

Abstract
Objective
To place imaging–genetics research in the context of child psychiatry.
Method
A conceptual overview is provided, followed by discussion of specific research examples.
Results
Imaging–genetics research is described linking brain function to two specific genes, for the serotonin-reuptake-transporter protein and a monoamine oxidase enzyme. Work is then described on phenotype selection in imaging genetics.
Conclusions
Child psychiatry applications of imaging genetics are only beginning to emerge. The approach holds promise for advancing understandings of pathophysiology and therapeutics.
Keywords: imaging, genetics, anxiety, children and adolescents
Genetics and imaging have the potential to generate fundamental insights on mental illness pathophysiology. Studies melding these two approaches, through imaging–genetics, are particularly promising. This report reviews research applications of imaging–genetics in child psychiatry. The first section provides a brief conceptual overview, delineating the promise and limitations of imaging genetics and highlighting topics relevant to child psychiatry. Next, we review findings from studies that begin by selecting specific genes and then use imaging to relate these genes to brain function and ultimately to clinical phenotypes. In this section, we discuss two specific genes, focusing in most depth on the gene for the serotonin-transporter protein. We necessarily focus on this particular narrow example so that we can illustrate in some depth the complexities that arise when applying imaging–genetics to child psychiatry. In the final section, we briefly discuss a complementary approach, which begins by selecting a clinical phenotype and relating it first to imaging data and then to genes.
Conceptual Overview
The field of “imaging–genetics” emerged over the past decade, motivated by two forces. The first reflected technical advances, allowing investigation of increasingly large samples.1,2 The second related to limits in psychiatric genetics and brain imaging research, where inconsistent findings continue to accumulate regarding genetic or neural correlates of individual differences in behavior, due to a host of methodologic factors.1,2 This led to suggestions that current nosology does not reflect accurately pathophysiologic associations between behavior and either genetics or brain function.2 The imaging–genetics approach attempts to capitalize on technical successes in imaging and genetics to quantify precisely relations among behavior, genes, and brain activity.
In addition to acknowledging the promise of imaging genetics and other translational approaches, it is important to consider their limitations. Work in other areas of medicine reveals the slow pace of progress in translational research.3,4 Because imaging–genetics and other neuroscience-based strategies are relatively new in psychiatry, it will be years, if not decades, before these techniques produce anything resembling diagnostic tests. However, for complex syndromes, such as mental illnesses, history shows quicker progress in translational work on novel treatments than diagnostic tests.3-5 Hence, neuroscience-based strategies may generate ideas for novel treatments relatively soon. For example, imaging–genetics might identify genes that contribute to anxiety through their effects on amygdala function. Ideas for novel treatments then might emerge from research in animal models using pharmacologic or experiential manipulations that affect these genes and their associated effects on the amygdala.5
Using Imaging Genetics to Understand Pathophysiology
Psychiatric applications of genetics and imaging share the goal of delineating pathophysiology. Research in both areas seeks to define causal chains of events that culminate in manifestations of mental illness. Attempts to identify “ultimate causes” invariably encounter the “chicken-and-egg” problem because behavioral variation reflects the end result of a complex interplay among factors intrinsic to the individual, such as genes, factors in the environment, and interactions between the two, each with reverberating impact on the other. A focus on genetics at least partly solves this problem because scientists have agreed that genes represent a useful starting point for understanding these chains of events when genes are studied in isolation and when they are studied in the context of gene-by-environment interactions.1,2,6-9 Figure 1 illustrates the framework from which imaging–genetics arises. Pathophysiology begins with genes and ends with individual differences in thought and behavior. Between these points, however, the effects of genes emerge through circuitous paths. Figure 1 shows the target of imaging–genetics: the arrow connecting genes to measurements of brain structure and function.
FIGURE 1.
