity |
| NIH headache | Symptoms causing no or minimal interferences with usual social & functional activities | Symptoms causing greater than minimal interferences with usual social and functional activities | Symptoms causing inability to perform usual social and functional activities |
| Pain (whatever the location) | Transient | Several hours but less <12 h. Interferes with activity, no treatment except paracetamol | >12 h, presence during the night Prevents daily activity or requires treatment |
| FDA (Myalgia) | Does not interfere with activity | Interferes with activity | Significant; prevents daily activity |
| NIH (Myalgia) | Muscle pain causing no or minimal interferences with usual social and functional activities | Muscle pain causing greater than minimal interferences with usual social and functional activities | Muscle pain causing inability to perform usual social and functional activities |
| Fatigue | Does not interfere with usual and social activities | Interferes with usual andsocial activity, no treatment | Prevents daily usual and social activity or requires treatment |
| FDA fatigue | Does not interfere with activity | Interferes with activity | Significant; prevents daily activity |
| NIH fatigue (or malaise) | Symptoms causing no or minimal interferences with usual social and functional activities | Symptoms causing greater than minimal interferences with usual social and functional activities | Symptoms causing inability to perform usual social and functional activities |
| Cognitive disturbances Concentration or memory disorder | Does not interfere with usual and social activities | Interferes with usual and social activity, no treatment | Prevents daily usual and social activity or requires treatment |
| Confusion or disorientation or attention disorder | Does not interfere with usual and social activities | Interferes with usual and social activity, no treatment | Prevents daily usual and social activity or requires treatment |
| Somnolence/drowsiness | Does not interfere with usual and social activities | Interferes with usual and social activity, no treatment | Prevents daily usual and social activity or requires treatment |
| Dizziness or disequilibrium or vertigo or light headiness | Does not interfere with usual and social activities | Interferes with usual and social activity, no treatment | Prevents daily usual and social activity or requires treatment |
| Malaise/syncope† | Does not interfere with activity | Interferes with activity, no treatment | Syncope†, or prevents daily activity, or requires treatment |
| Nausea‡ | Keep normal intake | Intake significantly decreased | No intake. Requires treatment |
| Vomiting | 1 episode | 2 to 4 episodes/day, or 2/day x 2 days | >4 episodes per day, or 2 or more per day prolonged on several days |
| Diarrhoea | Increase of 2–3 stools/day over normal pre-study flow | Increase of 4–5 stools/day or moderate cramping | Increase of 6–8 or severe cramping or incontinence or requires i.v. fluid replacement |
| FDA Nausea/vomiting | No interference with activity or 1–2 episodes | Some interference with activities or >2 episodes/24 h | Prevents daily activity or requires outpatient i.v. hydration |
| FDA diarrhoea | 2–3 loose stools or <400 g per day | 4–5 loose stools or 400–800 g per day | 6 or more watery stools or >800 g per days or requires outpatient i.v. hydration |
| NIH Nausea | Transient (<24 h) or intermittent with no or minimal interference with normal intake | Persistent nausea resulting in decreased oral intake for 24–48 h | Minimal oral intake for >48 h or aggressive rehydration (i.v. fluids) |
| NIH vomiting | Transient or intermittent vomiting with no or minimal interference with normal intake | Frequent episodes of vomiting with no or mild dehydration | Persistent vomiting resulting in orthostatic hypotension or aggressive rehydration indicated (e.g. i.v. fluids) |
| NIH diarrhoea | Transient or intermittent episodes of unformed stools or increase of < or = 3 stools over baseline per 24 h period | Persistent episodes of unformed watery stools or increase of 4–6 stools over baseline per 24 h period | Bloody diarrhoea or increase of = or >7 stools per 24-hour period or i.v. fluid replacement indicated |
| Muco-cutaneous AE (excluding local reaction to topical or injected compound) | Transient erythema or pruritus | Rash (limited) | Rash (extensive), vesiculation, dry or moist desquamation, or ulceration. |
Modulation and upgrading based on number of episodes and/or duration of symptoms and/or significant associated general effects.
See definition from European Society of Cardiology: ‘Transient self-limited loss of consciousness’[22].
The same criteria are applicable to dyspepsia, heartburn.
Table 2.
