

Abstract
AIMS
To compare the pharmacokinetics (PK) of a single-dose of liraglutide in subjects with hepatic impairment.
METHODS
This parallel group, open label trial involved four groups of six subjects with healthy, mild, moderate and severe hepatic impairment, respectively. Each subject received 0.75 mg of liraglutide (s.c., thigh), and blood samples were taken over 72 h for PK assessment. Standard laboratory and safety data were collected. The primary endpoint was area under the plasma liraglutide concentration–time curve from time zero to infinity (AUC(0,∞)).
RESULTS
Exposure to liraglutide was not increased by hepatic impairment. On the contrary, mean AUC(0,∞) was highest for healthy subjects and lowest for subjects with severe hepatic impairment (severe/healthy: 0.56, with 90% CI 0.39, 0.81) and equivalence in this parameter across groups was not demonstrated. Cmax also tended to decrease with hepatic impairment (severe/healthy: 0.71, with 90% CI 0.52, 0.97), but tmax was similar across groups (11.3–13.2 h). There were no serious adverse events, hypoglycaemic episodes or clinically significant changes in laboratory parameters and liraglutide was considered well tolerated.
CONCLUSIONS
This study indicated no safety concerns regarding use of liraglutide in patients with hepatic impairment. Exposure to liraglutide was not increased by impaired liver function; rather, the results suggest a decreased exposure with increasing degree of hepatic impairment. However, data are not conclusive to suggest a dose increase of liraglutide. Thus, the results indicate that patients with type 2 diabetes mellitus and hepatic impairment can use standard treatment regimens of liraglutide. There is, however, currently limited clinical experience with liraglutide in patients with hepatic impairment.
Keywords: DPP-IV, exenatide, glucagon-like peptide-1, incretin, liver disease, once-daily, type 2 diabetes mellitus
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
Liraglutide is a novel human GLP-1 analogue approved for treatment of type 2 diabetes mellitus. Since there is a high prevalence of co-morbid hepatic impairment in this population it is of clinical importance to establish whether the pharmacokinetics of liraglutide are affected by hepatic impairment.
WHAT THIS STUDY ADDS
This study shows that hepatic impairment does not increase liraglutide exposure. Rather, total exposure tends to decrease with increasing degree of hepatic impairment.
Introduction
The increasing prevalence of type 2 diabetes mellitus represents one of the major health-economic challenges of the 21st century [1, 2]. The aetiology of this disease is complex and involves increased insulin resistance coupled with progressive loss of beta-cell mass and function, resulting in chronic hyperglycaemia and associated multiple morbidities [1, 3]. There is evidence that nutrient-induced secretion of the glucose-regulating incretin hormone, glucagon-like peptide-1 (GLP-1), is impaired in type 2 diabetes mellitus [4].
The potential of GLP-1 as a therapy target in type 2 diabetes has gained increasing attention in recent years. This insulinotropic hormone is released from the L cells in the intestine in response to meals [5] to stimulate insulin secretion and decrease glucagon secretion [5, 6]. Both actions are glucose-dependent. Hence GLP-1 has the potential to be harnessed as a type 2 diabetes treatment that lowers blood glucose with a low risk of hypoglycaemia. Also GLP-1 decreases gastric emptying rate and appetite [5, 7], properties that are therapeutically desirable given the association of type 2 diabetes mellitus with obesity, and the association of obesity with insulin resistance [8, 9]. Furthermore, GLP-1 promotes beta-cell growth and rescue in animal models, offering the theoretical prospect of limiting or reversing disease progression [10–14].
Native GLP-1 is, however, rapidly metabolized by enzymes such as dipeptidyl peptidase-IV (DPP-IV), giving it a very short half-life (t1/2 < 2 min) [5, 15] and limiting its potential to be deployed as a therapeutic agent [10]. Analogues or mimetics of GLP-1 with a prolonged activity will therefore be required in order to exploit successfully the actions of GLP-1 in the treatment of type 2 diabetes. One such agent is liraglutide, a human GLP-1 analogue. The amino acid sequence of liraglutide has very high (97%) sequence identity to native GLP-1, but the peptide molecule is linked via a spacer to a 16-carbon fatty acid residue, which enables reversible binding to albumin [10]. This structural change results in slow release from the injection site and decreased susceptibility to degradation by DPP-IV when in the circulation. Liraglutide consequently has a plasma half-life of ∼13 h, making it suitable for once-daily dosing [16].
Clinical trials with liraglutide in patients with type 2 diabetes show many of the predicted therapeutic properties; blood glucose is lowered, with a low incidence risk of hypoglycaemia and a concomitant decrease in body weight [17–21]. Several other studies have shown liraglutide to improve beta-cell function in patients with type 2 diabetes [22–25], and its potential to inhibit beta-cell apoptosis and preserve beta-cell mass has been demonstrated in both in vitro[26–28] and in animal studies [29, 30].
As noted above, native GLP-1 is rapidly metabolized by DPP-IV, which is widely expressed and can be found in multiple tissues and cell types, as well as in the circulation [5]. Clearance of native GLP-1 and its metabolites is largely mediated via the kidneys [5]. The renal filtration and clearance of liraglutide is largely prevented by high binding to albumin and metabolic stabilization. Thereby liraglutide is fully metabolized in the body by sequential cleavage of small peptide fragments and amino acids, which involves DPP-IV and neutral endopeptidase (NEP), and clearance of liraglutide is suggested to take place by multiple organ/tissues (EMEA/379172/2009). A low potential for pharmacokinetic (PK) drug interactions related to cytochrome P450 and protein binding has been demonstrated (EMEA/379172/2009). Liraglutide is thus comparable with a large peptide, with no single organ acting as the major route of elimination [31], and hence hepatic impairment is not expected to lead to increased exposure.