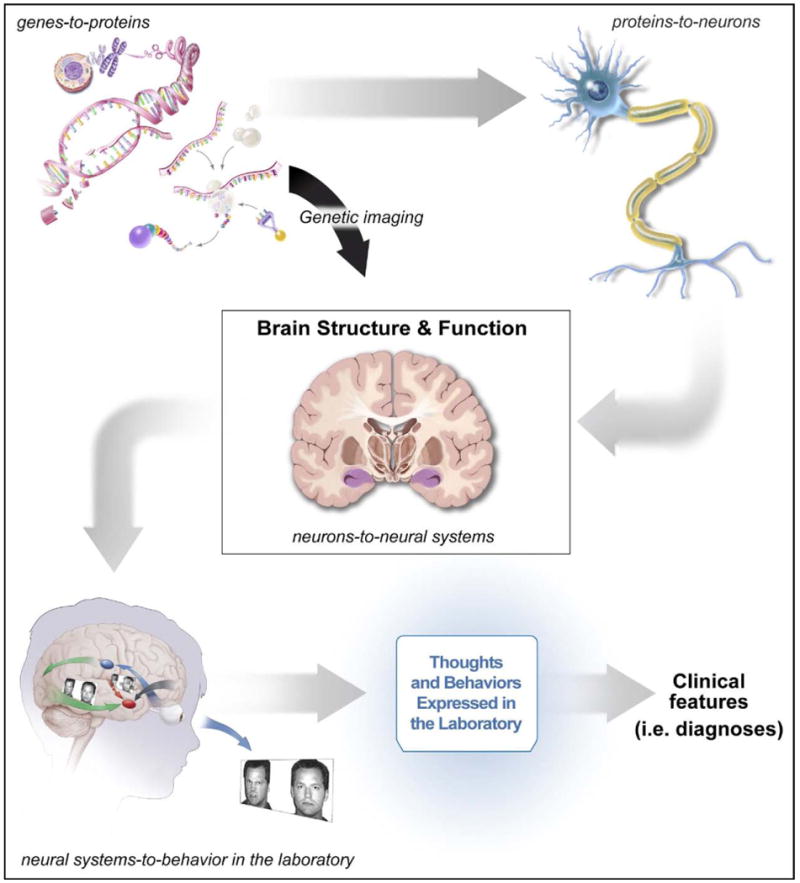
Conceptual framework from which imaging–genetics emerges. Note: This framework views pathophysiology as beginning in processes depicted in the upper left-hand corner. These processes then lead to influences on neurons, depicted in the upper right-hand corner, neural systems, as depicted in the middle box, and their relations to behaviors expressed in the laboratory, before manifesting as clinical features, as appears in the lower right-hand side. These constructs are connected by multiple, complex, nested relationships. The figure illustrates key processes connecting the constructs, including gene regulation, individual neuron formation, neural system configuration, and engagement of specific neural systems through imaging tasks that present emotional faces to children. Imaging–genetics research focuses relatively narrowly on the arrow connecting the upper left-hand portion of the figure to the middle box, connecting genes to constructs assessed through imaging.
Of course, Figure 1 simplifies complex relations. Genes encode neither individual differences in behavior nor the constructs assessed by imaging; instead, they encode and regulate protein expression. Thus, the figure spans many complex nested relations: those connecting genes to proteins, proteins to neurons, neurons to neural systems, and neural systems to behaviors manifest in the laboratory or brain scanner, before finally manifesting as individual differences classifiable as mental illnesses. Moreover, further complications arise through epigenetics, a particular focus in developmental neuroscience. Work in epigenetics examines how particular deoxyribonucleic acid (DNA) sequences produce varied proteins under different circumstances, when they interact with the environment or features of DNA extending beyond nucleic acid sequences. A focus on such processes in developmental research reflects the view that epigenetics vitally shapes brain development.
The major advance afforded by imaging–genetics lies in its ability to ground research on the genetic basis of human behavior in the rich context of neuroscience. By integrating findings from invasive genetic manipulation experiments in animal models with those from observational work in humans, neuroscience demonstrates how genes affect brain function, which in turn affects thought and behavior. Studies in animals link genes to mechanisms instantiated in neural systems, which can then be linked to behavior. As discussed below, these features might allow integration of imaging with genetics to capture larger genotype–phenotype correlations than possible in work relating genes to behavior in the absence of imaging data. Basic-to-clinical translation thereby emerges from work that measures highly similar processes and their relation to genes in humans and other species. Neuroimaging provides psychiatry with heretofore unseen opportunities to assess such highly similar processes, because processes assessed in humans with imaging are similar to those assessed with invasive techniques in rodents and nonhuman primates, i.e., current imaging techniques assess neural system function in humans with resolution comparable to that in many invasive techniques used in other species. For example, basic work shows that some forms of stimulus-reinforcement learning, such as fear conditioning and extinction, are mediated by functional changes in rodent amygdala-based circuitry. Human imaging studies link individual differences in amygdala function, studied with temporal and spatial resolutions similar to those in basic work, to large individual differences in fear-related processes, thereby allowing basic-to-clinical translation.5
Phenotype–Genotype Orientation
Imaging–genetics studies in child psychiatry can be classified based on their starting point. Using this approach, the current review summarizes recent imaging–genetics findings organized into two groups.