Determination of grade 1 threshold based on ‘Combined method’ use and application to NEJM normal range
| Usable healthy young male data [11] | ‘Combined method’ derived threshold Grade 1 limit: data expressed as relative to upper or lower | Usable general application | |||||
|---|---|---|---|---|---|---|---|
| Laboratory parameter | Units | Normal range Upper and lower limit of normal range* | Spontaneous Variation Limit to define grade 1 Limit Decrease: – Increase: +† | Limit of NR | NEJM normal range 2004 [12] | Derived threshold value Grade 1 limit | Suggested corrected value adapted to healthy young male |
| ALT | IU l−1 | 10–58 | +10 | 1.2*ULNR | 0–35 | 42 | |
| AST | IU l−1 | 10–43 | +9 | 1.2*ULNR | 0–35 | 42 | |
| Bilirubin‡ | µmol l−1 | 5–27 | +12 | 1.3*ULNR | 5–17 (f) | 22 | 35 |
| Alkaline. phosphatases | IU l−1 | 46–117 | +16 | 1.1*ULNR | 30–120 | 132 | |
| Creatinine | µmol l−1 | 78–113 | +15 | 1.1*ULNR | <133 (f) | 146 | 125 |
| Potassium | mmol l−1 | 3.5–4.9 | −0.2 | (−0.95*LLNR) | 3.5–5 | 3.3 | |
| No data§ | 1.05 ULNR | 5.2 | |||||
| Glucose | mmol l−1 | 3.8–5.9 | −0.4 | (−0.9*LLNR) | 4.2**–6.4 | 3.8 | 3.4 |
| Haemoglobin (male) | g dl−1 | 13.4–17.5 male | −0.8 | (−0.95*LLNR) | 13.5–17.5 male | 12.5 | |
| PMN – white people | 109 l−1 | 1.7–6.5 | −0.5 | (−0.7*LLNR) | 1.8–7.7 | 1.3 | |
| +1.8 | 1.3*ULNR | 10 | |||||
| Eosinophils | 109 l−1 | <or = 0.480 | +0.15 | 1.3*ULNR | No data | 0.6 | |
| Platelets | 109 l−1 | 153–324 | −20 | (−0.85*LLNR) | 150–350 | 130 | |
| CPK (male) | IU l−1 | 53–400 | +72 | 1.2*ULNR | 60–400 | 480 | |
| aPTT | s | 28–43 | +5 | 1.1*ULNR | 22–35 | 38.5 | |
| ECG¶ | |||||||
| PR interval | ms | 120–196 | −21 | (−0.8*LLNR) | No data | No data | 100 ms |
| +23 | 1.1*ULNR | No data | No data | 220 ms | |||
| QTcB interval | ms | 357–425 | +42 | 1.1*ULNR | No data | No data | 460 ms |
| QTcF interval | ms | 261–422 | +42 | 1.1*ULNR | No data | No data | 460 ms |
| Vital signs¶ | |||||||
| Supine heart rate | beats min–1 | 44–82 | −20 | not applicable | No data | NO data | Not applicable |
| +18 | 1.2*ULNR | No data | No data | 100 beats min–1 | |||
| Supine systolic blood pressure | mmHg | 102–146 | −25 | not applicable | No data | No data | Not applicable |
| +18 | 1.1*ULNR | No data | No data | 160 mmHg | |||
| Supine diastolic blood pressure | mmHg | 53–84 | −15 | not applicable | No data | No data | Not applicable |
| +11 | 1.1*ULNR | No data | No data | 90 mmHg | |||
Young male healthy subject data. Normal range values determined by non-parametric procedure (2.5–97.5% of the distribution of data) [11].
Young male healthy subject data. Spontaneous variation limits (changes) determined by the 2.5–97.5% interval of variation in phase I conditions [11].
27 µmol l−1 is also the accepted limit of inclusion [11].
No robust available data due to haemolysis bias on potassium values.
Un-published data from the same healthy male population (same clinical pharmacology unit, see ref. [11].).
NR of general population without relevance to healthy young male subjects. Note:It is recommended that each clinical pharmacology unit defines its own normal ranges and changes based on their own population.
Table 3.
Grade thresholds for liver function tests
| Grades | ||||
|---|---|---|---|---|
| Parameter | Origin | 1 | 2 | 3 |
| ALT (ULNR) | CPI | 1.2 (or # 70 IU l−1) to 3 ULNR | 3 to 5* | 5 to 10 |
| FDA | 1.1 to 2.5 ULNR | 2.6 to 5 | 5 to 10 | |
| NIH | 1.25 to 2.5 ULNR | 2.5 to 5 | 5 to 10 | |
| AST (ULNR) | CPI | 1.2 (or # 70 IU l−1) to 3 ULNR | 3 to 5* | 5 to 10 |
| FDA | 1.1 to 2.5 ULNR | 2.6 to 5 | 5 to 10 | |
| NIH | 1.25 to 2.5 ULNR | 2.5 to 5 | 5 to 10 | |
| Bilirubin (ULNR) | CPI | 1.3 (or # 35 IU l−1) to 2 ULNR if change from baseline >10 µmol l−1 | 2 to 2.5* | 2.5 to 3 |
| FDA, if LFT normal† | 1.1 to 1.5 ULNR | 1.6 to 2 | 2 to 3 | |
| FDA, if increase of LFT† | 1.1 to 1.25 | 1.26 to 1.5 | 1.51 to 1.75 | |
| NIH | 1.25 to 2.5 ULNR | 2.5 to 5 | 5 to 10 | |
| Alkaline Phosphatases (ULNR) | CPI | 1.1 (or # 132 IU l−1) to 2 ULNR | 2.1 to 3 | 3.1 to 10 |
| FDA | 1.1 to 2 ULNR | 2.1 to 3 | 3.1 to 10 | |
| NIH | 1.25 to 2.5 ULNR | 2.6 to 5 | 5 to 10 | |
Use Hy's law: ALT >3 ULNR and bilirubin >2 ULNR induces upgrading to level 3 [9.]
The appearance of worsening of fatigue, nausea, vomiting, fever, rash eosinophilia or right upper quadrant pain or tenderness or the association to INR superior to 1.5 induces an upgrading [17]. ULNR, Upper limit of normal range; LLNR, Lower limit of normal range; CPI, Club phase I task force.