Furthermore, type 2 diabetes and hepatic disease are associated, there being a higher frequency of hepatic impairment in patients with diabetes than in the general population [32].
At present there are no clinical data on the effect of impaired hepatic function on the PK of liraglutide. The present study therefore sought to provide information on the PK of single dose liraglutide in subjects with varying levels of hepatic impairment. The safety profiles of liraglutide were also assessed and compared by degree of hepatic impairment.
Methods
Design of study
This was a single centre, single dose, parallel group, open label trial investigating the PK profile of liraglutide in four groups of six subjects stratified by hepatic function: one healthy group with normal function, and the others with mild, moderate and severe impairment, respectively. The trial was conducted according to guidelines for trials in subjects with impaired hepatic function [33, 34].
Subjects
Inclusion criteria for subjects in the trial were men or women aged 18–75 years, with a body mass index (BMI) of 18.5–40.0 kg m−2. Subjects had either normal hepatic function or stable hepatic impairment classified according to the Child-Pugh scheme [35]: five parameters are assessed and 1–3 points given for each one, as indicated in Table 1. Subjects are then classified with impairment of grade A (mild hepatic impairment, 5–6 points), B (moderate hepatic impairment, 7–9 points) or C (severe hepatic impairment, 10–15 points).
Table 1.
Child-Pugh criteria for assessment of impaired liver function
| Points scored for observed findings | |||
|---|---|---|---|
| 1 | 2 | 3 | |
| Encephalopathy grade* | 0 | 1 or 2 | 3 or 4† |
| Ascites‡ | Absent | Slight | Moderate |
| Serum bilirubin (µmol l−1) | <34.2 | 34.2–51.3 | >51.3 |
| Serum albumin (g l−1) | >35 | 28–35 | <28 |
| Prothrombin time (s prolonged) | <4 | 4–6 | >6 |
Grade 0: normal consciousness, personality, neurological examination, electroencephalogram. Grade 1: restless, sleep disturbed, irritable/agitated, tremor, impaired handwriting, 5 cps waves. Grade 2: lethargic, time-disoriented, inappropriate, asterixis, ataxia, slow triphasic waves. Grade 3: somnolent, stuporous, place-disoriented, hyperactive reflexes, rigidity, slower waves. Grade 4: unrousable coma, no personality/behaviour, decerebrate, slow 2–3 cps delta activity.
Patients with encephalopathy grade 3 and 4 were excluded from the study.
Patients with advanced ascites were excluded from the study.
Exclusion criteria included liver transplanted subjects or signs of acute hepatic insufficiency, HIV-positive antibody status, major cardiac morbidity or hypertension, any clinically significant disease (other than conditions associated with hepatic impairment) that could interfere with the results of the trial and impaired renal function (serum creatinine ≥130 µmol l−1). Medications that were considered as possibly able to interfere with the trial results were not allowed and included some antidiabetic drugs (tolbutamide, glipizide), H2-receptor antagonist, cardiac glycosides, cimetidine, digoxin and non-steroidal anti-inflammatory drugs. Furthermore, the use of any medicine that the investigator considered could interfere with the trial results was also disallowed.
All subjects gave their written consent prior to any trial-related procedures. The study was conducted in accordance with the Declaration of Helsinki and Good Clinical Practice. The study protocol and informed consent information were approved by the Bioethics Committee of the Medical Academy in Warsaw, Poland.
Liraglutide administration and sample collection
Subjects attended the clinic for a screening visit (visit 1) to assess their eligibility, and were scheduled an appointment for an in-house visit (visit 2) within the next 4 weeks. For visit 2, subjects were admitted to the hospital for approximately 84 h (4 days and 3 nights). A single dose of 0.75 mg liraglutide was administered subcutaneously in the thigh in the evening on the first day (between 21.00–22.00 h), using a pen injector.
A total of 20 blood samples were collected for PK analysis: at 30 and 15 min prior to dosing, and at 2, 4, 6, 8, 9, 10, 11, 12, 13, 14, 15, 16, 21, 24, 36, 48, 60 and 72 h post dosing. The frequency of these sample times was designed to capture multiple readings around the known tmax of liraglutide [16]. For safety, plasma glucose concentration was measured before breakfast, lunch and dinner and at bedtime throughout the in-house period and any hypoglycaemic episodes were recorded. Hepatic blood flow and portal vein diameter were assessed by ultrasonography. All subjects continued their usual drug treatments during the in-house period and were served standard meals and beverages. A follow-up visit (visit 3) was scheduled 1–2 weeks after visit 2 in order to evaluate safety parameters. Vital signs, standard 12–lead electrocardiogram (ECG), physical examination and blood and urine sampling for standard laboratory assessments were performed at all three visits.
Laboratory assessments
The bioanalysis of liraglutide in plasma was performed using a validated enzyme-linked immunosorbent assay (ELISA) [16] by Capio Diagnostik a.s., Copenhagen, Denmark. The assay measures total liraglutide plasma concentration and is specific for intact liraglutide. The repeatability is 2.4–6.5%, the day-to-day variation is 3.7–10%, the lower limit of quantification is 18 pm, and dilution is documented up to 16-fold while maintaining linearity.