One set of studies begins with observations on genetic variation, which then are related to phenotypes; the other set begins with observations on phenotypes, which then are related to genes. In many instances, these two approaches are complementary, demonstrating mutually-reinforcing, reverberating research cycles. For example, when classic Mendelian syndromes are linked to genes, imaging studies then can map relations between variation in these genes and brain structure or function. This approach is relevant to child psychiatry because it is used with developmental syndromes, such as Williams and velocardiofacial syndromes.6-8
Most common pediatric mental illnesses do not exhibit Mendelian inheritance but rather are viewed as genetically complex phenotypes, potentially arising from small effects of multiple genes interacting with environmental risks. The current review focuses on imaging–genetics of these complex phenotypes, which often use tests of genetic association. With this approach, genetic associations are mapped onto phenotypes derived from brain imaging.
Imaging Phenotypes
Imaging–genetics maps individual differences in the brain using a variety of techniques, each with unique advantages and disadvantages in terms of safety, applicability, and spatial or temporal resolution. Functional magnetic resonance imaging (fMRI), structural MRI (sMRI), and magnetic resonance spectroscopy are particularly relevant to child psychiatry due to their noninvasive nature.
Functional Magnetic Resonance Imaging
Functional MRI capitalizes on the fact that changes in blood flow alter the magnetic susceptibility properties of tissue, and these changes are detected as a brightening of the fMRI signal. In this way, the brain is armed with its own endogenous contrast agent that can be used to map the relation between changes in psychological processes and changes in blood flow.
Considerable methodologic variation exists in fMRI. A frequently employed imaging–genetics strategy involves selecting a psychological process whose underlying neural architecture can be mapped. In essence, this approach requires investigators to link behavioral variation to specific psychological processes that can be engaged reliably in the fMRI scanner. The main advantage of fMRI for imaging–genetics relates to the technique's ability to assess brain functions that, when assessed in rodents and nonhuman primates, show very large associations with behavior. This creates the potential for identifying correspondingly large associations between brain functions and individual differences in behavior and genes.
Structural Magnetic Resonance Imaging
Structural MRI provides images of brain structures with precise anatomic detail. This allows investigators to quantify the size and shape of structures with submillimeter resolution. Because sMRI generates such images with outstanding reliability, the technique is ideally suited for repeated measurements over development, charting brain growth trajectories. Because more research in pediatric mental illness relies on sMRI than fMRI, methods in sMRI have become more standardized. This in turn has generated more data on heritability in twin samples and far more longitudinal research in large samples.10
Magnetic Resonance Spectroscopy
Magnetic resonance spectroscopy quantifies levels of brain chemicals, capitalizing on the fact that distinct chemicals generate unique magnetic resonance spectra based on their atomic constituents and configuration. Available magnetic resonance spectroscopic techniques can quantify an important but relatively limited set of chemicals, which somewhat decreases its applicability, although chemicals relevant to child psychiatry can be quantified. In imaging–genetics, the only relevant pediatric study examines associations in obsessive–compulsive disorder (OCD) between spectra influenced by glutamate and polymorphisms in genes that regulate glutamate function,11 a finding that may also relate to morphometric abnormalities in OCD.12 Such findings could be clinically relevant, because work on glutamate generates insights on novel therapeutics, including in the pharmacologic treatment of pediatric OCD.13 This in turn illustrates how basic science advances shape therapeutics relatively early in the phase of basic-to-clinical translation.
Studying Genes and their Association with Brain Function
One set of imaging–genetics studies begin by selecting particular genes and then relating them to brain function. The present section discusses research in this area. The section focuses in most depth on a gene for the serotonin-transporter (5HTT) protein but also briefly discusses a gene for monamine oxidase (MAO). Brain-derived neurotrophic factor is another protein subjected to considerable work in brain development and imaging–genetics.14 This section does not discuss brain-derived neurotrophic factor or other relevant proteins, due to space restrictions, coupled with the need to discuss data on at least one particular gene in some depth. An in-depth discussion is needed to illustrate the complexities in developmental applications of imaging genetics.
Serotonin-Transporter Polymorphisms
The 5HTT and Behavior
Primates exhibit polymorphisms in the gene for 5HTT (SLC6A4), based on the number of variable repeat sequences appearing in the promoter region of the gene.1,9,15-18 In humans, these polymorphisms include short (S) and long alleles (L), recognized more than a decade ago, as well as more recently discovered long-allele variants, based on single-nucleotide A→G substitution yielding two variants (LG and LA). One variant (LG) behaves physiologically like the S allele, whereas the other (LA) behaves differently from the other two alleles (S and LG). Relative to the LA allele, the S and LG alleles are associated with a decreased capacity for serotonin reuptake, the main function of the 5HTT protein.
