

Abstract
To identify new genetic regulators of cellular aging and senescence, we performed genome-wide comparative RNA profiling with selected human cellular model systems, reflecting replicative senescence, stress-induced premature senescence, and distinct other forms of cellular aging. Gene expression profiles were measured, analyzed, and entered into a newly generated database referred to as the GiSAO database. Bioinformatic analysis revealed a set of new candidate genes, conserved across the majority of the cellular aging models, which were so far not associated with cellular aging, and highlighted several new pathways that potentially play a role in cellular aging. Several candidate genes obtained through this analysis have been confirmed by functional experiments, thereby validating the experimental approach. The effect of genetic deletion on chronological lifespan in yeast was assessed for 93 genes where (i) functional homologues were found in the yeast genome and (ii) the deletion strain was viable. We identified several genes whose deletion led to significant changes of chronological lifespan in yeast, featuring both lifespan shortening and lifespan extension. In conclusion, an unbiased screen across species uncovered several so far unrecognized molecular pathways for cellular aging that are conserved in evolution.
Keywords: aging, evolution, replicative lifespan, replicative senescence, senescence, yeast
Introduction
A major goal in current aging research is to understand genetic regulation of aging in humans. However, many recent insights into genetic determinants of aging have been gained from studies with short-lived eukaryotic model systems of aging [for recent review, see (Partridge, 2009)], which, unlike human beings, are amenable to molecular genetic analysis in the laboratory. Extrapolation of results obtained in lower model organisms identified cellular pathways such as the insulin/IGF pathway (Holzenberger, 2004; Katic & Kahn, 2005), which are conserved in evolution and appear to modulate aging in a variety of organisms including mammals (Bartke, 2005). Although it is very likely that these very same mechanisms also modulate aging in humans, this has not yet been formally proven. Moreover, extrapolating findings in lower model organisms to more complex organisms, such as humans, bears the inherent risk that some pathways, which are less important or absent there, may be overlooked.
To circumvent this potential problem, we applied an alternative strategy, namely to employ human model systems of cellular aging and senescence as the primary screening system. There is now increasing evidence that the appearance of cellular senescence contributes to the aging of tissues, such as the skin (Dimri et al., 1995; Ressler et al., 2006), the vascular system (Vasile et al., 2001; Minamino et al., 2002; reviewed by Erusalimsky, 2009), and the kidney (Melk & Halloran, 2001). We performed genome-wide RNA profiling on several experimental models for cellular senescence, including stress-induced premature senescence (SIPS), replicative senescence, and other distinct forms of cellular aging, as proposed recently (Lepperdinger et al., 2008; Hackl et al., 2010) and collected the data in a novel database referred to as the GiSAO (genes involved in senescence, apoptosis, and oxidative stress) database. Comparison of the differential gene expression patterns of the models should in turn unveil distinct novel genetic regulators of cellular aging. Potentially, the information obtained from these experiments, when filtered accordingly, should provide molecular information about highly conserved gene expression patterns related to cellular aging. Because of the genome-wide approach, the data collected from cell types as diverse as those employed herein should also uncover recurrent cellular alterations indicative for hitherto unknown biological aging mechanisms. As a further step in evaluation of the functional role of the individually selected genes, those that were also found to be highly conserved during phylogeny were taken to functional tests in yeast deletion strains thereby monitoring the respective lifespan of the mutant organisms.
Results
Comparative RNA profiling of aging and senescence in human cellular models
Accumulation of damage is considered a key factor driving cellular aging, and oxidative stress is a common denominator of stress that induces cellular senescence in vitro in a variety of cell types (Dumont et al., 2000). To identify key transcriptional events preceding senescence induced by oxidative stress, a comprehensive set of human cellular models were subjected to oxidative stress at doses known to elicit a senescence response. The corresponding model systems are referred to as experimental group 1 (EG1) (Table 1). For comparison, other model systems of cellular aging were also analyzed collectively referred to as experimental group 2 (EG2) (Table 1). On the one hand, we used models of in vitro cellular senescence not triggered by oxidative stress, namely replicative senescence of human umbilical vein endothelial cells (HUVEC) and renal proximal tubular epithelial cells (RPTEC). Similarly, we applied primary foreskin fibroblasts (PFF) in which premature senescence was induced by mitochondrial dysfunction (Passos et al., 2007), which is largely independent of oxidative stress (Stockl et al., 2007). On the other hand, three alternative models of cellular aging (included in experimental group 2, see Table 1) were also analyzed: first, we compared CD28− and CD28+ CD8+ T lymphocytes, based on the assumption that the increased percentage of CD28+ T cells result from high level cell proliferation and hence represent a form of in vivo senescence (Rufer et al., 2003; Lazuardi et al., 2009). Furthermore, we also analyzed differences in the gene expression profiles of mesenchymal stem cells (MSC) obtained from young and old human donors, another system that permits the impact of human donor age on cellular phenotype to be assessed (Fehrer et al., 2007). Finally, we also analyzed gene expression profiles of prostate stromal cells (PrSC), which were driven to transdifferentiate into premature senescent myofibroblasts by TGFβ1, a process considered a hallmark of the aging human prostate, leading to benign prostatic hyperplasia (Untergasser et al., 2005). RNA was prepared from all samples and analyzed on Affymetrix arrays for whole-genome expression profiling. For validation purposes, the expression levels of 22 genes, which were found to be differentially expressed in a high number of individual experiments, were determined by quantitative reverse transcription–PCR (qPCR). Good concordance between data of the microarray and qPCR analyses was obtained. Linear regression yielded an r-value of 0.68 and 362 of 380 individual experimental data pairs sitting within the 95% prediction interval (Fig. S1).
Table 1.
Overview microarray experiments: experimental model systems: HUVEC, human umbilical vein epithelial cells; PFF, primary foreskin fibroblasts; PrSC, primary prostatic stromal fibroblasts; RPTEC, renal proximal tubular epithelial cells; CD8, CD8+ T lymphocytes; MSC, mesenchymal stem cells; reagents: t-BHP (tert-butylhydroperoxide), FCCP (carbonyl cyanide p-(trifluoromethoxy)phenylhydrazone), AMP (adenosine monophosphate); number of array hybridizations/samples; number of array experiments for senescence and oxidative stress; EMBL-EBI Array Express accession number
| Controls |
Experimental group 1 (oxidative stress) |
Experimental group 2 (cellular aging) |
||||
|---|---|---|---|---|---|---|
| Celltype | No. of arrays | Treatment | No. of arrays | Treatment | No. of arrays | Array Express accession number |
| HUVEC | 6 | t-BHP | 2 | RS | 2 | E-MEXP-2283 |
| PFF | 8 | ND | ND | FCCP | 2 | E-MEXP-2285 |
| AMP | 2 | |||||
| Oligomycin | 2 | |||||
| RPTEC | 4 | ND | ND | RS | 1 | E-MEXP-2683 |
| High/low ROS | 1 | |||||
| PrSC | 10 | t-BHP | 2 | TGF-β (trans-differentiation) | 2 | E-MEXP-2167 |
| 20% vs. 3% O2 | 4 | |||||
| CD8 | 15 | t-BHP | 7 | CD28−/CD28+ | 4 | E-MEXP-2345 |
| MSC | 4 | 20% vs. 3% O2 | 2 | Young vs. old donor | 2 | E-MEXP-1506 |
| Total | 47 | 17 | 18 | |||
RS, replicative senescence; ND, not determined.
Identification of transcriptional signatures of oxidative stress and cellular aging
For computational comparison, normalized whole-genome expression profiles from 47 independent experimental samples were established (Table 1), and hierarchical clustering of the gene expression profiles derived from all experimental human cell models was performed (Fig. S2). Differential expression values were calculated and compiled for further analysis by assembling 35 pairs of individual experiments with regard to cellular aging or oxidative stress (Fig. 1; see also Table S1). Considerable resemblance with respect to sets of differentially expressed genes was observed between HUVEC and RPTEC that have undergone replicative senescence, in vivo aged CD8 T cells and MSC, as well as in a compilation of these experimental model systems (Fig. S3).
Fig. 1.
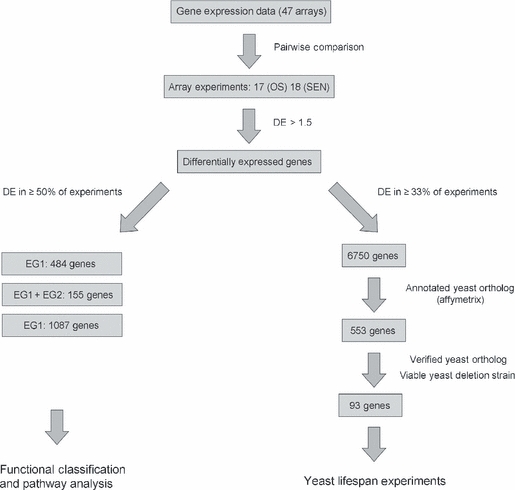
Workflow for candidate gene identification and functional classification. First, gene expression data from 47 microarrays were processed to identify differentially expressed (DE) genes in experimental group 1 (EG1) and experimental group 2 (EG2). The selection of genes for functional classification and pathway analysis, starting from a total of 1566 genes, is shown (left arm). In the right arm of the diagram, the strategy to identify yeast genes for testing in lifespan analysis is depicted.
Several methods have been established to identify genes that are differentially expressed in a homologous set of biological replicates. However, complex datasets such as those described herein, which comprise transcriptional profiles from different cell types derived from differently aged, nonrelated human individuals, often show highly variable transcript levels with regard to the expression of individual genes (Lu et al., 2004; Rodwell et al., 2004; Tan et al., 2005; Lener et al., 2006). This can be either because of variant genetic background or the redundant use of closely related homologous genes. Therefore, we employed a value counting method.