Table 4.
Grade thresholds for creatinine and electrolytes
| Grades | ||||
|---|---|---|---|---|
| Parameter | Origin | 1 | 2 | 3 |
| Creatinine (µmol l−1) | CPI | (or # 125 µmol l−1) to 1.3 ULNR assuming an increase superior to 10% | 1.3 to 1.5 | 1.5 to 2 |
| FDA | 1.5 to 1.7 ULNR | 1.8 to 2 | 2.1 to 2.5 | |
| NIH | 1.1 to 1.3 | 1.4 to 1.8 | 1.9 to 3.4 | |
| Potassium hypokalaemia (mEq)* | CPI | Below 0.95 LLNR and decrease exceeding minus 0.2 mEq (or 3.3 to 3.1 mEq) | No relevance | ECG signs or value < or = 3 mEq |
| FDA | 3.6 to 3.5 mEq | 3.4 to 3.3 | 3.2 to 3.1 | |
| NIH | 3.4 to 3 mEq | 2.9 to 2.5 | 2.4 to 2 | |
| Potassium hyperkalaemia (mEq)* | CPI | >to ULNR and increase exceeding 0.4 mEq | No relevance | ECG signs or value >5.5 mEq |
| FDA | 5.1 to 5.2 mEq | 5.3 to 5.4 | 5.5 to 5.6 | |
| NIH | 5.6 to 6 mEq | 6.1 to 6.5 | 6.6 to 7 | |
| Glucose hypoglycaemia (mmol l−1)* | CPI | < to 0.9 ULNR and decrease exceeding minus 0.5 mmol l−1 (or <3.4 mmol l−1) | No relevance | Clinical signs or value <3 mmol l−1 |
| FDA | 3.8 to 3.5 (65−69 mg dl−1) | 3.4 to 3 (55–64) | 2.5 to 2.9 (45–54) | |
| NIH | 3.55 to 3.05 mmol l−1 | 3 to 2.2 | 2.16 to 1.67 | |
Conditions: sampling without tourniquet, sample immediately assessed by the laboratory, blood glucose and not dextrostix, plus fast control confirming the persistence of the abnormality. ULNR, Upper limit of normal range; LLNR, Lower limit of normal range; CPI, Club phase I task force;
Table 5.
Grade thresholds for haematology
| Grades | ||||
|---|---|---|---|---|
| Parameter | Origin | 1 | 2 | 3 |
| Haemoglobin decrease (male g dl−1) | CPI | 12.5 to 12 and decrease exceeding –1.5 | 11.9 to 10 | <10 |
| FDA | 13.5 to 12.5 and change <1.5 | 12.4 to 10.5 and change from 1.6 to 2 | 10.4 to 8.5 and change from 2.1 to 5 | |
| NIH (HIV negative) | 10.9 to 10 and decrease 2.5 to 3.4 | 9.9 to 9 and decrease 3.5 to 4.4 | 8.9 to 7 and decrease >4.5 | |
| Haemoglobin decrease (women g dl−1) | CPI | 11.5 to 11 and decrease exceeding minus 2 | 10.9 to 9.5 | <9.5 |
| FDA | 12 to 11 and change <1.5 | 10.9 to 9.5 and change from 1.6 to 2 | 9.4 to 8 and change from 2.1 to 5 | |
| NIH | No data | No data | No data | |
| PMN decrease Caucasian people (109 l−1) | CPI | LLNR to 0.7 LLNR(or # 1.3) and decrease exceeding minus 0.5 109 | 0.7 LLNR (or # 1.3 giga) to 1 109 | <1 109 |
| FDA | 3.5 to 2.5 109 (Caucasian and Black people) | 2.4 to 1.5 109 | 1.4 to 1 109 | |
| NIH | 2.5 to 2 109 | 1.9 to 1.5 109 | 1.5 to 1.1 | |
| PMN decrease Black people (109 l−1) | CPI | LLNR to 0.7 LLNR(or # 1.1) and decrease exceeding minus 0.5 109 | 0.7 LLNR (or # 1.1 109) to 0.8 109 | <0.8 109 |
| FDA | No data | No data | No data | |
| NIH | No data | No data | No data | |
| Eosinophils (109 l−1) * | CPI | 0.5 109* to 1.5 ULNR (or 0.75 109) and increase exceeding 0.15 109 | 1.5 to 3 ULNR (or 0.75 to 1.5 109) | >3 ULNR or >1.5 109* |
| FDA | 0.65 to 1.5 109 | 1.5 to 5 109 | >5 109 | |
| NIH | No data | No data | No data | |
| Platelets decrease (109 l−1) assuming no platelet cluster | CPI | 0.85 LLNR (or 130 109) to 0.8 LLNR (or 120 109) | 0.7 LLNR to 100 109 | <100 109 |
| FDA | 140 to 125 109 | 124 to 100 109 | 99 to 25 109 | |
| NIH | 125 to 100 109 | 99 to 50 109 | 50 to 25 109 | |
Definition of eosinophilia is 0.5 109 l−1 and hypereosinophilic syndrom is 1.5 109 l−1[19].ULNR, Upper limit of normal range; LLNR, Lower limit of normal range; CPI, Club phase I task force.
Table 6.