The plasma protein binding (fb) of liraglutide was measured in vitro on spiked samples (liraglutide concentrations: 1000, 10 000 and 100 000 pmol l−1) by reiterated stepwise equilibrium dialysis by Novo Nordisk A/S, Måløv, Denmark. Unbound fraction of liraglutide (fu) was calculated as (100 – fb)%. For this, an additional amount of blood (approximately 27 ml) was taken from each trial subject before liraglutide administration.
Plasma glucose was measured for safety reasons when subjects were at the site using a glucometer. Analysis for clinical chemistry and haematology were performed by MDS Pharma Services Central Lab, Hamburg, Germany.
Endpoints and statistical methods
The primary PK endpoint derived from the liraglutide plasma profiles was the area under the concentration–time curve from time zero (dosing) to infinity (AUC(0,∞)), estimated by standard non-compartment methods. It was derived as the sum of two areas; the area under the curve from zero to last valid measurement t (AUC(0,t)) and the area under the curve from the last valid measurement to infinity. AUC(0,t) was calculated by the trapezoidal method. The last area was approximated by the area from t to infinity under an exponential curve with the terminal rate (λz). In addition secondary standard PK endpoints derived from the individual liraglutide plasma profiles included maximum concentration (Cmax), time for maximum concentration (tmax), total apparent clearance (CL/F), terminal half-life (t1/2) and apparent volume of distribution (Vz/F).
Safety endpoints were adverse events, laboratory parameters (haematology, biochemistry (including alanine aminotransferase [ALAT] and aspartate aminotransferase [ASAT], urinalysis and plasma glucose), physical examination, vital signs and ECG. Diameter of the portal vein and hepatic blood flow were measured, and the aetiology of liver disease was registered.
The between-group comparison was performed using a linear normal analysis of variance (anova) for the log transformed values of AUC(0,∞). The model included fixed effects of hepatic group and gender, whereas age and log(weight) were included as covariates. The groups of healthy subjects and of subjects with severe hepatic impairment were to be declared equivalent with respect to an endpoint if the 90% confidence interval (CI) of the ratio between hepatic impaired vs. healthy subjects was fully contained within the interval (0.70, 1.43). This no effect boundary was chosen instead of the standard range (0.8, 1.25) based on the broad therapeutic range of liraglutide. Analyses of secondary endpoints were performed as described for the primary endpoint. The analysis of tmax was performed by non-parametric methods for independent samples (Hodges-Lehmann estimator).
Regression analysis of log AUC was performed in which log(albumin), log(weight), log(portal vein diameter), age and gender were accounted for. Statistical analyses were performed using SAS V.8.0, Proc MIXED.
Results
Study groups
A total of 25 subjects were screened, with one subject withdrawing consent prior to the commencement of any trial-related activities. Twenty-four subjects were exposed to trial product and provided PK data, but one of them did not attend visit 3 (follow-up). The PK and safety populations both comprised the 24 subjects.
Baseline characteristics by hepatic group are presented in Table 2. All of the 24 enrolled subjects (14 male, 10 female) were Caucasians aged 21–61 years. The groups were well balanced in terms of gender, weight and BMI, although subjects with moderate and severe hepatic impairment tended to be older than subjects in the other two groups. None of the subjects had clinically significant abnormal hepatic blood flow. Five subjects had type 2 diabetes mellitus, three in the mildly, one in the moderately and one in the severely impaired hepatic group.
Table 2.
Baseline demographics and characteristics
| Hepatic impairment group | ||||
|---|---|---|---|---|
| Healthy | Mild | Moderate | Severe | |
| Male/female (n) | 3/3 | 4/2 | 3/3 | 4/2 |
| Age (years) | 43.8 (13.5) | 44.5 (12.7) | 53.2 (6.6) | 49.8 (10.4) |
| Weight (kg) | 79.0 (11.9) | 75.4 (18.0) | 77.5 (21.6) | 74.8 (15.5) |
| BMI (kg m−2) | 27.0 (1.7) | 27.1 (5.2) | 27.9 (5.2) | 25.9 (4.4) |
| Albumin (g dl−1) | 38.2 (2.7) | 37.2 (2.6) | 35.3 (4.3) | 27.8 (4.4) |
| Bilirubin (µmol l−1) | 11.67 (3.33) | 15.00 (8.72) | 40.83 (12.64) | 67.00 (24.41) |
| PTT (s) | 0.17 (0.41) | 1.17 (1.17) | 2.83 (0.75) | 4.83 (1.72) |
| Encephalopathy (n) (None / 1 and 2) | 6/0 | 6/0 | 3/3 | |
| Ascites (n) | ||||
| Absent/slight/moderate | – | 6/0/0 | 4/2/0 | 2/0/4 |
| Portal vein (mm) | 10.3 (0.8) | 12.5 (1.0) | 13.6 (1.9) | 11.8 (0.8) |
Data are presented as mean (SD), or as number of patients, denoted (n). BMI, body mass index; PTT, partial thromboplastin time.
Liraglutide pharmakokinetics
The liraglutide concentration profiles (Figure 1) showed a higher average concentration from ∼5–25 h for the healthy subjects compared with subjects with hepatic impairment. Subjects with severe hepatic impairment had the lowest average concentration during the entire period. Overall, all groups demonstrated a comparable concentration vs. time profile, with liraglutide concentrations approaching zero approximately 72 h after administration.
Figure 1.