The number a particular gene was found under- or overexpressed was counted, and the list of all genes was sorted according to occurrence. Using this methodology, we identified 586 probe sets representing 484 genes across EG1 with differential expression (DE) higher than ±1.5-fold in more than 50% of individual pair-wise comparisons, and 1423 probe sets representing 1087 genes across EG2, again with the same criteria of DE > ±1.5 in > 50% of experiments, whereas 180 probe sets representing 155 genes were present in both data sets. With respect to a cumulative binomial distribution (CBD), we additionally calculated P-values together with the corresponding q-values (Table S2) yielding a probability of 0.12 for any probe set to be differentially expressed with senescence and a probability of 0.11 for a probe set to be differentially expressed with oxidative stress. The q-values were ≤ 0.0030 in both EG1 and EG2, and P-values were ≤ 0.000028 in EG1 and ≤ 0.000085 in EG2 (Table S2). When evaluating both experimental groups along these lines, we revealed a set of 335 candidate genes with DE > ±1.5 in at least 18 pair-wise comparisons (Table S3).
The highest overall score (with most occurrences of DE) was observed for PAF (PCNA associated factor) (Table 2A) (Yu et al. 2001), a gene recently reported to be involved in DNA repair in HeLa cells (Turchi et al., 2009). Another top scoring gene was SKP2 (Table 2A), encoding a regulatory subunit of an ubiquitin ligase complex, which was recently identified as playing a key role in setting the threshold for stress-induced cellular senescence (Lin et al., 2010). Several additional genes were identified with high scores, such as SLC1A4, a member of the solute carrier family 1 (Table 2A), which so far have not been linked to the senescent phenotype. Of note, four genes involved in the regulation of transcription (BHLHB2, TCF8, RUNX1, and TCEA3) were included in the top 15 genes common to both experimental groups (Table 2A).
Table 2.
(A) Genes that were found differentially expressed with highest occurrence in both experimental groups, (B) group 1 (oxidative stress), (C) group 2 (cellular aging)
| Symbol | Description | Unigene | GenBank | EG1 up | EG1 down | EG2 up | EG2 down | EG1 + EG2DE |
|---|---|---|---|---|---|---|---|---|
| (A) | ||||||||
| KIAA0101 | KIAA0101/PAF | Hs.81892 | NM_014736 | 7 | 7 | 6 | 7 | 27 |
| APOBEC3G | Apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like 3G | Hs.660143 | NM_021822 | 0 | 10 | 8 | 8 | 26 |
| MEST | Mesoderm-specific transcript homolog (mouse) | Hs.270978 | NM_002402 | 4 | 5 | 8 | 8 | 25 |
| RRM2 | Ribonucleotide reductase M2 polypeptide | Hs.226390 | BE966236 | 7 | 5 | 4 | 9 | 25 |
| SYDE2 | Synapse defective 1, Rho GTPase, homolog 2 (C. elegans) | Hs.718601 | N90719 | 7 | 5 | 5 | 8 | 25 |
| TXNIP | Thioredoxin interacting protein | Hs.715525 | AA812232 | 2 | 9 | 7 | 7 | 25 |
| BHLHB2 | Basic helix-loop-helix domain containing, class B, 2 | Hs.719093 | NM_003670 | 5 | 6 | 10 | 3 | 24 |
| BICD1 | Bicaudal D homolog 1 (Drosophila) | Hs.505202 | BC010091 | 10 | 3 | 7 | 4 | 24 |
| CYP51A1 | Cytochrome P450, family 51, subfamily A, polypeptide 1 | Hs.417077 | U40053 | 10 | 1 | 10 | 3 | 24 |
| HMGCS1 | 3-hydroxy-3-methylglutaryl-Coenzyme A synthase 1 (soluble) | Hs.397729 | NM_002130 | 9 | 3 | 9 | 3 | 24 |
| SKP2 | S-phase kinase-associated protein 2 (p45) | Hs.23348 | BC001441 | 5 | 9 | 3 | 7 | 24 |
| SLC1A4 | Solute carrier family 1 (neutral amino acid transporter), member 4 | Hs.654352 | W72527 | 6 | 1 | 11 | 6 | 24 |
| TCF8 | Transcription factor 8 (represses interleukin 2 expression) | Hs.282113 | NM_030751 | 6 | 6 | 6 | 6 | 24 |
| C20orf129 | Chromosome 20 open reading frame 129 | Hs.472716 | BC001068 | 11 | 1 | 7 | 4 | 23 |
| CD302 | CD302 antigen | Hs.130014 | NM_014880 | 7 | 4 | 6 | 6 | 23 |
| IFI44L | Interferon-induced protein 44-like | Hs.389724 | NM_006820 | 3 | 7 | 8 | 5 | 23 |
| PDGFD | Platelet derived growth factor D | Hs.352298 | NM_025208 | 5 | 4 | 6 | 8 | 23 |
| RUNX1 | Runt-related transcription factor 1 (acute myeloid leukemia 1) | Hs.675708 | BU789637 | 2 | 9 | 9 | 3 | 23 |
| TCEA3 | Transcription elongation factor A (SII), 3 | Hs.446354 | AI675780 | 2 | 8 | 4 | 9 | 23 |
| TSPAN2 | Tetraspanin 2 | Hs.310458 | AI743596 | 4 | 6 | 7 | 6 | 23 |
| Symbol | Description | Unigene | GenBank | EG1 up | EG1 down | EG1 DE |
|---|---|---|---|---|---|---|
| (B) | ||||||
| KIAA0101 | KIAA0101/PAF | Hs.81892 | NM_014736 | 7 | 7 | 14 |
| RRM2 | Ribonucleotide reductase M2 polypeptide | Hs.226390 | BC001886 | 8 | 6 | 14 |
| SKP2 | S-phase kinase-associated protein 2 (p45) | Hs.23348 | BC001441 | 5 | 9 | 14 |
| TRPV2 | Transient receptor potential cation channel, subfamily V, member 2 | Hs.279746 | NM_015930 | 4 | 10 | 14 |
| BICD1 | Bicaudal D homolog 1 (Drosophila) | Hs.505202 | BC010091 | 10 | 3 | 13 |
| EIF4A2 | Eukaryotic translation initiation factor 4A, isoform 2 | Hs.518475 | AI332397 | 10 | 3 | 13 |
| NQO1 | NAD(P)H dehydrogenase, quinone 1 | Hs.406515 | NM_000903 | 9 | 4 | 13 |
| C20orf129 | Chromosome 20 open reading frame 129 | Hs.472716 | BC001068 | 11 | 1 | 12 |
| CASP1 | Caspase 1, apoptosis-related cysteine peptidase (interleukin 1, beta, convertase) | Hs.2490 | U13699 | 2 | 10 | 12 |
| CCNB2 | Cyclin B2 | Hs.194698 | NM_004701 | 5 | 7 | 12 |
| CIRBP | Cold-inducible RNA-binding protein | Hs.634522 | AL565767 | 8 | 4 | 12 |
| CKAP2 | Cytoskeleton-associated protein 2 | Hs.444028 | NM_018204 | 11 | 1 | 12 |
| CYBA | Cytochrome b-245, alpha polypeptide | Hs.513803 | NM_000101 | 3 | 9 | 12 |
| DAAM1 | Disheveled-associated activator of morphogenesis 1 | Hs.19156 | AA890373 | 8 | 4 | 12 |
| EGR1 | Early growth response 1 | Hs.326035 | NM_001964 | 6 | 6 | 12 |
| EIF5A | Eukaryotic translation initiation factor 5A | Hs.534314 | NM_001970 | 11 | 1 | 12 |
| ERRFI1 | ERBB receptor feedback inhibitor 1 | Hs.658370 | AW612461 | 7 | 5 | 12 |
| FBXO9 | F-box protein 9 | Hs.216653 | AK095307 | 8 | 4 | 12 |
| HMGCS1 | 3-hydroxy-3-methylglutaryl-Coenzyme A synthase 1 (soluble) | Hs.397729 | NM_002130 | 9 | 3 | 12 |
| IGFBP3 | Insulin-like growth factor binding protein 3 | Hs.450230 | BF340228 | 4 | 8 | 12 |
| Symbol | Description | Unigene | GenBank | EG2 up | EG2 down | EG2 DE |
|---|---|---|---|---|---|---|
| (C) | ||||||
| SLC1A4 | Solute carrier family 1 (neutral amino acid transporter), member 4 | Hs.654352 | BF340083 | 11 | 7 | 18 |
| ARRB1 | Arrestin, beta 1 | Hs.503284 | BE207758 | 8 | 9 | 17 |
| APOBEC3G | Apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like 3G | Hs.660143 | NM_021822 | 8 | 8 | 16 |
| MEST | Mesoderm-specific transcript homolog (mouse) | Hs.270978 | NM_002402 | 8 | 8 | 16 |
| CTSC | Cathepsin C | Hs.128065 | AI246687 | 10 | 5 | 15 |
| METTL7A | Methyltransferase like 7A | Hs.716437 | NM_014033 | 10 | 5 | 15 |
| GLIPR1 | GLI pathogenesis-related 1 (glioma) | Hs.205558 | AV682252 | 7 | 8 | 15 |
| RAB27B | RAB27B, member RAS oncogene family | Hs.25318 | BF438386 | 11 | 4 | 15 |
| TRIB3 | Tribbles homolog 3 (Drosophila) | Hs.516826 | NM_021158 | 10 | 5 | 15 |
| ABL2 | v-abl Abelson murine leukemia viral oncogene homolog 2 | Hs.159472 | AK025877 | 9 | 5 | 14 |
| EIF4EBP1 | Eukaryotic translation initiation factor 4E binding protein 1 | Hs.411641 | AB044548 | 8 | 6 | 14 |
| ETV1 | ets variant gene 1 | Hs.22634 | BE881590 | 9 | 5 | 14 |
| IFIT3 | Interferon-induced protein with tetratricopeptide repeats 3 | Hs.714337 | AI075407 | 7 | 7 | 14 |
| KLF9 | Kruppel-like factor 9 | Hs.150557 | AI690205 | 3 | 11 | 14 |
| MCC | Mutated in colorectal cancers | Hs.593171 | BE967311 | 3 | 11 | 14 |
| PDGFD | Platelet derived growth factor D | Hs.352298 | NM_025208 | 6 | 8 | 14 |
| PHGDH | Phosphoglycerate dehydrogenase | Hs.487296 | NM_006623 | 4 | 10 | 14 |
| PHLDA1 | Pleckstrin homology-like domain, family A, member 1 | Hs.602085 | AK026181 | 10 | 4 | 14 |
| PTGS2 | Prostaglandin-endoperoxide synthase 2 | Hs.196384 | NM_000963 | 8 | 6 | 14 |
| RAB1A | RAB1A, member RAS oncogene family | Hs.709257 | BG530481 | 4 | 10 | 14 |
When both experimental groups were treated separately, the highest scoring genes (with most occurrences of DE) for experimental group 1 (oxidative stress) were genes involved in regulating cell proliferation, such as RRM2, cyclin B2, EGR1, and IGFBP-3 (Table 2B), consistent with the induction of rapid growth arrest by oxidative stress (Chen & Ames, 1994). In addition, several genes were identified for which a role in stress-induced senescence was not known before, such as the translation initiation factors EIF4A and EIF5A (Table 2B). The highest scoring genes (with most occurrences of DE) for experimental group 2 (cellular aging) are shown in Table 2C. Surprisingly, the top 15 genes in this group were distinct from the top scorers obtained in experimental group 1. Moreover, most of the genes identified in this approach were not previously associated with cellular senescence. Hence, this list of genes may contain novel information about potential unidentified regulators of cellular aging, probably including genes that are relevant for cellular aging in vivo. The regulation of selected genes was also addressed at the protein level. On the one hand, selected genes were analyzed across all cellular model systems. For example, protein levels were determined for the conserved candidate gene EZH2 (DE = 19; Table S2), coding for a histone methyltransferase that plays a major role in epigenetic regulation of senescence (Bracken et al., 2007.). Consistent with data obtained by RNA profiling, EZH2 protein level was considerably reduced in senescent RPTEC and HUVEC, as well as in PFF after treatment with adenosine monophosphate (AMP) and carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP) but not oligomycin (Fig. 2A). EZH2 levels were not significantly regulated in MSC, PrSC, and CD8 T lymphocytes (Fig. 2B, C) consistent with the results of RNA profiling. Additional protein analyses were performed for relevant proteins selectively in particular model systems. Thus, we confirmed by Western blot upregulation of IGFBP-3 in trans-differentiated PrSC and MSC grown in a hyperoxic atmosphere (Fig. 2B). In additional experiments, we confirmed the upregulation of IGFBP-3 and IGFBP-6 in cellular supernatants by ELISA and the downregulation of IGF1R in CD8+CD28− T lymphocytes by antibody assisted flow cytometry (data not shown).