Grade thresholds for muscle and coagulation
| Grades | ||||
|---|---|---|---|---|
| Parameter | Origin | 1 | 2 | 3 |
| CPK* (IU l−1) | CPI | 1.2 to 2.5 ULNR (or 480 to 1000 IU l−1) | 2.5 to 5 ULNR (or 1000 to 2000 IU l−1) | 5 to 10 ULNR (or 2000 to 5000 IU l−1) |
| FDA | 1.25 to 1.5 ULNR | 1.6 to 3 ULNR | 3.1 to 10 ULNR | |
| NIH | 3 to 5.9 ULNR | 6 to 9.9 ULNR | 10 to 19.9 ULNR | |
| aPPT† (ULNR) | CPI | 1.1 to 1.3 ULNR | 1.3 to 1.5 ULNR | >1.5 ULNR or minor bleeding |
| FDA | 1.1 to 1.2 ULNR | 1.2 to 1.4 ULNR | 1.4 to 1.5 ULNR | |
| NIH | 1.1 to 1.66 ULNR | 1.67 to 2.33 ULNR | 2.34 to 3 ULNR | |
| INR† (ULNR) | CPI | 1.1 to 1.3 ULNR | 1.3 to 1.5 ULNR | >1.5 ULNR or minor bleeding |
| FDA | No data | No data | No data | |
| NIH | No data | No data | No data | |
Table 7.
Grade thresholds for ECG
| Grades | ||||
|---|---|---|---|---|
| Parameter | Origin | 1 | 2 | 3 |
| PR interval increase (ms)* | CPI | 220 to 250 and increase exceeding 20 ms | >250 | Mobitz 2 or syncope [21] |
| FDA | No data | No data | No data | |
| NIH | 210 to 250 | >250 | Mobitz 2 or ventricular pause >3 s | |
| QTc interval increase (young male) using the most accurate QTc formula (ms)* | CPI | ULNR to 475 ms and increase exceeding 40 ms | 476 to 499 | >500 ms, or QTC over 460 and increase exceeding 60 ms |
| FDA | No data | No data | No data | |
| NIH | No data | No data | No data | |
| QTc interval increase (women)* | CPI | Same as male plus 20 ms | ||
| FDA | No data | No data | No data | |
| NIH | No data | No data | No data | |
Assuming supine position, 10 min at rest conditions, not sleeping subjects and several concordant results. ULNR, Upper limit of normal range; LLNR, Lower limit of normal range; CPI, Club phase I task force.
Table 8.
Grade thresholds for vital signs
| Grades | ||||
|---|---|---|---|---|
| Parameter | Origin | 1 | 2 | 3 |
| Heart rate – bradycardia (beats min–1)* | CPI | Not applicable | <40 if decrease from baseline exceeding –20 | Clinical lack of tolerability and /or ECG abnormalities |
| FDA | 54 to 50 | 49 to 45 | <45 | |
| NIH | No data | No data | No data | |
| Heart rate – tachycardia (beats min)* | CPI | 100 to 115 | 116 to 130 | >131 or ventricular dysrhythmias |
| FDA | 100 to 115 | 116 to 130 | >130 | |
| NIH | No data | No data | No data | |
| Supine systolic blood pressure increase (mmHg) * | CPI | ULNR to 150 | 150 to 160 | >160 or headache or clinical signs |
| FDA | 141 to 150 | 151 to 155 | >155 | |
| NIH | 140 to 159 | 160 to 179 | >180 | |
| Supine diastolic blood pressure increase (mmHg) * | CPI | 95 to 99 and increase exceeding 10 | 100 to 110 | >110 or headache or clinical signs |
| FDA | 91 to 95 | 96 to 100 | >100 | |
| NIH | 90 to 99 | 100 to 109 | >110 | |
| Supine systolic blood pressure decrease (mmHg) * | CPI | LNNR to 80 and decrease exceeding –25 | 80 to 70 | <70 or symptomatic |
| FDA | No data | No data | No data | |
| NIH | No data | No data | No data | |
| Postural hypotension – systolic blood pressure 2 to 3 min after standing (mmHg)* | CPI | Decrease exceeding –20 and association to reflex tachycardia | Cannot stay standing | Syncope or prevents daily activity or requires treatment |
| FDA | Not applicable | No data | No data | |
| NIH | NA | Symptomatic corrected by oral fluid replacement | Symptomatic i.v. fluid indicated | |
Assuming supine position, 10 min at rest conditions, not sleeping subjects, measurements on the same arm and several concordant results. ULNR, Upper limit of normal range; LLNR, Lower limit of normal range; CPI, Club phase I task force.
Stopping rules
The grade, the frequency of AEs and the blindness are taken in account.
At the individual level, basically, the grade 2 is at least a safety alert leading to caution and closer assessment of safety in other subjects. An upgrading applies if rapid worsening, concomitant findings, clinical symptoms and signs occur. The grade 3 always supports stopping that suffering subject.
At the cohort level, the algorithm supporting the stopping rules is presented in Figure 1. An un-blinding, limited to suffering subjects, is applied. Basically, a 50% frequency of AEs or findings (grade 3, or 2, as explained above) of same types, is proposed to support the stopping decision rules. However, depending on the type and potential risk of AE, any modulation may be decided, for example, for safety concerns, a lower frequency level.