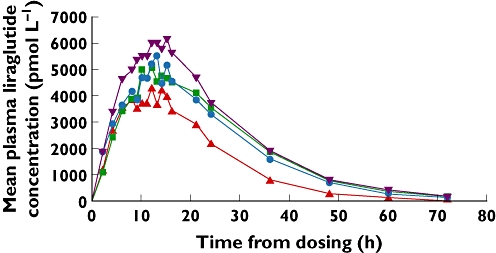
Mean plasma liraglutide concentration–time profile in healthy and hepatically impaired subjects.  healthy;
healthy;  mild;
mild;  moderate and
moderate and  severe hepatic impairment
severe hepatic impairment
The mean values for AUC(0,∞) were highest for healthy subjects and lowest for those with severe hepatic impairment (Table 3). The 90% CI for the ratio of this parameter between the group of severely impaired hepatic subjects and the healthy subject group (0.39, 0.81) was not fully contained in the pre-specified interval (0.70, 1.43; Table 4). Equivalence for AUC(0,∞) was therefore not demonstrated for this group comparison. This was also the case for the other hepatically impaired groups, where hepatic impairment was associated with a lower estimated AUC(0,∞) compared with that of healthy subjects (Tables 3 and 4).
Table 3.
Pharmacokinetic characteristics of liraglutide by degree of hepatic impairment
| Hepatic impairment group | ||||
|---|---|---|---|---|
| Healthy | Mild | Moderate | Severe | |
| AUC (0,∞) (pmol l−1 h) | ||||
| Mean (SD) | 179 641 (43 154) | 149 812 (70 007) | 154 615 (47 939) | 105 158 (40 843) |
| Min–Max | 114 195–218 365 | 87 851–273 622 | 91 623–208 707 | 54 724–153 211 |
| Cmax (pmol l−1) | ||||
| Mean (SD) | 6 746 (1 534) | 6 433 (3 859) | 5 593 (1 558) | 4 872 (1 637) |
| Min–Max | 4 852–8 978 | 4 162–14 202 | 4 003–7 952 | 2 769–7 506 |
| tmax (h) | ||||
| Mean (SD) | 12.3 (2.3) | 11.3 (3.8) | 12.7 (2.3) | 13.2 (2.9) |
| Min–Max | 9–15 | 4–14 | 10–15 | 8–16 |
| t1/2 (h) | ||||
| Mean (SD) | 11.2 (1.0) | 10.7 (1.1) | 11.4 (2.2) | 9.5 (1.0) |
| Min–Max | 9.9–12.3 | 9.1–12.0 | 9.7–15.6 | 8.1–10.7 |
| CL/F (l h−1) | ||||
| Mean (SD) | 1.18 (0.33) | 1.55 (0.59) | 1.42 (0.51) | 2.21 (0.99) |
| Min–Max | 0.92–1.75 | 0.73–2.28 | 0.96–2.18 | 1.30–3.65 |
| Vz/F (l) | ||||
| Mean (SD) | 18.7 (3.8) | 23.5 (7.9) | 23.1 (8.2) | 30.2 (13.5) |
| Min–Max | 15.6–25.0 | 11.9–32.3 | 14.1–33.9 | 16.8–50.1 |
AUC(0,∞), area under the liraglutide plasma concentration–time curve from time zero to infinity. The measurements were performed up to 72 h after liraglutide dosing. Cmax, maximum liraglutide plasma concentration. tmax, time to reach maximum liraglutide concentration. t1/2, terminal plasma half-life calculated as t1/2 = ln2/λz. λz, terminal elimination rate constant, calculated by log-linear regression on the log linear phase of the plasma concentration profile. CL/F, total apparent clearance, estimated as CL/F = dose/AUC. Vz/F, apparent volume of distribution, estimated as Vz/F = (CL/F)/λz.
Table 4.
Between-group comparison of liraglutide pharmacokinetics. Ratios and 90% CI comparing hepatically impaired groups to the healthy subject group
| Hepatic impairment group | |||
|---|---|---|---|
| Mild/healthy | Moderate/healthy | Severe/healthy | |
| AUC (0,∞) Estimate (90% CI) | 0.77 (0.53, 1.11) | 0.87 (0.60, 1.25) | 0.56 (0.39, 0.81) |
| Cmax Estimate (90% CI) | 0.89 (0.65, 1.21) | 0.80 (0.59, 1.09) | 0.71 (0.52, 0.97) |
| t1/2 (h) Estimate (90% CI) | 0.95 (0.83, 1.10) | 1.01 (0.88, 1.17) | 0.85 (0.73, 0.98) |
| CL/F Estimate (90% CI) | 1.30 (0.90, 1.87) | 1.15 (0.80, 1.66) | 1.78 (1.23, 2.58) |
| Vz/F Estimate (90% CI) | 1.23 (0.86, 1.77) | 1.17 (0.82, 1.67) | 1.51 (1.05, 2.17) |
AUC(0,∞), area under the liraglutide plasma concentration–time curve from time zero to infinity. The measurements were performed up to 72 h after liraglutide dosing. Cmax, maximum liraglutide plasma concentration. tmax, time to reach maximum liraglutide concentration. t1/2, terminal plasma half-life calculated as t1/2 = ln2/λz. λz, terminal elimination rate constant, calculated by log-linear regression on the log linear phase of the plasma concentration profile. CL/F, total apparent clearance, estimated as CL/F = dose/AUC. Vz/F, apparent volume of distribution, estimated as Vz/F = (CL/F)/λz.