Fig. 2.
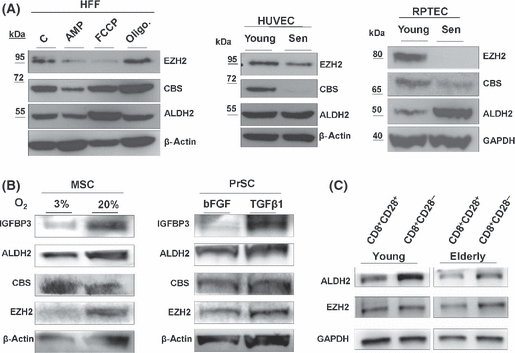
Western blot analysis. Cell lysates were prepared from various model systems employed in this study: young and senescent human umbilical vein endothelial cells and renal proximal tubular epithelial cells, respectively; PFF treated with FCCP, AMP, and oligomycin for three days (A); mesenchymal stem cells cultured at atmospheric conditions of 3% or 20% oxygen; prostate stromal cells treated with either basic fibroblast growth factor (bFGF) or transforming growth factor beta 1 (TGFβ1) (B); CD8 T cells either isolated from a young or an elderly donor and sorted with respect to CD28 (C); Proteins were analyzed by immunoblotting using antibodies to EZH2, ALDH2, cystathionine beta-synthase, and IGFBP3, as indicated. For loading control, antibodies to β-actin and GAPDH were used as indicated.
Pathway analysis
To analyze the large amount of gene expression data in a user-friendly way and for optimal data mining, a database was constructed referred to as the GiSAO database (‘Genes Involved in Senescence, Apoptosis and Oxidative stress’ (Lepperdinger et al., 2008); for details, see ‘Experimental procedures’). Currently, more than a hundred whole-genome array data sets fulfilling MIAME criteria from cellular aging model systems were deposited by this consortium in the GiSAO database (https://gisao.genome.tugraz.at/gisao_web/), also including the datasets of miRNA expression published recently (Hackl et al., 2010). To identify principal biological processes associated with age-related transcriptional changes, we first evaluated top-ranked genes via Pathway Explorer (Mlecnik et al., 2005), a web-accessible tool that allows life scientists to infer the biological meaning behind large gene lists. Pathway analysis for genes that are DE in both experimental groups highlighted the p53 signaling pathway, apoptosis, and cell cycle regulation as the most relevant pathways. The general importance of pathways controlling cell proliferation and cell death for cellular senescence is well established (Mallette & Ferbeyre, 2007). Thus, these observations validate our experimental approach. The analysis identified additional pathways that were significantly overrepresented (P < 0.001) in the group of differentially regulated genes, which have not previously been linked to cellular senescence, such as cholesterol biosynthesis, IL-18 signaling, and nitrogen metabolism (Table 3).
Table 3.
Pathway analysis: functional classification of genes that are differentially expressed in experimental groups 1, 2 or both, using Pathway Explorer (Mlecnik et al., 2005; available online: https://pathwayexplorer.genome.tugraz.at/), gene number attributed to pathway, P-values were calculated from complete expression value dataset (54 675 probe sets) with Fisher’s exact test, where gene numbers are given, all P-values are < 0.05
| Pathway | Gene number EG1 | Gene number EG2 | Gene number EG1 + EG2 |
|---|---|---|---|
| Apoptosis | 17 | 39 | 7 |
| Cell cycle | 15 | 27 | 7 |
| p53 signaling pathway | 7 | 32 | 7 |
| Cell proliferation | * | 32 | 7 |
| Chromosome | 14 | * | 5 |
| Lipid biosynthesis | 13 | * | 4 |
| Cytokinesis | 10 | * | 5 |
| Pyrimidine metabolism | 9 | * | 3 |
| G1 to S cell cycle control | 8 | * | 3 |
| mRNA metabolism | 10 | * | * |
| ATP-dependenthelicase activity | 9 | * | * |
| Fatty acid metabolism | 9 | * | * |
| mRNA processing | 9 | * | * |
| D4-GDI signaling pathway | 7 | * | * |
| DNA replication | 7 | * | * |
| MAPK signaling pathway | * | 36 | * |
| Extracellular matrix | * | 33 | * |
| Focal adhesion | * | 29 | * |
| Enzyme inhibitor activity | * | 28 | * |
| Cytokine–cytokinereceptor interaction | * | 27 | * |
| Cell growth | * | 24 | * |
| Cell surface receptor linked signal transduction | * | 24 | * |
| Wnt signaling pathway | * | 24 | * |
| Cholesterol biosynthesis | * | * | 3 |
| Fructose and mannose metabolism | * | * | 3 |
| IL 18 signaling pathway | * | * | 2 |
| Nitrogen metabolism | * | * | 3 |
| Synthesis and degradation of ketone bodies | * | * | 2 |
No genes were found for particular pathway or P > 0.05.
Pathway data source: KEGG pathway database (http://www.genome.jp/kegg/pathway.html), GenMapp (http://www.genmapp.org/), Biocarta (http://www.biocarta.com/genes).
Pathway analysis was also separately applied to experimental groups 1 and 2. For genes differentially expressed in experimental group 1 (oxidative stress), the highest ranked clusters were related to the regulation of apoptosis and cell cycle, in particular the G1/S transition. Additionally, chromosome maintenance, lipid biosynthesis, D4-GDI signaling, and DNA replication scored highly significant (P < 0.001). Among the genes differentially expressed in experimental group 2, top clusters related to apoptosis, MAPK signaling, and extracellular matrix. In addition, highly significant scores (P < 0.001) were observed for p53 signaling, enzyme inhibitor activity, cell cycle regulation, cell surface receptor linked signal transduction, and cell growth. In this group, we also noted a high representation of the Wnt signaling pathway and cytokine–cytokine receptor interactions, two pathways previously linked with cellular senescence (Ye et al., 2007; Acosta et al. 2008; Coppe et al., 2010b); however, in both cases, statistical significance was rather weak.