Figure 1.
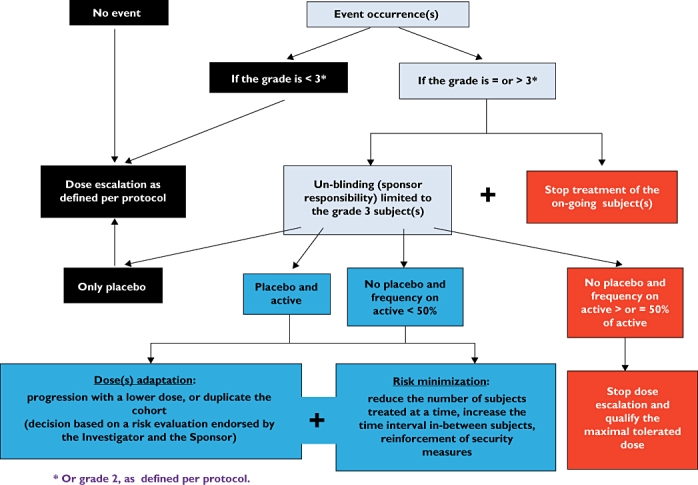
Algorithm for decision making for dose escalation at the cohort level based on severity grading of adverse events and findings
Discussion
Some points to consider, but not a guideline
The proposed thresholds and the stopping rules are to be used as ‘points to consider’ to facilitate the decision making process and not as mandatory guidelines. This grading is appropriate for a young healthy male population; an adaptation is needed for female, elderly or patient populations if they are involved in early development trials. For example this has already been done for cancer patients: CTCAE by the NCI and the NIH [2, 3] and WHO toxicity criteria [4]. A certain amount of flexibility needs to be maintained in order to adapt these criteria to different healthy study populations, specific study conditions and local normal laboratory ranges. Such adaptations are usually proposed by agreement between sponsor and investigator. Modification of the proposed thresholds can be made based on the type of event (or finding) and on the risk assessment, i.e. stopping dose escalation could be based on a different frequency or can be based on the occurrence of certain grade 2 ‘precursor’ events. These rules are proposed to standardize better and to help decision making process, knowing the final responsibility belongs to the investigator medical judgement.
The suggested three step grading scale is simple, based on clinical common sense and easily applicable; moreover, this type of approach is already in action, in the recent NIH or FDA guidelines [5, 6]. They all use a grading scale based on the consequences to ‘activity or daily life’ and on the need for concomitant rescue medication.
The fairly common approach of grading AEs in patients is to consider grade 1 as ‘mild’, grade 2 as ‘moderate’, grade 3 as ‘severe’ and grade 4 as ‘life-threatening’. However CP1 would not recommend using this wording for healthy subjects. Furthermore, the grades 0 ‘No AE or within normal limits’ and the grade 5 ‘death related to adverse event’, as proposed by CTCAE [2], have unequivocal impact on the course of a FIM study in healthy subjects (i.e. grade 0: no effect; grade 5: stop dosing).
This grading system does not include the ‘serious’ criteria. A serious adverse event is based on subject/patient outcome or action criteria, described in its Good Clinical Practice definition. Seriousness serves as a guide for defining regulatory reporting obligations, but does not necessarily refer to the severity of the event.
For grade 1 determination the CPI selected ‘combined method’ is designed to discriminate between common spontaneous variations and significant abnormalities in healthy subjects and therefore gives robustness to the grade 1 threshold. This approach offers a solution to the discussion and disagreement between the NIH and the FDA on the definition of ‘healthy’. The application of this method is also compatible with phase I clinical trial conditions since the supporting data was collected from comparable study conditions and populations. Use of normal ranges and variations of each clinical pharmacology unit would permit the best possible determination of thresholds, mainly for the grade 1. However, adjustments of these thresholds are necessary if they are to be applied to a different ‘healthy’ population (e.g. postmenopausal women or the elderly) or to different study conditions (e.g. ambulatory trial) or population as underlined above. Moreover, if there is a new published disease definition providing well-defined safety thresholds, this will take precedent over the limit derived using the combined method (to date, this applies to a limited number of cases for example anaemia, hypoglycaemia, hypercholesterolemia, and hyper- or hypo- tension, etc.).
Grade 1 accuracy
Some of the limits proposed by the FDA or the NIH may not necessarily apply to healthy subjects. In some cases this is due to the inappropriate normal range values and/or to a specific bias induced by the patient population and/or treatment i.e. AIDS patients, AIDS treatments or vaccine conditions and effects. Several examples are discussed below:
Bilirubin. The incidence of Gilbert disease is 10–12% in the young male population. Subjects with Gilbert's disease are usually accepted as healthy subjects in phase 1 clinical trials. Therefore, it makes sense to use a higher bilirubin upper limit, ULNR to 27 µmol l−1, as a threshold value, as proposed by CPI [11]. Such a value is also supported by the reference values published by Siest [18].
Creatinine. The FDA proposed grade 1 limit is 1.5 times the ULNR, which in our opinion is too high and would even be considered as significant if observed in a phase I healthy subject [18]. This limit does not seem appropriate for YMHS since in young healthy subjects there is little spontaneous variation in this parameter (inferior or equal to 15 mmol l−1 i.e. approximately 15%), which corresponds to no more than 1.1 times the ULNR [11].