The group comparisons of subjects with severe, moderate and mild hepatic impairment vs. healthy subjects were found to be equivalent with respect to t1/2 but not for any other PK endpoints (Cmax, CL/F, Vz/F; Table 4).
Overall, tmax was similar across groups (11.3–13.2 h) and there was no statistically significant difference between healthy subjects and subjects with hepatic impairment. In subjects with severe hepatic impairment tmax occurred 1 h later, but was similar in subjects with moderate impairment and occurred 0.5 h earlier in subjects with mild impairment compared with healthy subjects.
A multiple regression analysis was performed with the primary endpoint as the dependent variable and albumin as an independent variable. In addition the analysis was adjusted for age, gender, weight and diameter of the portal vein. This analysis showed that albumin concentration had a statistically significant effect on AUC(0,∞) (P = 0.041, data not shown): the higher the albumin concentration, the higher the estimated AUC. This tendency is also apparent in Figure 2. However, as albumin concentration declined with increasing degree of hepatic impairment, and due to the small size of this trial, it is not possible to state if the observed relationship is solely attributed to lower albumin concentrations or the other aspects of hepatic impairment. This was illustrated further by a multiple regression analysis where both albumin and hepatic group were included, and neither the effect of hepatic group (P = 0.53) nor the effect of albumin (P = 0.54) was statistically significant.
Figure 2.
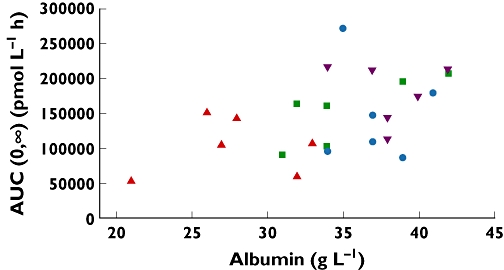
Scatter plot AUC(0,∞) vs. albumin concentrations.  healthy;
healthy;  mild;
mild;  moderate and
moderate and  severe hepatic impairment
severe hepatic impairment
The unbound fraction of liraglutide was very low in all groups, and no clear association between unbound fraction of liraglutide and hepatic group was found. At a liraglutide concentration of 1000 pmol l−1, mean (SD) unbound percentages were (respectively for the healthy subjects and mild, moderate and severe hepatic groups): 0.53% (0.46), 0.59% (0.61), 0.68% (0.45) and 0.40% (0.20). At a liraglutide concentration of 100 000 pmol l−1, the respective unbound fractions were: 0.71% (0.56), 0.52% (0.51), 0.48% (0.30) and 0.35% (0.24). Thus, the observed mean fraction unbound in the group of subjects with severe hepatic impairment was, in fact, lower than the mean fraction unbound in the healthy group.
Safety and tolerability
No serious adverse events were reported. Three adverse events were reported in this trial, two of which, nausea and headache, were experienced by two different subjects in the moderate hepatic impairment group. These events were considered possibly related to liraglutide administration, and both subjects recovered within 1 day. The third adverse event was an episode of acute bronchitis, experienced 9 days post dosing with liraglutide. No hypoglycaemic episodes were reported during the trial period. No clinically significant changes were reported for any of the laboratory parameters investigated, and there were no trends for changes by hepatic group evident on liraglutide administration. This was also true for vital signs, the physical examination and ECG.
Discussion
The present study has provided reassuring results concerning the use of liraglutide in patients with hepatic impairment, suggesting that drug exposure to liraglutide is not increased but may rather be somewhat decreased as a result of impaired liver function. Liraglutide was well tolerated and it can, based on PK variables, be concluded that patients with type 2 diabetes mellitus and concomitant hepatic impairment are not expected to require dose adjustment compared with subjects with normal hepatic function, when treated with liraglutide. Similarly, renal dysfunction is found not to increase exposure of liraglutide and no dose adjustment is expected in this patient group with type 2 diabetes, treated with liraglutide [36].
The general concern with hepatic disease is that exposure to a drug that is extensively metabolised in the liver will be increased, thereby potentially exaggerating the pharmacodynamic effects and/or side effects. In the case of liraglutide, clearance was not expected to be hepatically mediated. Its property of reversible albumin binding has nevertheless been a contributing factor of this investigation being undertaken since hepatic impairment is often associated with reduced albumin concentrations. The vast majority of liraglutide molecules are circulated reversibly bound to plasma albumin. As it is the free fraction of liraglutide that interacts with GLP-1 receptors, it is possible that the pharmacodynamic effect will be enhanced in the setting of reduced circulating albumin concentrations. On the other hand, a decrease in albumin concentration may result in an increased rate of metabolism of liraglutide by DPP-IV and NEP. It is also possible that hepatocyte destruction in liver disease might result in an increased enzyme outflow [37, 38] with a potential impact on liraglutide metabolism. However, no association was found between liraglutide exposure and ALAT or ASAT in this study.
In fact, the present study showed that hepatic impairment tended to decrease exposure to liraglutide. Considering the primary endpoint, the ratio of AUC(0,∞) in severely hepatically impaired subjects was estimated at 0.56 (90% CI 0.39, 0.81) versus that of healthy subjects with normal hepatic function, and reduced estimated ratios were also shown when subjects with moderate and mild hepatic impairment were compared with the healthy subjects. It is noteworthy that the half-life of liraglutide was not affected by hepatic impairment, and this suggests that the differences in the AUC (0,∞) of liraglutide might result primarily from differences in absorption of the drug from the subcutaneous depot rather than differences in its subsequent metabolism.