Functional validation of candidate genes by lifespan analysis in yeast mutants
Next, potential homologues of the human candidate genes were identified in the genome of budding yeast, Saccharomyces cerevisiae. To this end, an extended list of human genes was compiled with differential expression (DE > 1.5) in at least 6 of 17 experiments in experimental group 1 or at least 6 of 18 experiments in experimental group 2 (Fig. 1). Both upregulated and downregulated genes were considered. Starting with a total of 6750 human genes, 553 yeast orthologs were identified, of which only the nonessential genes were considered for further analysis. For the top ranking human genes, 93 nonessential yeast orthologs were identified (Table S4). Subsequently, functional tests regarding long-term proliferation were performed, and the viability of the respective mutant yeast strain was validated. The deletion mutants of the corresponding S. cerevisiae homologues were obtained from the EUROSCARF knockout strain collection. A panel of yeast mutant cells in stationary phase was analyzed by chronological lifespan experiments (MacLean et al., 2001; Piper, 2006) involving 22-day culturing in stationary phase as described (Fabrizio et al., 2004; Herker et al., 2004). As control, wild-type strains were also analyzed, which have a mean lifespan of about 11 days under these conditions (Fabrizio et al., 2004; Herker et al., 2004). Twenty-two days after the start of the experiment, the wild-type strain completely ceased growth and was all found dead. The 93 selected yeast mutant strains displayed significantly different survival rates (Table 4), ranging from drastic lifespan shortening to lifespan extension relative to wild-type cells. For a more detailed analysis, the lifespan data were repeated for the nine most short-lived strains containing mutations in ATG18, GCM5, KGD1, LYS69, MSW1, NCR1, TIM1, RAD27, and SHM1. Lifespan shortening relative to the wild-type was highly reproducible (Fig. 3A), with the disruption of KGD1, MSW1, and TIM1 having the greatest effect on lifespan, suggesting that these genes play important roles for survival in stationary culture. Similarly, yeast mutants with extended lifespan were also identified (Table 4), and a significant extension of lifespan was obtained for the mutants ΔCYS4, ΔALD4, and ΔPDX3 (Fig. 3B). Whereas yeast strains mutated in UBC12 and PAS3 showed a trend for extended lifespan, this did not reach statistical significance. The CYS4 gene encodes a cystathionine beta-synthase involved in the first step of cysteine biosynthesis while Ald4p is the major aldehyde dehydrogenase isoform. PDX3, encoding pyridoxine (pyridoxamine) 5′-phosphate oxidase, is involved in the salvage pathway of pyridoxal 5′-phosphate. As a first step to address the relevance of the human orthologs in our cellular senescence models, expression and regulation of ALDH2 and cystathionine beta-synthase (CBS) was analyzed by Western blot. ALDH2 protein level was upregulated in senescent RPTEC and in FCCP-treated PFF (Fig. 2), but remained unchanged in the other experimental models (Fig. 2 and data not shown). Cystathionine beta-synthase protein level was downregulated with senescence in both HUVEC and RPTEC, as well as in AMP-treated PFF (Fig. 2), consistent with the data obtained by RNA profiling. In contrast, CBS was not significantly regulated in the other cellular model systems (data not shown). Together, the data suggest that the levels of ALDH2 and CBS gene products are indeed regulated by the various treatments, in most cases reflecting the differential expression values as elucidated by our initial genomic screening method. The pattern of regulation is complex in both cases, and more work is required to understand the exact contribution of these genes to cellular senescence.
Table 4.
Effects on chronological aging observed in 93 single deletion strains: deletion strains were assigned to five categories, depending on the effects on survival during aging when compared to the wild-type
| Survival during chronological aging (compared to WT) | Single deletion of |
|---|---|
| Strongly reduced (< 50% of wt) | KGD1, SOD2, RPL31A, NCR1, LYS9, SHM1, GCN5, HAP3, PIM1, PRY1, ATG18, CYC1, RPL13B, MSW1, RAD27, YDC1, TIF3 |
| Slightly reduced(> 50% and < 85%) | CLB1, CLB2, APM1, SYM1, IDP2, GSY2, ALT2, SIZ1, TIF1, TIF2, NPL3, PTC5, RNR3, ERG24, MRT4, MSH6, NUP170, HAT1, INP53, SSO1, TWF1, SGT2, LHS1, RPL37A, ARF1, DHP5, CHL1, TRX1, UBC8, CHD1, SPE2, HRT3, MRE11, MSH2, DBP1, SSN3, SSF1 |
| Not affected(same as wt ±15%) | HTA2, AIP1, HXK1, SER1, YPK2, UBC11, ENO1, RPL22B, RPL43B, PEX13, REV3, SYG1, MSH5, ERP6, TPK1, PER1, URA2, SLH1, CHK1, ALD5, CAF40 |
| Slightly increased (> 115% and < 150%) | MAD2, PCH2, YTA7, PHO89, UGA1,DIE2, CAF17, HMT1, ALG5, SSA3, LSB6 |
| Strongly increased (> 150%) | CYS4, DNF1, ALD4, PDX3, RNH201, UBC12, PRS3 |
Fig. 3.
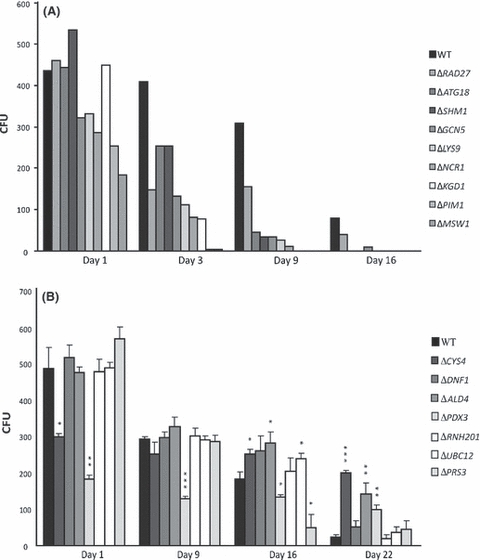
Functional analysis of selected candidate genes in yeast. Lifespan data for the nine yeast mutants with shortest lifespan (A) and for the seven mutants with lifespan extension (B) are shown. (*P < 0.05; **P < 0.01; ***P < 0.001).
Discussion
The here presented analysis has identified gene expression signatures of cellular aging, which are conserved between different human tissues. The experimental design was based on the assumptions that (i) genes that are differentially regulated in human cellular aging and senescence are trustworthy candidates for modulators of aging and that (ii) the ability of a certain gene to restrict lifespan in yeast suggests that this gene has a role in modulating the rate of aging that potentially extends beyond yeast. This analysis revealed several genes and molecular pathways already known to be involved in aging, thereby validating the screen. In addition, a reasonable number of novel genes and a few novel pathways were identified that have not been linked to aging before. Based on these findings, new experimental approaches to study human aging will become possible.
Model systems for cellular aging
According to current hypotheses, aging at the cellular level contributes significantly to organismic aging, and for a better understanding of human aging, cellular model systems are indispensible (Campisi, 2005; Toussaint et al., 2002a, 2002b). Several model systems for cellular aging have been developed, and a representative selection of these models was applied for the present study: replicative senescence, a process of cellular aging in vitro that is primarily driven by telomere erosion, is a classic experimental system considered relevant for the aging of tissues with high regenerative potential (von Figura et al., 2009). In addition to replicative senescence, most if not all primary human cells can be driven into premature senescence by various stressors, in a process which does not necessarily involve telomere shortening. It is assumed that SIPS is relevant for aging of both mitotic and postmitotic tissues (Toussaint et al., 2002a, 2002b). The importance of SIPS for in vivo aging is supported by the fact that SIPS can be induced in vitro by factors that are well-known risk factors for age-associated degeneration of the corresponding tissue in vivo. For example, vascular endothelial cells undergo SIPS in response to oxidative stress, high glucose, shear stress, and incubation with oxidized LDL, all of which are known risk factors for age-associated vascular dysfunction and disease (Erusalimsky & Kurz, 2006; Goligorsky et al., 2009). Three additional model systems of cellular aging, with a particular focus on aging in vivo, were included in the analysis: first, we compared CD28− and CD28+ CD8+ T lymphocytes, described as one of the rare models for in vivo senescence (Rufer et al., 2003; Lazuardi et al., 2009); second, we compared MSC freshly obtained from young and old human donors (Fehrer et al., 2007); finally, PrSC were included, which can be transdifferentiated into premature senescent myofibroblasts, a process considered a hallmark of the aging human prostate (Untergasser et al., 2005). These cellular model systems were developed by the participating laboratories and are not easily available elsewhere. Information on genetic regulators of cellular aging obtained from these systems is considered particularly valuable, given the scarcity of knowledge on molecular genetic processes involved in human aging.
The GiSAO database
Other resources have been created to systematically compare the influence of the genome on aging processes. Of note, the Human Ageing Genomic Resources (HAGR) (de Magalhaes et al., 2005) represent a systematic collection of genomic data relevant for aging processes. It consists of the database GenAge featuring genes associated with aging and longevity in short-lived model organisms and humans, and AnAge, a database of aging in animals, featuring information on aging processes in over 4000 animal species (de Magalhaes et al., 2009a, 2009b). The GiSAO database established in the current communication focuses on genetic regulation of aging at the cellular level, based on human gene expression data, and allows the identification of evolutionarily conserved regulators of cellular aging. In our view, information contained in the GiSAO database conveniently complements the data on organismic aging available in the HAGR. We have compared data sets in both studies. Indeed, several genes with a high score in the meta-analysis described earlier (de Magalhaes et al., 2009a, 2009b), including TXNIP, S100A4, and MT1F, featured prominently in our analysis described here (Table S3). In turn, 15 genes taken from Table S3, including IGFBP-3, PTGS2, EGR1, FOS, ATM, DDIT3 (DE ≥ 20) are also highlighted as potential regulators of aging in the HAGR database. Hence, future attempts to link both resources are warranted.
Confirmation of known association of genes/pathways with aging
Several of the pathways highlighted in the current analysis have been associated with aging in cellular senescence and/or animal models before. For example, apoptosis regulation (Campisi, 2003), MAP kinase signaling (Maruyama et al., 2009), p53 signaling (Zuckerman et al., 2009), control of cell proliferation and cell cycle (Chen, 2000), cytokine–cytokine receptor interaction (Coppe et al., 2010a), and Wnt signaling (Ye et al., 2007) are thought to contribute to cellular senescence and aging. In addition, several of the genes identified in the unbiased screen described here were found to play important roles as regulators of senescence. Thus, Skp2 (Lin et al., 2010) and EZH2 (Bracken et al., 2007) are known to contribute to the senescence response in various cell types. Work in the consortium established the genes coding for IGFBP-3 (Kim et al., 2007; Muck et al., 2008), glutaminase (Unterluggauer et al., 2008), and IGFBP-6 (Micutkova et al., submitted) as functional regulators of cellular senescence. The prominent appearance of these pathways and genes in our screening results serves as positive control and indicates that the screening procedure was adequate.