Hypokalaemia. The FDA grade 1 limit seems too restrictive since the whole interval of FDA grade 1 (3.6 to 3.5 mEq) is actually included within the limit of NR for YMHS [11] or even for the whole population (NEJM [12]).
Hyperkalaemia. The NIH grade 1 limit, 5.6 mEq, being already potentially at a level of risk for healthy young subjects is questionable.
Hypoglycaemia. The FDA grade 1 limit, 3.8 mmol l−1, is far above the LLNR in YMHS (3 mmol l−1) [11]. Moreover, as there is a high stability of glucose in healthy subjects (reference change is –0.4 mmol l−1[11]), it makes sense to take into consideration together the magnitude of the variation (i.e. <0.9 LLNR) and a decrease (exceeding –0.5 mmol l−1) to define the threshold, as proposed by CPI.
Haemoglobin. The NIH limit (10.9 g dl−1) is too low for YMHS (reference values: 13.4 g dl−1 and reference changes: –0.8 g dl−1[11]). Furthermore, the definition of anaemia in males (<13 g dl−1) make use of the NIH limit of 10.9 g dl−1 questionable. Interestingly the CPI, FDA, and NIH proposals all suggest considering the variation (magnitude of the decrease) to determine the grade 1. Moreover, blood collection for pharmacokinetics should also be taken into account in such trials as it may be associated with a mild decrease in haemoglobin.
PMN (polymorphonuclear leucocytes) or neutrophils. The PMN values are low in YMHS (reference value: 1.7 109 l−1[11, 18]). Therefore, the limits for grade 1 proposed by the FDA (3.5 109 l−1) and the NIH (2.5 109 l−1) are not appropriate for YMHS. Such a low LLNR supports the CPI proposal to associate LLNR and the magnitude of variation from baseline i.e. exceeding minus 0.5 109 l−1. Furthermore, the difference between the white and black population, where PMN values for black subjects can be 40% lower than white [18], also requires that for black subjects the limit is lowered by 0.2 109 l−1.
CPK. There is a specific difficulty in setting a grade 1 limit for CPK due to the different values for the ULNR. In many laboratories this is around 150–200 IU l−1. This is not an appropriate limit for YMHS (reference value is 400 IU l−1[11]), which is confirmed by the NEJM NR (also 400 IU l−1[12]). It therefore makes sense to propose the grade 1 limit both as relative to the ULNR and as an absolute value. It is, however, well known that exercise in healthy individuals can cause important CPK and myoglobin elevations without renal impairment. It is therefore mandatory for an appropriate interpretation of any CPK increase to eliminate any intensive exercise (sport, etc.), and to control physical activity even in confined studies. Due to the lack of the absolute values in the FDA and NIH proposals it is difficult to make a comparison between these approaches.
For most of the other laboratory tests, the grade 1 limits reported by FDA, NIH and CPI are similar. The combined method using both the limits of the normal range as well as the variability of the parameter helps to exclude the non significant spontaneous variations and leads to a reduction in the number of reported not significant events. One must again specify that grade 1 does not correspond to a screening threshold for subject inclusion/exclusion criteria.
Grade 3 events
Grade 3 events are based on the potential safety outcomes in healthy subjects. There are several issues concerning a number of the NIH grade 3 proposals due to the potential risk for healthy subjects, e.g. bilirubin: five times the ULNR, hyperkalaemia: 6.6 mEq, hypoglycaemia: 2.16 mmol l−1, haemoglobin: 8.9 g dl−1, platelet: 50 109 l−1 and aPPT: 2.34 times the ULNR. These NIH grade 3 proposals which are potentially risky for a healthy population were possibly the consequences of the necessary adaptations to AIDS therapy. The hypereosinophilic disease definition being 1.5 109 l−1[19] means that the FDA grade 3 limit of 5 109 l−1 is questionable knowing it has been proposed due to vaccine reactions. However, these NIH or FDA grade 3 thresholds are not appropriate for use in FIM studies assessing new molecule for other indications with healthy young subjects.
Liver injury
A specific new FDA draft guideline completes the recommendation of the way to improve detecting liver toxicity within drug development [17]. Passing the already prudent Hy's law, this guideline proposed to consider the clinical symptoms and the association to concomitant modifications of other laboratory parameters to reinforce the signal and to induce an upgrading. Moreover, in the case of possible liver injury, we recommend to prohibit the use of paracetamol knowing its toxic effect on the liver [20, 21].
Allowed concomitant medication
In this paper, ‘requires a treatment’ induces a level 3 grading. This is also the criterion of the FDA or NIH scaling. This means no treatment except paracetamol is acceptable in FIM. Actually, there are two different rationale to such choice: the first one is really related to the grading – requiring treatment means the AE is severe enough and that a treatment is needed to avoid deleterious consequences; the second one is justified by caution – at the time of the FIM study, a possible drug–drug interaction is not specified yet. For example, in a repeated dose study, use of even over-the-counter remedies, for example an antacid, which could modify drug absorption, usually is one of the exclusion criteria. Thus, the FDA, NIH or CPI very conservative recommendation is meaningful as a general rule. However, depending on the tested molecule, some medications could be acceptable and then be specified in the protocol.