A statistically significant positive association between albumin concentration and liraglutide AUC(0,∞) was observed (P = 0.041). Albumin concentration in the group with severe hepatic impairment was lower than that of healthy subjects (mean 27.8 mmol l−1, range 21–33 mmol l−1vs. mean 38.2 mmol l−1, range 34–42 mmol l−1), but as albumin is part of the Child-Pugh classification of the hepatic impaired subjects it is not possible, based on a study of this size, to state if the relationship between liraglutide AUC and hepatic impairment is solely attributable to lower albumin concentrations or other aspects of hepatic impairment. This was confirmed by further statistical analysis where no significant observation was found when both albumin and hepatic group were included in the analysis.
A similar methodology was used in the present trial to one previously reported that was designed to assess the impact of renal impairment on liraglutide kinetics [36]. This provides an opportunity to compare data across the healthy control groups. In fact, this comparison suggests that healthy controls in the present study had a somewhat lower (∼25%) overall and maximal exposure compared with healthy controls from the renal impairment study. The reason for this is not altogether clear, but probably relates to the choice of the thigh (as opposed to the abdomen) as the injection site in this study compared with the renal study [36, 39]. The tmax (median 12.5 h) was similar to that reported here.
No safety concerns were raised during the trial. Both of the reported adverse events considered possibly related to liraglutide administration were experienced by subjects in the moderate hepatic impairment group. These events, a moderate episode of nausea and one of headache, were reported 12 and 18 h post dosing, respectively, and resolved within a day of reporting. They cannot be attributed to increased exposure to liraglutide as exposure was not higher in these subjects than in the healthy controls. No hypoglycaemic events were reported, and the low number of adverse events reported overall indicated that liraglutide was well tolerated at the dose given in all groups.
In conclusion, the results of this study indicate no safety concerns regarding the use of liraglutide in patients with hepatic impairment. Exposure to liraglutide is not increased by impaired liver function. Rather, the results suggest a decreased exposure with increasing degree of hepatic impairment. However, data are not conclusive to suggest a dose increase of liraglutide in patients with hepatic impairment and the degree of hepatic impairment did not appear to be associated with increased risk of adverse events. Thus, the results indicate that patients with type 2 diabetes mellitus who also suffer from hepatic impairment can use standard treatment regimens of liraglutide. There is, however, currently limited clinical experience with liraglutide in this patient group.
Acknowledgments
The authors accept direct responsibility for this paper and are grateful for the contribution made by Watermeadow Medical (supported by Novo Nordisk A/S, Bagsvaerd, Denmark) in developing the draft manuscript from an agreed outline and in collating comments.
Competing interests
AF, CH and MZ are employed by Novo Nordisk and hold stocks in the company. KN and PJ received financial support for conducting this study from Novo Nordisk A/S, the developer of liraglutide.
REFERENCES
- 1.Freeman H, Cox RD. Type-2 diabetes: a cocktail of genetic discovery. Hum Mol Genet. 2006;15:R202–9. doi: 10.1093/hmg/ddl191. [DOI] [PubMed] [Google Scholar]
- 2.Naser KA, Gruber A, Thomson GA. The emerging pandemic of obesity and diabetes: are we doing enough to prevent a disaster? Int J Clin Pract. 2006;60:1093–7. doi: 10.1111/j.1742-1241.2006.01003.x. [DOI] [PubMed] [Google Scholar]
- 3.Kasuga M. Insulin resistance and pancreatic beta cell failure. J Clin Invest. 2006;116:1756–60. doi: 10.1172/JCI29189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vilsbøll T, Krarup T, Deacon CF, Madsbad S, Holst JJ. Reduced postprandial concentrations of intact biologically active glucagon-like peptide 1 in type 2 diabetic patients. Diabetes. 2001;50:609–13. doi: 10.2337/diabetes.50.3.609. [DOI] [PubMed] [Google Scholar]
- 5.Baggio LL, Drucker DJ. Biology of incretins: GLP-1 and GIP. Gastroenterology. 2007;132:2131–57. doi: 10.1053/j.gastro.2007.03.054. [DOI] [PubMed] [Google Scholar]
- 6.Larsson H, Holst JJ, Ahren B. Glucagon-like peptide-1 reduces hepatic glucose production indirectly through insulin and glucagon in humans. Acta Physiol Scand. 1997;160:413–22. doi: 10.1046/j.1365-201X.1997.00161.x. [DOI] [PubMed] [Google Scholar]