Identification of novel genes/pathways related to aging
Some interesting human genes, e.g. genes related to cholesterol biosynthesis, came up in our screen, which have not been related to aging before. Similarly, genes related to the regulation of focal adhesion, extracellular matrix, mRNA metabolism, fatty acid metabolism, and lipid biosynthesis were not known to be associated with aging processes. Although many of these genes could not be analyzed in yeast because there are no obvious orthologs in the yeast genome, these observations will stimulate future research on the role of the respective genes/pathways both in short-lived model organisms of aging and in human cellular aging.
Using extension of the chronological lifespan in yeast, which depends far less on cell-to-cell communication compared to higher organisms, as an additional screening tool, we expected to identify primarily basic cell autonomous mechanisms of aging. In this category, three genes particularly stood out, as follows: (i) ALD4, the major (mitochondrial) aldehyde dehydrogenase isoform, which is involved in the degradation of various amino acids and fatty acids in yeast, whereas the human homolog ALDH2 acts in the major oxidative pathway of alcohol metabolism; (ii) CYS4, cystathionine beta-synthase, that carries out the first committed step in cysteine biosynthesis, whereas the human homolog CBS catalyzes the conversion of homocysteine to cystathionine, the first step in the trans-sulfuration pathway; (iii) Pyridoxine (pyridoxamine) phosphate oxidase (PDX3), known to play a role in the synthesis of pyridoxal 5′-phosphate in yeast, similar to the human homolog pyridoxamine 5′-phosphate oxidase (PNPO), which catalyzes the terminal, rate-limiting step in the synthesis of pyridoxal 5′-phosphate, also known as vitamin B6. Interestingly, CBS requires vitamin B6 as a cofactor for the conversion of homocysteine to cystathionine in human cells, suggesting that CBS and PNPO influence aging via the same pathway. Importantly, none of these pathways have been linked to aging before, and future studies are required to address the role of these proteins in multi-cellular aging model systems including humans. As a first step to validate their relevance in human cellular senescence, expression levels were analyzed in cellular extracts where antibodies were available. We found differential regulation of ALDH2 and CBS protein levels in several of our model systems (Fig. 2). Accordingly, further studies are warranted to determine their role in cellular senescence.
Experimental procedures
Cell isolation, cultivation, and characterization
Human diploid fibroblasts from foreskin were seeded in 10-cm dishes at a density of 200,000 cells per dish and treated in parallel with the following compounds: untreated, vehicle treated (ethanol), oligomycin (8 μmol L−1 final concentration), FCCP (3 μmol L−1 final concentration), and AMP (3 μmol L−1 final concentration). Every day, medium was replaced and inhibitory compounds freshly added. This treatment leads to premature senescence within 14 days (data not shown, see also (Stockl et al., 2006, 2007; Zwerschke et al., 2003). RNA was prepared after 72 h of incubation and analyzed by RNA profiling as described earlier.
Human umbilical vein endothelial cells were isolated from human umbilical veins and cultured in Endothelial Cell Basal Medium (Lonza, Basel, Switzerland) supplemented with endothelial cell growth medium (EGM) Single Quots (Lonza), containing hEGF 0.5 mL, hydrocortisone 0.5 mL, GA-1000 0.5 mL, BBE 2.0 mL, and FBS 10.0 mL. The cells were subcultured by trypsination with trypsin-EDTA (Gibco Life Technologies, Vienna, Austria), seeded on cell culture dishes coated with 0.2% gelatine and grown at 37°C at ambient atmosphere containing 5% CO2. Cells were passaged at a ratio of 1:5 in regular intervals. At later passages, the splitting ratio was reduced to 1:3 and 1:2, respectively. Cells were passaged before reaching 70–80% confluence. Population doublings (PDL) were estimated using the following equation: n= (log10 F − log10 I)/0.301 (where n is the population doublings, F, number of cells at the end of one passage, and I, number of cells that were seeded at the beginning of one passage). After 50 PDL, the cells reached growth arrest, and the senescent phenotype was verified by staining for senescence-associated beta galactosidase, which was positive for ≥ 95% of cells.
Human primary PrSC were established and trans-differentiation induced by TGF-beta1 treatment, as described (Untergasser et al., 2005). Briefly, PrSC were derived from prostate cancer patients who have not received hormonal therapy (n = 3, age 65–72) after obtaining written informed consent. After radical prostatectomy and inspection by the pathologist, two tissue wedges showing no histological signs of malignancy were removed from the transition zone. These explants were minced into organoids of ∼ 1 mm3 and seeded on uncoated plastic material in stromal cell growth medium containing insulin, human basic fibroblast growth factor, 5% fetal calf serum, and gentamycin and amphotericin-B as antibiotics (SCGM; Lonza). Explants were maintained at 37°C in a humidified atmosphere of 5% CO2. These conditions produce a homogeneous fibroblast cell population after 7 days of culture. When cells reached 70% confluence, they were split at a 1:3 ratio using trypsin-EDTA to expand the population. In all experiments, cells of passage 2–4 were used directly from culture (not previously frozen).
Renal proximal tubular epithelial cells were cultivated as recently reported (Wieser et al., 2008). In brief, within 24 h after surgery, tissue from the renal cortex was fragmented and incubated at 37°C for 15–20 min in DMEM/Ham’s F12 (1:1) (Biochrom KG, Berlin, Germany) containing 1 mg mL−1 collagenase type IV (PAN-BioTech GmbH, Aidenbach, Germany) and 1 mg mL−1 trypsin-inhibitor (Sigma, Vienna, Austria). After being passed through a 105-μm nylon mesh, the filtrate was centrifuged, washed twice with phosphate-buffered saline (PBS), resuspended in medium, and dispensed into roux-flasks (Nunc, Wiesbaden, Germany). 24 hours thereafter medium was changed. The initial passage of confluent cells after 3–5 days was considered as PDL zero. Cells were passaged (1:2 to 1:4) at confluence, using 0.25% trypsin/0.02% EDTA, which was inactivated with 1 mg mL−1 trypsin-inhibitor. Cumulative PDL was calculated as a function of passage number and split ratio (4). Medium consisted of DMEM/Ham’s F12 (1:1) supplemented with 4 mm l-glutamine, 10 mm HEPES buffer, 5 pm tri-iodothyronine, 10 ng mL−1 recombinant human EGF, 3.5 μg mL−1 ascorbic acid, 5 μg mL−1 transferrin, 5 μg mL−1 insulin, 25 ng mL−1 prostaglandin E1, 25 ng mL−1 hydrocortisone, and 8.65 ng mL−1 sodium selenite (all from Sigma). After 24 PDL, the cells reached growth arrest, and the senescent phenotype was verified by staining for senescence-associated beta galactosidase, which was positive for ≥ 95% of cells. Cells at intermediate passage could be divided into two groups concerning their redox status, as monitored by dichlorofluorescein diacetate (DCFDA) staining. To address the significance of this distinction, RPTECs were sorted into DCFDAbright and DCFDAdim subpopulations, which were analyzed separately.
Mesenchymal stem cells were isolated from the iliac crest of systemically healthy individuals (young donor, age 5; elderly donor, age 56), which had been harvested for reconstructive bone surgery of defects within other areas of the body as described previously (Fehrer et al., 2007). Briefly, a small biopsy of substantia spongiosa osseum, which otherwise would have been discarded based on necessary bone for molding and re-contouring prior to insertion into the recipient site, was taken to further investigation under an Institutional Review Board-approved protocol after having obtained the parents’ and the respective patient’s written consent. After surgery, the bone was transferred into minimal essential medium (MEM) supplemented with 20% heat-inactivated fetal calf serum, 100 units mL−1 penicillin, and 100 μg mL−1 streptomycin (growth medium) for transportation from the operation theater to the clean room at room temperature. The biopsies were fragmented, and marrow cells were isolated from pieces (20–100 mm3) by centrifugation (400 g, 1 min). After centrifugation, the remaining pieces were treated with collagenase (2.5 mg mL−1 in MEM) for 2–3 h at 37°C, 20% O2, and 5% CO2. Thereafter, the specimen was again centrifuged (400 g, 1 min). Cells were resuspended and loaded on a Ficoll-Paque Plus® gradient and centrifuged at 2500 g for 30 min. Cells were harvested from the interphase (density < 1.075 g mL−1), washed, and collected by centrifugation (1500 g, 15 min). Cells were cultured at a density of 0.2–0.5 × 106 cells cm−2 at either 20% or 3% O2 in combination with 5% CO2 and 37°C (HeraCell240 – Heraeus, Thermo Scientific, Vienna, Austria; Thermo Electron Forma Series II, 3110). After 24 h, the nonadherent cell fraction was removed by washing twice with PBS. After the primary culture had reached approximately 30–50% confluence, cells were washed twice with PBS and subsequently treated with 0.05% trypsin/1 mm EDTA for 3–5 min at 37°C. Cells were harvested, washed in MEM, and further expanded at a density of 50 cells cm−2.