Stopping rules – individual level
The main point of the discussion concerns the decision to stop as soon as grade 3 is reached or exceeded. This is a recognized rule, but is may be not prudent enough for healthy subjects where certain significant or adverse effects are unacceptable, since healthy subjects have no direct therapeutic benefit from participating in a FIM trial. For healthy subjects a high level of risk management is required including a risk minimization plan. Thus, there is a need to consider grade 2 at least as a safety ‘alert’. A safe approach in such a case would be to review all available data, not only for the subject himself, but also data from all the other subjects of the same dose group. Concomitant findings, rapid worsening of a finding, association with clinical signs and also the occurrence of safety signals in other subjects of the same cohort should prompt increased caution. This is the reason why the concept of ‘upgrading’ as used in Hy's law [9] and in the recent FDA guideline [17] on liver injury was proposed here, allowing for an upgrading of events or findings under such circumstances. Sometimes, grade 2 may also be selected as the relevant stopping criterion, for example if a risky and drug specific AE is expected.
Stopping rules algorithm – cohort (dose group) level
Strict formal criteria for stopping dose escalation are sometimes challenging to establish and may need to be combined with flexibility in the definition of a stopping rule at a cohort level. Any adaptation to the number of events and subjects, and the nature of the events is acceptable as long as it fits to the type of study and the characteristics of the tested drug. Breaking the code is a frequent point of discussion as the respect of blinding is so important in clinical trials. However, in phase I, it is not ethical to continue a study if there is a high risk, as there is no therapeutic benefit for the healthy subjects participating in the trial. On the other hand, it is not justifiable to stop the dose escalation if the ‘risky event’ occurred after placebo administration. Limited un-blinding of subjects experiencing grade 3 events or findings is a satisfactory way to resolve these issues.
Specific stopping criteria may in addition be defined in the protocol, for example an expected event based on study drug non-clinical safety data (toxicology or safety pharmacology), previous experience with a similar compound, pharmacological activity or pharmacodynamic limit or, pharmacokinetic criteria (Cmax or AUC limits). For example, it is of common use to define a pharmacokinetic stopping criterion for each dose escalation in the event of pharmacokinetic dose supra-proportionality or for certain types of toxicity (life-threatening, non-monitorable, etc. …).
Conclusion
In the context of the increasing need for a risk minimization strategy for FIM trials the method and the results presented in this paper provide a simple safety grading system derived from relevant criteria, for use by investigators and sponsors to support and rationalize dose escalation decisions for young healthy subjects.
Two approaches for grading adverse events and safety findings have been proposed. A direct grading based on the ‘observed severity’ or impact on daily activities and on the need for concomitant rescue medication for the clinical adverse events, and the ‘likelihood of risk or consequence’ for continuous variables, e.g. routine laboratory results. CPI recommendation is that a ‘combined method’ be used for continuous variables to determine safety thresholds using the normal range limits and the normal spontaneous variations for each parameter under phase I study conditions.
Some grading limits are proposed for helping the dose escalation process, rather than as mandatory limits. Some adaptation of the proposed limits can be decided by the investigator based on the type of event (or finding) and on the risk assessment, i.e. dose escalation could be stopped based on a different grade, or a different grading limit, or on the occurrence of certain ‘precursor’ events. Concomitant changes to additional safety parameters, clinical signs, rapid worsening or the occurrence of safety signals in other subjects of the same cohort are integrated into the final safety grading as part of a risk minimization strategy.
An algorithm is proposed to direct the decision process at group level: dose escalation or study termination. The proposed algorithm is based on the number of subjects in phase I, the rarity of events, the possible high potential of risk. It also considers the different conclusions if the event occurred under an active drug or a placebo, thus defining the usefulness of limited un-blinding.
These proposals are in line with the European authority's recent reinforcements of the safety requirements for FIM studies.
As these propositions are still ‘points to consider’ and not yet a guideline, the CPI working group would be delighted to receive comments and suggestions supported by relevant data to confirm, to complete or to modify certain grades or rules.
Acknowledgments
We thank Suzan Baird-Bellaire who mainly contributed to the manuscript preparation and Stephan Chalon who kindly added valuable comments.
Competing interests
There are no competing interests to declare.
REFERENCES
- 1.Suntharalingam G, Perry MR, Ward S, Brett SJ, Castello-Cortes A, Brunner MD, Panoskaltsis N. Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. NEJM. 2006;355:1018–28. doi: 10.1056/NEJMoa063842. [DOI] [PubMed] [Google Scholar]
- 2.Cancer Therapy Evaluation Program. Common terminology criteria for adverse events (CTCAE) v4.0. 2009. Available at http://evs.nci.nih.gov/ftp1/CTCAE/CTCAE_4.02-2009-09-15_QuickReference_8.5x11.pdf (last accessed 10 September 2010.
- 3.National Cancer Institute. Cancer Therapy Evaluation Program. Common Terminology Criteria for Adverse Events (CTCAE) and Common Toxicity Criteria (CTC). Available at http://ctep.cancer.gov/protocolDevelopment/electronic_applications/ctc.htm (last accessed 10 September 2010.