- 7.Verdich C, Flint A, Gutzwiller JP, Naslund E, Beglinger C, Hellstrom PM, Long SJ, Morgan LM, Holst JJ, Astrup A. A meta-analysis of the effect of glucagon-like peptide-1 (7-36) amide on ad libitum energy intake in humans. J Clin Endocrinol Metab. 2001;86:4382–9. doi: 10.1210/jcem.86.9.7877. [DOI] [PubMed] [Google Scholar]
- 8.Haffner SM. Relationship of metabolic risk factors and development of cardiovascular disease and diabetes. Obesity (Silver Spring) 2006;14:121S–7S. doi: 10.1038/oby.2006.291. [DOI] [PubMed] [Google Scholar]
- 9.Muoio DM, Newgard CB. Obesity-related derangements in metabolic regulation. Annu Rev Biochem. 2006;75:367–401. doi: 10.1146/annurev.biochem.75.103004.142512. [DOI] [PubMed] [Google Scholar]
- 10.Vilsbøll T. Liraglutide: a once-daily GLP-1 analogue for the treatment of type 2 diabetes mellitus. Expert Opin Investig Drugs. 2007;16:231–7. doi: 10.1517/13543784.16.2.231. [DOI] [PubMed] [Google Scholar]
- 11.Nauck MA, Meier JJ, Creutzfeldt W. Incretins and their analogues as new antidiabetic agents. Drug News Perspect. 2003;16:413–22. doi: 10.1358/dnp.2003.16.7.829353. [DOI] [PubMed] [Google Scholar]
- 12.Cohen A, Horton ES. Progress in the treatment of type 2 diabetes: new pharmacologic approaches to improve glycemic control. Curr Med Res Opin. 2007;23:905–17. doi: 10.1185/030079907x182068. [DOI] [PubMed] [Google Scholar]
- 13.Gallwitz B. Glucagon-like peptide-1 as a treatment option for type 2 diabetes and its role in restoring beta-cell mass. Diabetes Technol Ther. 2005;7:651–7. doi: 10.1089/dia.2005.7.651. [DOI] [PubMed] [Google Scholar]
- 14.Wajchenberg BL. Beta-cell failure in diabetes and preservation by clinical treatment. Endocr Rev. 2007;28:187–218. doi: 10.1210/10.1210/er.2006-0038. [DOI] [PubMed] [Google Scholar]
- 15.Deacon CF, Nauck MA, Toft-Nielsen M, Pridal L, Williams B, Holst JJ. Both subcutaneously and intravenously administered glucagon-like peptide 1 are rapidly degraded from the NH2-terminus in type II diabetic patients and in healthy subjects. Diabetes. 1995;44:1126–31. doi: 10.2337/diab.44.9.1126. [DOI] [PubMed] [Google Scholar]
- 16.Agersø H, Jensen LB, Elbrond B, Rolan P, Zdravkovic M. The pharmacokinetics, pharmacodynamics, safety and tolerability of NN2211, a new long-acting GLP-1 derivative, in healthy men. Diabetologia. 2002;45:195–202. doi: 10.1007/s00125-001-0719-z. [DOI] [PubMed] [Google Scholar]
- 17.Madsbad S, Schmitz O, Ranstam J, Jakobsen G, Matthews DR. Improved glycemic control with no weight increase in patients with type 2 diabetes after once-daily treatment with the long-acting glucagon-like peptide 1 analog liraglutide (NN2211): a 12-week, double-blind, randomized, controlled trial. Diabetes Care. 2004;27:1335–42. doi: 10.2337/diacare.27.6.1335. [DOI] [PubMed] [Google Scholar]
- 18.Harder H, Nielsen L, Tu DT, Astrup A. The effect of liraglutide, a long-acting glucagon-like peptide 1 derivative, on glycemic control, body composition, and 24-h energy expenditure in patients with type 2 diabetes. Diabetes Care. 2004;27:1915–21. doi: 10.2337/diacare.27.8.1915. [DOI] [PubMed] [Google Scholar]
- 19.Feinglos MN, Saad MF, Pi-Sunyer FX, An B, Santiago O, Liraglutide Dose-Response Study Group Effects of liraglutide (NN2211), a long-acting GLP-1 analogue, on glycaemic control and bodyweight in subjects with Type 2 diabetes. Diabet Med. 2005;22:1016–23. doi: 10.1111/j.1464-5491.2005.01567.x. [DOI] [PubMed] [Google Scholar]
- 20.Nauck MA, Hompesch M, Filipczak R, Le TD, Zdravkovic M, Gumprecht J, NN2211-1499 Study Group Five weeks of treatment with the GLP-1 analogue liraglutide improves glycaemic control and lowers body weight in subjects with type 2 diabetes. Exp Clin Endocrinol Diabetes. 2006;114:417–23. doi: 10.1055/s-2006-924230. [DOI] [PubMed] [Google Scholar]
- 21.Vilsbøll T, Zdravkovic M, Le-Thi T, Krarup T, Schmitz O, Courrèges JP, Verhoeven R, Bugánová I, Madsbad S. Liraglutide, a long-acting human glucagon-like peptide-1 analog, given as monotherapy significantly improves glycemic control and lowers body weight without risk of hypoglycemia in patients with type 2 diabetes. Diabetes Care. 2007;30:1608–10. doi: 10.2337/dc06-2593. [DOI] [PubMed] [Google Scholar]
- 22.Degn KB, Juhl CB, Sturis J, Jakobsen G, Brock B, Chandramouli V, Rungby J, Landau BR, Schmitz O. One week's treatment with the long-acting GLP-1 derivative, liraglutide (NN2211), markedly improves 24-h glycaemia, alpha- and beta-cell function and reduces endogenous glucose release in patients with type 2 diabetes. Diabetes. 2004;53:1187–94. doi: 10.2337/diabetes.53.5.1187. [DOI] [PubMed] [Google Scholar]