Isolation of CD8+CD28+ and CD8+CD28− T cells from peripheral blood of apparently healthy young (< 35 year, n = 6, mean age 29, range 26–35) and elderly (> 65 year, n= 10, mean age 72, range 66–87) donors was performed by preparing peripheral blood mononuclear cells (PBMCs) by Ficoll-Paque PLUS (Amersham Biosciences) density gradient centrifugation as approved by the Ethics committee of Innsbruck Medical University. CD8+ T cells were negatively selected from the obtained PBMC fraction by applying the magnetic separation protocol CD8+ T cell isolation kit II (depleting CD4, CD14, CD16, CD19, CD36, CD56, CD123, TCRγ/δ, and CD235a; Miltenyi Biotec, Bergisch Gladbach, Germany) according to the manufacturer’s instructions. Subsequently, purified CD8+ T cells were stained with an allophycocyanin (APC)-conjugated αCD28 monoclonal antibody (mAb) and split into CD8+CD28+ and CD8+CD28− T-cell populations using αAPC MicroBeads (Miltenyi Biotec) by passing the cell suspension through a positive selection column (LS; Miltenyi Biotec) mounted in a magnetic field. The CD8+CD28− T-cell fraction was then re-incubated with αAPC MicroBeads and run over a fresh LS-column to increase purity. For phenotypic analysis, purified T-cell fractions were labelled with a combination of mAbs (αTCRαβ-FITC, αCD16-PE, αCD4-PerCP, αCD8-PE-Cy7, αCD28-APC, and αCD3-APC-Cy7; all BD Biosciences, Heidelberg, Germany) and analyzed on a FACSCanto II (BD Biosciences) revealing that the described isolation protocol yields population homogeneities of > 95%.
Applying oxidative stress by tBHP treatment
Human umbilical vein endothelial cells, PrSC, RPTEC, and CD8+ T lymphocytes were treated by exposure to sublethal doses of tBHP (Unterluggauer et al., 2003). Induction of senescence by repeated exposure to sublethal levels of tBHP takes 10–14 days (Unterluggauer et al., 2003, and data not shown). To identify early transcriptional events involved in the senescence response, cells were analyzed 3 days after starting the treatment.
RNA Isolation, whole-genome array analysis of mRNA expression, and quantitative PCR
RNA was isolated using either Tri Reagent (Sigma-Aldrich) or performing homogenization in 4.2 m guanidinium thiocyanate before phenol extraction and ethanol precipitation (Chomczynski & Sacchi, 1987), followed by LiCl precipitation at a final concentration of 4.5 m. Whole-genome expression analysis was carried out on GeneChip HG-U133 Plus 2.0 Arrays (Affymetrix) by MFT Services, Tuebingen, Germany.
Bioinformatics, clustering, and visualization of array data
Array raw data were normalized via CARMAweb (Rainer et al., 2006) using the gcrma algorithm (Wu et al., 2004). Hierarchical clustering of samples and genes (Euclidian distance, average linkage) was performed after filtering out genes with low variance on a subset of 20 000 genes with the MeV program package (Saeed et al., 2006) available online: http://www.tm4.org/mev/).
To evaluate the probability of observing an elevated number of under- or overexpressed gene occurrences, the distinct number of occurrences in which the gene is under- or overexpressed in a respective group of experiments was determined. The resulting gene list was ranked according to occurrences with respect to either age/senescence-related model systems or oxidative stress–derived data sets as well as a gene list of highly under- or overrepresented candidates present in both cases.
Statistical analysis to identify significant probe sets was performed as described by de Magalhaes et al. (2009a,b);: For each probe set, a P-value was calculated with the cumulative binomial distribution (CBD):
providing the probability P for a probe set to be as often or more often differentially expressed than the times k, it was actually differentially expressed in n experiments. The threshold for differential expression of a probe set was defined as a fold change between samples ≥ ±1.5. Applying statistical analysis based on CBD, the probability P that any probe set is differentially expressed with senescence or oxidative stress was defined as the average of differentially expressed probe sets in the experimental group senescence or oxidative stress divided by the number of all probe sets per whole-genome analysis.
The q-value was calculated for each probe set using Storey’s false discovery rate (FDR) approach with the bootstrapping method (Storey et al., 2004). The robust parameter was used to make the q-values more accurate for small P-values (Storey, 2002). Statistical computation was carried out using the statistical framework R (R-Team, 2007) version 2.5.1. Bioconductor q-value package version 1.10.0 (Storey & Tibshirani, 2003) was used to calculate the FDR.
Differentially expressed genes were grouped into protein families associated with characterized pathways applying Pathway Explorer ((Mlecnik et al., 2005), available online: https://pathwayexplorer.genome.tugraz.at/), P-values were calculated from the complete expression value dataset (54 675 probe sets) with Fisher’s exact test, pathway data sources: KEGG pathway database (http://www.genome.jp/kegg/pathway.html), GenMapp (http://www.genmapp.org/), Biocarta (http://www.biocarta.com/genes/).
Database
GiSAO.db (https://gisao.genome.tugraz.at) is a web-based database system for storing and retrieving data of genes involved in senescence, apoptosis, and oxidative stress. The application is based on a three-tier architecture consisting of a web interface, business logic, and a database. The web interface enables data input and presentation. It was implemented by using Struts framework (http://struts.apache.org/) with Java Server Pages (http://java.sun.com/products/jsp/). The business logic, which is responsible for data processing is an Enterprise JavaBeans 3 (http://java.sun.com/products/ejb/) application deployed on JBoss (http://www.jboss.org/jbossas/) application server. The data are stored in an Oracle database, a relational database management system.
The GiSAO database contains normalized gene expression values obtained from experiments evaluated with the aid of Affymetrix arrays. Gene expression values of each experiment can be displayed and compared with the gene expression values of two or more microarray experiments. Additionally, experimental data of follow-up experiments regarding candidate genes, such as qPCR or Western Blot analysis, are entered into the GiSAO.db. Furthermore, GiSAO.db contains two types of orthologue data: orthologue data provided and updated by Affymetrix from cross reference tables linking Saccharomyces cerevisiae, Caenorhabditis elegans, Drosophila melanogaster, Mus musculus and Homo sapiens facilitate comparative genomic analyses, as well as orthology data computed and entered manually. Besides mRNA expression profiles, also data from proteome analysis and further validation with respect to functional analyses together with information from public resources are available for distinct candidate genes. Moreover, external links lead to orthologs of HomoloGene (http://www.ncbi.nlm.nih.gov/homologene) and InParanoid (http://inparanoid.sbc.su.se/cgi-bin/index.cgi). The gene IDs are linked to their respective database, such as Entrez Gene or RefSeq. GiSAO.db also provides gene annotation (Gene Symbol, Gene Name, etc.) and GO terms (http://www.geneontology.org/). Finally, KEGG pathways (http://www.genome.jp/kegg/) can be displayed and data can be exported in various formats.
Identification of human orthologues in Sacharomyces cerevisiae
Either previously assigned cross references distinctly referring to homologous gene identifiers from other species provided in the array annotation tables accompanying the Affymetrix gene chip or data from reciprocal-best-fit-protein-homology searches (Moreno-Hagelsieb & Latimer, 2008) were employed to define the closest homologous gene pair between man and yeast. Only orthologous genes, which were found nonessential after genomic deletion in yeast laboratory strains (http://mips.helmholtz-muenchen.de/proj/eurofan/eurofan_1/b0/search/simpleSearch.html) were selected for further functional analyses.
Lifespan analysis of gene disruption mutants of Sacharomyces cerevisiae
Experiments were carried out in BY4741 (MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0) as the wild-type strain and respective null mutants, obtained from Euroscarf. Strains were grown at 28°C on SC medium containing 0.17% yeast nitrogen base (Difco), 0.5% (NH4)2SO4 and 30 mg L−1 of all amino acids (except 80 mg L−1 histidine and 200 mg L−1 leucine), 30 mg L−1 adenine, and 320 mg L−1 uracil with 2% glucose. For all experiments, yeast cells were grown at 28°C and 145 rpm. For chronological lifespan experiments, cultures were inoculated at an OD600 of 0.1, and aliquots were taken to perform survival plating at indicated time points.
Statistics
All values were expressed as means ± standard deviation of the mean. Statistical differences of experimental scores were evaluated using Student’s t tests. Differences were considered significant when P was < 0.05.
Acknowledgments
We thank Michael Neuhaus for excellent technical assistance. This work was supported by the Austrian Science Fund (FWF, NFNS93) and the Austrian Ministry of Science and Research (GEN-AU project BIN). N.S. is a recipient of a Lise Meitner Scholarship and D.H. is supported by a European FLARE fellowship.
Author contributions
G.T.L., E.H., C.M., L.M., R.B., A.J., D.T., D.H., S.B., C.Z., N.S. and M.W. performed the cell culture work and contributed RNAs for the microarray analysis; D.R., D.C., J.R., and C.R. performed lifespan analysis in yeast; G.T.L., G.L., E.H., G.G.T., and Z.T. did the bioinformatic analysis and constructed the GiSAO database; G.T.L., M.B., K.F., B.G., P.B., R.G., J.G., Z.T., F.M., G.L., and P.J. conceived the experiments, interpreted the results, and wrote the manuscript.
Supporting Information
Additional supporting information may be found in the online version of this article:
Fig. S1 Validation of microarray analysis: expression values of 22 genes exhibiting differential expression of > 1.5-fold in more than 18 of 37 experimental pairs were assessed by quantitative reverse transcription–PCR.
{kind=link}
Fig. S2 Hierarchical clustering of genome-wide gene expression profiles derived from all experimental human cell models (47 individual datasets).
{kind=link}
Fig. S3 Comparison of numbers of differentially expressed probe sets (DE > 1.5) between individual members of EG2.