- 4.WHO. Recommendations for grading of acute and subacute toxicity: reporting the results of cancer treatment. Cancer. 1981;47:207–14. [Google Scholar]
- 5.National Institutes of Health. Division of AIDS table for grading the severity of adult and paediatric adverse events. 2004. Available at http://www.ucdmc.ucdavis.edu/clinicaltrials/documents/DAIDS_AE_GradingTable_FinalDec2004.pdf (last accessed 10 September 2010.
- 6.US Food and Drug Administration. Guidance for industry: Toxicity grading scale for healthy adult and adolescent volunteers enrolled in preventive vaccine clinical trials. 2007. Available at http://www.fda.gov/BiologicsBloodVaccines/GuidanceComplianceRegulatoryInformation/Guidances/Vaccines/ucm074775.htm (last accessed 10 September 2010. [DOI] [PubMed]
- 7.National Institutes of Health. Comments on FDA draft guidance for industry: toxicity grading scale for healthy adult and adolescent volunteers enrolled in preventive vaccine clinical trials (April 2005). Docket No. 2005D-0155. August 2005. Available at http://www.fda.gov/ohrms/dockets/dockets/05d0155/05D-0155_emc-000002-02.pdf.
- 8.ICH Harmonised Tripartite Guideline. Post-approval safety data management: definitions and standards for expedited reporting. E2D Current Step 4 version. 12 November 2003. Available at http://www.ich.org/LOB/media/MEDIA631.pdf.
- 9.Lewis JH. ‘Hy's Law’, the ‘Rezulin rule’, and other predictors of severe drug-induced hepatotoxicity: putting risk benefit into perspective. Pharmacoepidemiol Drug Saf. 2006;15:221–9. doi: 10.1002/pds.1209. [DOI] [PubMed] [Google Scholar]
- 10.Sibille M, Bresson V, Janin A, Boutouyrie B, Rey J, Vital Durand D. Critical limits to define a laboratory adverse event during phase I studies. Eur J Clin Pharmacol. 1997;52:81–6. doi: 10.1007/s002280050254. [DOI] [PubMed] [Google Scholar]
- 11.Sibille M, Deigat N, Durieu I, Guillaumont M, Morel D, Bienvenu J, Massignon D, Vital Durand D. Laboratory data in healthy volunteers: reference values, reference changes, screening and laboratory adverse event limits in Phase I clinical trials. Eur J Clin Pharmacol. 1999;55:13–9. doi: 10.1007/s002280050586. [DOI] [PubMed] [Google Scholar]
- 12.Kratz A, Ferraro M, Sluss PM, Lewandrowski KB. Normal reference laboratory values. NEJM. 2004;351:1548–63. doi: 10.1056/NEJMcpc049016. [DOI] [PubMed] [Google Scholar]
- 13.Sibille M, Deigat N, Janin A, Kirkesseli S, Vital Durand D. Adverse events in phase I studies – a report in 1015 healthy volunteers. Eur J Clin Pharmacol. 1998;54:13–20. doi: 10.1007/s002280050413. [DOI] [PubMed] [Google Scholar]
- 14.Rosenzweig P, Miget N, Brohier S. Transaminase elevation on placebo during phase I trials: prevalence and significance. Br J Clin Pharmacol. 1999;45:19. doi: 10.1046/j.1365-2125.1999.00952.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lutfullin A, Kuhlmann J, Wensing G. Adverse events in volunteers participating in phase I clinical trials: a single-center five-year survey in 1559 subjects. Int J Clin Pharmacol Ther. 2004;43:217–26. doi: 10.5414/cpp43217. [DOI] [PubMed] [Google Scholar]
- 16.Sibille M, Donazzolo Y, Lecoz F, Krupka E. After the London tragedy, is it still possible to consider phase I is safe? Br J Clin Pharmacol. 2006;62:502–3. doi: 10.1111/j.1365-2125.2006.02740.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.U.S. Department of Health and Human Services Food and Drug Administration, Center for Drug Evaluation and Research (CDER), Center for Biologics Evaluation and Research (CBER) 2009. Guidance for industry drug-induced liver injury: premarketing clinical evaluation. July. Drug Safety. Available at http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM174090.pdf. [DOI] [PubMed]
- 18.Siest G, Henny J, Schiele F. Interpretation Des Examens De Laboratoire: Valeurs De Reference Et Variations Biologiques. Basel: Karger; 1981. [Google Scholar]
- 19.Chusid MJ, Dal DC, Wet BC, Wolff SM. The hypereosinophilic syndrome. Medicine. 1975;54:1–27. [PubMed] [Google Scholar]
- 20.Benichou C. Criteria of drug-induced liver disorders. Report of an international consensus meeting. J Hepatol. 1990;11:272–6. doi: 10.1016/0168-8278(90)90124-a. [DOI] [PubMed] [Google Scholar]
- 21.Danan G, Benichou C. Causality assessment of adverse reactions to drugs – I. A novel method based on the conclusions of international consensus meetings: application to drug-induced liver injuries. J Clin Epidemiol. 1993;46:1323–30. doi: 10.1016/0895-4356(93)90101-6. [DOI] [PubMed] [Google Scholar]
- 22.ESC Guidelines. Guideline on management (diagnosis and treatment) of syncope. Update 2004. Europace. 2004;6:467–537. doi: 10.1016/j.eupc.2004.08.008. [DOI] [PubMed] [Google Scholar]