- 23.Juhl CB, Hollingdal M, Sturis J, Jakobsen G, Agersø H, Veldhuis J, Pørksen N, Schmitz O. Bedtime administration of NN2211, a long-acting GLP-1 derivative, substantially reduces fasting and postprandial glycemia in type 2 diabetes. Diabetes. 2002;51:424–9. doi: 10.2337/diabetes.51.2.424. [DOI] [PubMed] [Google Scholar]
- 24.Chang AM, Jakobsen G, Sturis J, Smith MJ, Bloem CJ, Galecki A, Halter JB. The GLP-1 derivative NN2211 restores beta-cell sensitivity to glucose in type 2 diabetic patients after a single dose. Diabetes. 2003;52:1786–91. doi: 10.2337/diabetes.52.7.1786. [DOI] [PubMed] [Google Scholar]
- 25.Mari A, Degn K, Brock B, Rungby J, Ferrannini E, Schmitz O. Effects of the long-acting human GLP-1 analogue liraglutide on beta-cell function in normal living conditions. Diabetes Care. 2007;30:2032–3. doi: 10.2337/dc07-0310. [DOI] [PubMed] [Google Scholar]
- 26.Bregenholt S, Moldrup A, Blume N, Karlsen AE, Nissen Friedrichsen B, Tornhave D, Knudsen LB, Petersen JS. The long-acting glucagon-like peptide-1 analogue, liraglutide, inhibits beta-cell apoptosis in vitro. Biochem Biophys Res Commun. 2005;330:577–84. doi: 10.1016/j.bbrc.2005.03.013. [DOI] [PubMed] [Google Scholar]
- 27.Piper Hanley K, Dijkstra I, Dunleavey L, Hanley NA. The long-acting GLP-1 analog, liraglutide, increases beta cell numbers during early human development. Diabetologia. 2005;48(Suppl. 1):A151. (Abstract) [Google Scholar]
- 28.Hui H, Wang C, Xu J, Hui Z, Perfetti R. Liraglutide inhibits cell death in human isolated islets by regulating the expression of apoptosis-related genes. Diabetes. 2006;55(Suppl. 1):A1993. (Abstract) [Google Scholar]
- 29.Sturis J, Gotfredsen CF, Rømer J, Rolin B, Ribel U, Brand CL, Wilken M, Wassermann K, Deacon CF, Carr RD, Knudsen LB. GLP-1 derivative liraglutide in rats with beta-cell deficiencies: influence of metabolic state on beta-cell mass dynamics. Br J Pharmacol. 2003;140:123–32. doi: 10.1038/sj.bjp.0705397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rolin B, Larsen MO, Gotfredsen CF, Deacon CF, Carr RD, Wilken M, Knudsen LB. The long-acting GLP-1 derivative NN2211 ameliorates glycemia and increases beta-cell mass in diabetic mice. Am J Physiol Endocrinol Metab. 2002;283:E745–52. doi: 10.1152/ajpendo.00030.2002. [DOI] [PubMed] [Google Scholar]
- 31.Bjørnsdottir I, Olsen AK, Larsen U, Helleberg H, Vanggaard J, OOsterhuis B, van Lier J, Zdravkovic M, Malm-Erjefält M. Metabolism and excretion of the once-daily human GLP-1 analogue liraglutide in human healthy subjects and its in vitro degradation by dipeptidyl peptidase IV and neutral endopeptidase. Diabetologia. 2008;51(Suppl. 1):S356. doi: 10.1124/dmd.110.034066. (Abstract) [DOI] [PubMed] [Google Scholar]
- 32.Bell DS, Allbright E. The multifaceted associations of hepatobiliary disease and diabetes. Endocr Pract. 2007;13:300–12. doi: 10.4158/EP.13.3.300. [DOI] [PubMed] [Google Scholar]
- 33.FDA. Guidance for industry – pharmacokinetics in patients with impaired hepatic function -trial design, data analysis, and impact on dosing and labelling. 2010. Available at: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm072127.pdf (Accessed 19 August 2010)
- 34.EMA. Guideline on the evaluation of the pharmacokinetics of medicinal products in patients with impaired hepatic function (CHMP/EWP/2339/02) 2005 Available at: http://www.ema.europa.eu/ema/index.jsp?curl = pages/regulation/general/general_content_000370.jsp&murl = menus/regulations/regulations.jsp&mid = WC0b01ac0580032ec5&jsenabled = true. (Accessed 19 August 2010) [Google Scholar]
- 35.Pugh RN, Murray-Lyon IM, Dawson JL, Pietroni MC, Williams R. Transection of the oesophagus for bleeding oesophageal varices. Br J Surg. 1973;60:646–9. doi: 10.1002/bjs.1800600817. [DOI] [PubMed] [Google Scholar]
- 36.Jacobsen LV, Hindsberger C, Robson R, Zdrackovic M. Effect of renal impairment on the pharmacokinetics of the GLP-1 analogue liraglutide. Br J Clin Pharmacol. 2009;68:898–905. doi: 10.1111/j.1365-2125.2009.03536.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Skude G, Wadstein J. Amylase, hepatic enzymes and bilirubin in serum of chronic alcoholics. Acta Med Scand. 1977;201:53–8. doi: 10.1111/j.0954-6820.1977.tb15654.x. [DOI] [PubMed] [Google Scholar]
- 38.Karoui S, Hamzaoui S, Sahli F, Matri S, Boubaker J, Filali A. [Mortality in cirrhosis: prevalence, causes, and predictive factors] Tunis Med. 2002;80:21–5. [PubMed] [Google Scholar]
- 39.Kapitza C, Zdravkovic M, Zijlstra E, Segel S, Heise T, Flint A. Effect of three different injection sites on the pharmacokinetics of the once-daily human GLP-1 analogue liraglutide. J Clin Pharmacol. 2010 doi: 10.1177/0091270010374474. 0: 0091270010374474v1. [DOI] [PubMed] [Google Scholar]