{kind=link}
Table S1 Experimental systems: HUVEC (human umbilical vein epithelial cells), PFF (primary foreskin fibroblasts), PrSC (primary prostate stromal cells), RPTEC (renal proximal tubular epithelial cells), CD8 (CD8+ T lymphocytes), MSC (mesenchymal stem cells), Reagents: bFGF: basic fibroblast growth factor, TGFβ: transforming growth factor-β 1, t-BHP (tert-butylhydroperoxide), FCCP (carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone), AMP (adenosine monophosphate), DCFDA (2′,7′-dichlorofluorescein diacetate), number of up- and downregulated probe sets for each array experiment (DE > 1.5, total probe set number: 54 675 for Affymetrix U133 Plus 2.0 array)
Table S2 Genes that are differentially expressed (DE > 1.5) in ≥ 50% of the experiments for experimental groups 1 and 2, numbers for up- and downregulation in EG1 and EG2, P-values (cumulative binomial distribution), q-values (Storeys FDR method)
Table S3 Three hundred and thirty-four genes differentially expressed (DE > 1.5) with the highest combined score for both experimental groups (≥ 18), provided as Excel file
Table S4 Highest scoring genes, which were found differentially expressed (DE > 1.5) in both experimental groups, for which an orthologous nonessential gene in yeast could be identified (according to orthology information provided by Affymetrix Inc
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer-reviewed and may be re-organized for online delivery, but are not copy-edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
References
- Acosta JC, O’Loghlen A, Banito A, Raguz S, Gil J. Control of senescence by CXCR2 and its ligands. Cell cycle. 2008;7:2956–2959. doi: 10.4161/cc.7.19.6780. [DOI] [PubMed] [Google Scholar]
- Bartke A. Minireview: role of the growth hormone/insulin-like growth factor system in mammalian aging. Endocrinology. 2005;146:3718–3723. doi: 10.1210/en.2005-0411. [DOI] [PubMed] [Google Scholar]
- Bracken AP, Kleine-Kohlbrecher D, Dietrich N, Pasini D, Gargiulo G, Beekman C, Theilgaard-Mönch K, Minucci S, Porse BT, Marine JC, Hansen KH, Helin K. The Polycomb group proteins bind throughout the INK4A-ARF locus and are disassociated in senescent cells. Genes Dev. 2007;2007:21. doi: 10.1101/gad.415507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campisi J. Cellular senescence and apoptosis: how cellular responses might influence aging phenotypes. Exp. Gerontol. 2003;38:5–11. doi: 10.1016/s0531-5565(02)00152-3. [DOI] [PubMed] [Google Scholar]
- Campisi J. Senescent cells, tumor suppression, and organismal aging: good citizens, bad neighbors. Cell. 2005;120:513–522. doi: 10.1016/j.cell.2005.02.003. [DOI] [PubMed] [Google Scholar]
- Chen QM. Replicative senescence and oxidant-induced premature senescence. Beyond the control of cell cycle checkpoints. Ann. N Y Acad. Sci. 2000;908:111–125. doi: 10.1111/j.1749-6632.2000.tb06640.x. [DOI] [PubMed] [Google Scholar]
- Chen Q, Ames BN. Senescence-like growth arrest induced by hydrogen peroxide in human diploid fibroblast F65 cells. Proc. Natl. Acad. Sci. U S A. 1994;91:4130–4134. doi: 10.1073/pnas.91.10.4130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal. Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- Coppe JP, Desprez PY, Krtolica A, Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol. 2010a;5:99–118. doi: 10.1146/annurev-pathol-121808-102144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppe JP, Patil CK, Rodier F, Krtolica A, Beausejour CM, Parrinello S, Hodgson JG, Chin K, Desprez PY, Campisi J. A human-like senescence-associated secretory phenotype is conserved in mouse cells dependent on physiological oxygen. PLoS ONE. 2010b;5:e9188. doi: 10.1371/journal.pone.0009188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, Medrano EE, Linskens M, Rubelj I, Pereira-Smith O, Peacocke M, Campisi J. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. U S A. 1995;92:9363–9367. doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumont P, Burton M, Chen QM, Gonos ES, Frippiat C, Mazarati JB, Eliaers F, Remacle J, Toussaint O. Induction of replicative senescence biomarkers by sublethal oxidative stresses in normal human fibroblast. Free Radic. Biol. Med. 2000;28:361–373. doi: 10.1016/s0891-5849(99)00249-x. [DOI] [PubMed] [Google Scholar]
- Erusalimsky JD. Vascular endothelial senescence: from mechanisms to pathophysiology. J. Appl. Physiol. 2009;106:326–332. doi: 10.1152/japplphysiol.91353.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erusalimsky JD, Kurz DJ. Endothelial cell senescence. Handb Exp Pharmacol. 2006;176(Pt 2):213–248. doi: 10.1007/3-540-36028-x_7. [DOI] [PubMed] [Google Scholar]
- Fabrizio P, Battistella L, Vardavas R, Gattazzo C, Liou LL, Diaspro A, Dossen JW, Gralla EB, Longo VD. Superoxide is a mediator of an altruistic aging program in Saccharomyces cerevisiae. J. Cell Biol. 2004;166:1055–1067. doi: 10.1083/jcb.200404002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fehrer C, Brunauer R, Laschober G, Unterluggauer H, Reitinger S, Kloss F, Gully C, Gassner R, Lepperdinger G. Reduced oxygen tension attenuates differentiation capacity of human mesenchymal stem cells and prolongs their lifespan. Aging cell. 2007;6:745–757. doi: 10.1111/j.1474-9726.2007.00336.x. [DOI] [PubMed] [Google Scholar]
- von Figura G, Hartmann D, Song Z, Rudolph KL. Role of telomere dysfunction in aging and its detection by biomarkers. J. Mol. Med. 2009;87:1165–1171. doi: 10.1007/s00109-009-0509-5. [DOI] [PubMed] [Google Scholar]
- Goligorsky MS, Chen J, Patschan S. Stress-induced premature senescence of endothelial cells: a perilous state between recovery and point of no return. Curr. Opin. Hematol. 2009;16:215–219. doi: 10.1097/MOH.0b013e32832a07bd. [DOI] [PubMed] [Google Scholar]
- Hackl M, Brunner S, Fortschegger K, Schreiner C, Micutkova L, Muck C, Laschober GT, Lepperdinger G, Sampson N, Berger P, et al. miR-17, miR-19b, miR-20a, and miR-106a are down-regulated in human aging. Aging cell. 2010;9:291–296. doi: 10.1111/j.1474-9726.2010.00549.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herker E, Jungwirth H, Lehmann KA, Maldener C, Frohlich KU, Wissing S, Buttner S, Fehr M, Sigrist S, Madeo F. Chronological aging leads to apoptosis in yeast. J. Cell Biol. 2004;164:501–507. doi: 10.1083/jcb.200310014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holzenberger M. The role of insulin-like signalling in the regulation of ageing. Horm. Res. 2004;62(Suppl 1):89–92. doi: 10.1159/000080765. [DOI] [PubMed] [Google Scholar]
- Katic M, Kahn CR. The role of insulin and IGF-1 signaling in longevity. Cell. Mol. Life Sci. 2005;62:320–343. doi: 10.1007/s00018-004-4297-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KS, Seu YB, Baek SH, Kim MJ, Kim KJ, Kim JH, Kim JR. Induction of cellular senescence by insulin-like growth factor binding protein-5 through a p53-dependent mechanism. Mol. Biol. Cell. 2007;18:4543–4552. doi: 10.1091/mbc.E07-03-0280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazuardi L, Herndler-Brandstetter D, Brunner S, Laschober GT, Lepperdinger G, Grubeck-Loebenstein B. Microarray analysis reveals similarity between CD8+CD28- T cells from young and elderly persons, but not of CD8+CD28+ T cells. Biogerontology. 2009;10:191–202. doi: 10.1007/s10522-008-9167-1. [DOI] [PubMed] [Google Scholar]
- Lener T, Moll PR, Rinnerthaler M, Bauer J, Aberger F, Richter K. Expression profiling of aging in the human skin. Exp. Gerontol. 2006;41:387–397. doi: 10.1016/j.exger.2006.01.012. [DOI] [PubMed] [Google Scholar]
- Lepperdinger G, Berger P, Breitenbach M, Frohlich KU, Grillari J, Grubeck-Loebenstein B, Madeo F, Minois N, Zwerschke W, Jansen-Durr P. The use of genetically engineered model systems for research on human aging. Front Biosci. 2008;13:7022–7031. doi: 10.2741/3207. [DOI] [PubMed] [Google Scholar]
- Lin HK, Chen Z, Wang G, Nardella C, Lee SW, Chan CH, Yang WL, Wang J, Egia A, Nakayama KI, et al. Skp2 targeting suppresses tumorigenesis by Arf-p53-independent cellular senescence. Nature. 2010;464:374–379. doi: 10.1038/nature08815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu T, Pan Y, Kao SY, Li C, Kohane I, Chan J, Yankner BA. Gene regulation and DNA damage in the ageing human brain. Nature. 2004;429:883–891. doi: 10.1038/nature02661. [DOI] [PubMed] [Google Scholar]
- MacLean M, Harris N, Piper PW. Chronological lifespan of stationary phase yeast cells; a model for investigating the factors that might influence the ageing of postmitotic tissues in higher organisms. Yeast (Chichester, England) 2001;18:499–509. doi: 10.1002/yea.701. [DOI] [PubMed] [Google Scholar]
- de Magalhaes JP, Costa J, Toussaint O. HAGR: the Human Ageing Genomic Resources. Nucleic Acids Res. 2005;33:D537–D543. doi: 10.1093/nar/gki017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Magalhaes JP, Budovsky A, Lehmann G, Costa J, Li Y, Fraifeld V, Church GM. The Human Ageing Genomic Resources: online databases and tools for biogerontologists. Aging cell. 2009a;8:65–72. doi: 10.1111/j.1474-9726.2008.00442.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Magalhaes JP, Curado J, Church GM. Meta-analysis of age-related gene expression profiles identifies common signatures of aging. Bioinformatics. 2009b;25:875–881. doi: 10.1093/bioinformatics/btp073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallette FA, Ferbeyre G. The DNA damage signaling pathway connects oncogenic stress to cellular senescence. Cell cycle. 2007;6:1831–1836. doi: 10.4161/cc.6.15.4516. [DOI] [PubMed] [Google Scholar]
- Maruyama J, Naguro I, Takeda K, Ichijo H. Stress-activated MAP kinase cascades in cellular senescence. Curr Med Chem. 2009;16:1229–1235. doi: 10.2174/092986709787846613. [DOI] [PubMed] [Google Scholar]
- Melk A, Halloran PF. Cell senescence and its implications for nephrology. J. Am. Soc. Nephrol. 2001;12:385–393. doi: 10.1681/ASN.V122385. [DOI] [PubMed] [Google Scholar]
- Mlecnik B, Scheideler M, Hackl H, Hartler J, Sanchez-Cabo F, Trajanoski Z. PathwayExplorer: web service for visualizing high-throughput expression data on biological pathways. Nucleic Acids Res. 2005;33:W633–W637. doi: 10.1093/nar/gki391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minamino T, Miyauchi H, Yoshida T, Ishida Y, Yoshida H, Komuro I. Endothelial cell senescence in human atherosclerosis: role of telomere in endothelial dysfunction. Circulation. 2002;105:1541–1544. doi: 10.1161/01.cir.0000013836.85741.17. [DOI] [PubMed] [Google Scholar]
- Moreno-Hagelsieb G, Latimer K. Choosing BLAST options for better detection of orthologs as reciprocal best hits. Bioinformatics. 2008;24:319–324. doi: 10.1093/bioinformatics/btm585. [DOI] [PubMed] [Google Scholar]
- Muck C, Micutkova L, Zwerschke W, Jansen-Durr P. Role of insulin-like growth factor binding protein-3 in human umbilical vein endothelial cell senescence. Rejuvenation Res. 2008;11:449–453. doi: 10.1089/rej.2007.0628. [DOI] [PubMed] [Google Scholar]
- Partridge L. Some highlights of research on aging with invertebrates, 2009. Aging cell. 2009;8:509–513. doi: 10.1111/j.1474-9726.2009.00498.x. [DOI] [PubMed] [Google Scholar]
- Passos JF, Saretzki G, Ahmed S, Nelson G, Richter T, Peters H, Wappler I, Birket MJ, Harold G, Schaeuble K, et al. Mitochondrial dysfunction accounts for the stochastic heterogeneity in telomere-dependent senescence. PLoS Biol. 2007;5:e110. doi: 10.1371/journal.pbio.0050110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piper PW. Long-lived yeast as a model for ageing research. Yeast. 2006;23:215–226. doi: 10.1002/yea.1354. [DOI] [PubMed] [Google Scholar]
- R Development Core Team R: A language and environment for statistical computing. 2007. R Foundation for Statistical Computing, Vienna, Austria. ISBN 3-900051-07-0, URL http://www.R-project.org [accessed on 16 August 2010]
- Rainer J, Sanchez-Cabo F, Stocker G, Sturn A, Trajanoski Z. CARMAweb: comprehensive R- and bioconductor-based web service for microarray data analysis. Nucleic Acids Res. 2006;34:W498–W503. doi: 10.1093/nar/gkl038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ressler S, Bartkova J, Niederegger H, Bartek J, Scharffetter-Kochanek K, Jansen-Dürr P, Wlaschek M. p16INK4A is a robust in vivo biomarker of cellular aging in human skin. Aging Cell. 2006;5:379–389. doi: 10.1111/j.1474-9726.2006.00231.x. [DOI] [PubMed] [Google Scholar]
- Rodwell GE, Sonu R, Zahn JM, Lund J, Wilhelmy J, Wang L, Xiao W, Mindrinos M, Crane E, Segal E, et al. A transcriptional profile of aging in the human kidney. PLoS Biol. 2004;2:e427. doi: 10.1371/journal.pbio.0020427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rufer N, Zippelius A, Batard P, Pittet MJ, Kurth I, Corthesy P, Cerottini JC, Leyvraz S, Roosnek E, Nabholz M, et al. Ex vivo characterization of human CD8+ T subsets with distinct replicative history and partial effector functions. Blood. 2003;102:1779–1787. doi: 10.1182/blood-2003-02-0420. [DOI] [PubMed] [Google Scholar]
- Saeed AI, Bhagabati NK, Braisted JC, Liang W, Sharov V, Howe EA, Li J, Thiagarajan M, White JA, Quackenbush J. TM4 microarray software suite. Methods Enzymol. 2006;411:134–193. doi: 10.1016/S0076-6879(06)11009-5. [DOI] [PubMed] [Google Scholar]
- Stockl P, Hutter E, Zwerschke W, Jansen-Durr P. Sustained inhibition of oxidative phosphorylation impairs cell proliferation and induces premature senescence in human fibroblasts. Exp. Gerontol. 2006;41:674–682. doi: 10.1016/j.exger.2006.04.009. [DOI] [PubMed] [Google Scholar]
- Stockl P, Zankl C, Hutter E, Unterluggauer H, Laun P, Heeren G, Bogengruber E, Herndler-Brandstetter D, Breitenbach M, Jansen-Durr P. Partial uncoupling of oxidative phosphorylation induces premature senescence in human fibroblasts and yeast mother cells. Free Radic. Biol. Med. 2007;43:947–958. doi: 10.1016/j.freeradbiomed.2007.06.005. [DOI] [PubMed] [Google Scholar]
- Storey JD. A direct approach to false discovery rates. Journal of the Royal Statistical Society, Series B. 2002;64:479–498. [Google Scholar]
- Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc.Natl.Acad.Sci.U.S.A. 2003;100:9440–9445. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storey JD, Taylor JE, Siegmund D. Strong control, conservative point estimation, and simultaneous conservative consistency of false discovery rates: a unified approach. Journal of the Royal Statistical Society, Series B. 2004;66:187–205. [Google Scholar]
- Tan Q, Christensen K, Christiansen L, Frederiksen H, Bathum L, Dahlgaard J, Kruse TA. Genetic dissection of gene expression observed in whole blood samples of elderly Danish twins. Hum. Genet. 2005;117:267–274. doi: 10.1007/s00439-005-1308-x. [DOI] [PubMed] [Google Scholar]
- Toussaint O, Royer V, Salmon M, Remacle J. Stress-induced premature senescence and tissue ageing. Biochem. Pharmacol. 2002a;64:1007–1009. doi: 10.1016/s0006-2952(02)01170-x. [DOI] [PubMed] [Google Scholar]
- Toussaint O, Remacle J, Dierick JF, Pascal T, Frippiat C, Zdanov S, Magalhaes JP, Royer V, Chainiaux F. From the Hayflick mosaic to the mosaics of ageing. Role of stress-induced premature senescence in human ageing. Int. J. Biochem. Cell Biol. 2002b;34:1415–1429. doi: 10.1016/s1357-2725(02)00034-1. [DOI] [PubMed] [Google Scholar]
- Turchi L, Fareh M, Aberdam E, Kitajima S, Simpson F, Wicking C, Aberdam D, Virolle T. ATF3 and p15PAF are novel gatekeepers of genomic integrity upon UV stress. Cell Death Differ. 2009;16:728–737. doi: 10.1038/cdd.2009.2. [DOI] [PubMed] [Google Scholar]
- Untergasser G, Madersbacher S, Berger P. Benign prostatic hyperplasia: age-related tissue-remodeling. Exp. Gerontol. 2005;40:121–128. doi: 10.1016/j.exger.2004.12.008. [DOI] [PubMed] [Google Scholar]
- Unterluggauer H, Hampel B, Zwerschke W, Jansen-Durr P. Senescence-associated cell death of human endothelial cells: the role of oxidative stress. Exp. Gerontol. 2003;38:1149–1160. doi: 10.1016/j.exger.2003.08.007. [DOI] [PubMed] [Google Scholar]
- Unterluggauer H, Mazurek S, Lener B, Hütter E, Eigenbrodt E, Zwerschke W, Jansen-Dürr P. Premature senescence of human endothelial cells induced by inhibition of glutaminase. Biogerontology. 2008;2008:9. doi: 10.1007/s10522-008-9134-x. [DOI] [PubMed] [Google Scholar]
- Vasile E, Tomita Y, Brown LF, Kocher O, Dvorak HF. Differential expression of thymosin beta-10 by early passage and senescent vascular endothelium is modulated by VPF/VEGF: evidence for senescent endothelial cells in vivo at sites of atherosclerosis. FASEB J. 2001;15:458–466. doi: 10.1096/fj.00-0051com. [DOI] [PubMed] [Google Scholar]
- Wieser M, Stadler G, Jennings P, Streubel B, Pfaller W, Ambros P, Riedl C, Katinger H, Grillari J, Grillari-Voglauer R. hTERT alone immortalizes epithelial cells of renal proximal tubules without changing their functional characteristics. Am J Physiol Renal Physiol. 2008;295:F1365–F1375. doi: 10.1152/ajprenal.90405.2008. [DOI] [PubMed] [Google Scholar]
- Wu Z, et al. A model-based background adjustment for oligonucleotideexpression arrays. J. Am Stat. Assoc. 2004;99:909–917. [Google Scholar]
- Ye X, Zerlanko B, Kennedy A, Banumathy G, Zhang R, Adams PD. Downregulation of Wnt signaling is a trigger for formation of facultative heterochromatin and onset of cell senescence in primary human cells. Mol. Cell. 2007;27:183–196. doi: 10.1016/j.molcel.2007.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu P, Huang B, Shen M, Lau C, Chan E, Michel J, Xiong Y, Payan DG, Luo Y. p15(PAF), a novel PCNA associated factor with increased expression in tumor tissues. Oncogene. 2001;20:484–489. doi: 10.1038/sj.onc.1204113. [DOI] [PubMed] [Google Scholar]
- Zuckerman V, Wolyniec K, Sionov RV, Haupt S, Haupt Y. Tumour suppression by p53: the importance of apoptosis and cellular senescence. J. Pathol. 2009;219:3–15. doi: 10.1002/path.2584. [DOI] [PubMed] [Google Scholar]
- Zwerschke W, Mazurek S, Stockl P, Hutter E, Eigenbrodt E, Jansen-Durr P. Metabolic analysis of senescent human fibroblasts reveals a role for adenosine monophosphate in cellular senescence. Biochem. J. 2003;376:403–411. doi: 10.1042/BJ20030816. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.