Many scientists find the 5HTT alleles to be of considerable relevance for translational research on developmental psychopathology. Research reliably demonstrates in vitro functional differences among 5HTT alleles that parallel the effects of selective-serotonin reuptake inhibitors. This research in turn reliably extends considerable work linking 5HT to behavior. For example, manipulations of 5HTT function in rodents and nonhuman primates affect behaviors relevant to psychopathology, such as an organism's response to threats or rewards.9 Specifically, at least in rodents, reducing 5HTT activity early in life increases measurements of fear assessed much later in life; this effect does not occur in mature rodents, thereby linking 5HTT, behavior, and development. However, despite agreement on the importance of studying 5HTT, disagreement surrounds some aspects of the research.
One area of disagreement concerns the expected magnitude of associations between SLC6A4 and psychopathology. Initial findings did relate the 5HTT gene to marked variations in neuroticism or harm avoidance, and subsequent studies extended the findings to other phenotypes, including anxiety and major depressive disorders.9 However, recent meta-analyses suggested that associations are weaker than originally thought.15 This supports conclusions in other work noting that genetically complex phenotypes, such as mood and anxiety disorders, show small associations with individual genes.19-21 Subsequent work then suggested that large effects manifest in high-stress environments, due to powerful gene-by-environment interactions.9 However, here, too, recent meta-analyses suggested that this may not be the case.18
Imaging Genetics, Child Psychiatry, and the 5HTT
Most scientists agree that major advances can accrue from research in humans that directly links brain function to genes. Without measurements of brain function, work attempting to link genetically mediated variation in 5HTT function to behavior is not easily integrated with work in rodents and nonhuman primates, given inadequacies in animal models of psychopathology.9,19-21 Basic-to-clinical translation is easier when it is based on research in multiple species, each relating the 5HTT to amygdala–prefrontal cortex (PFC) function.1,2,5,21 The advantage of work in this area reflects the fact that highly similar phenotypes can be studied in different species. This approach is easier to apply for particular phenotypes, such as fear conditioning or attention orienting,5 where data demonstrate similar brain–behavior associations in rodents, nonhuman primates, and humans. Other phenotypes, such as those emphasizing data from verbal reports, are less easily applied across species. Moreover, when comparable phenotypes are studied, invasive experiments in animal models can probe the manner in which specific polymorphisms shape brain functions studied in imaging. Using such invasive techniques, neuroscientists can elucidate the manner in which functionality of polymorphisms maps onto behaviors and brain functions with precision that is not achievable in less invasive studies performed with humans.
Controversy has arisen about some aspects of 5HTT imaging–genetics.19,20,22 Data in healthy adults link the SLC6A4 genotype to an amygdala–PFC circuit.1,16,17 The most consistent finding17 is that healthy adults with low-activity variants of the 5HTT gene manifest greater amygdala threat-related reactivity than do those with high-activity variants. This led to suggestions that relations between the SLC6A4 genotype and diagnosis are weak because they are indirect and attenuated by diverse sources of noise. It was further suggested that the association between the SLC6A4 genotype and amygdala–PFC function is more direct and, hence, stronger. However, there is controversy about the reasonableness of these suggestions, specifically with regard to the expected magnitude of associations between genes and imaging.19-22 Initial work suggested the presence of large effects,1 but contrary views suggest that imaging–genetics is vulnerable to similar weaknesses plaguing other psychiatric–genetics studies.19-23 These weaknesses are attributed to incorrect assumptions about the simplicity of gene effects on neuroimaging and other neuroscience-based phenotypes, which may indeed exhibit genetic architectures that are as complex as clinical phenotypes, yielding small genotype–phenotype associations.
Clearly, imaging–genetics has promise and complexity, and important work focuses on structural and functional correlates of genetic variability in amygdala–PFC circuitry. In research on 5HTT, one set of complexities concerns interpretations of amygdala-activation data in adults. Initial interpretations emphasized the role of decreased 5HTT function in threat hypersensitivity, consistent with work in rodents and nonhuman primates showing that decreased 5HTT function produces threat hypersensitivity through effects on the amygdala.1,9,16 However, initial fMRI findings in humans were open to other interpretations, in part because fMRI lacks absolute quantification. Thus, for example, between-group differences in amygdala activity in response to neutral and threatening faces can reflect increased responses to one stimulus (e.g., threatening faces) or decreased responses to the other (e.g., neutral faces). Although the initial studies suggested that low-activity 5HTT alleles predict enhanced threat responsiveness, more recent work used additional fMRI conditions. These more recent studies showed that low-activity 5HTT alleles predict normal amygdala response to threats, in tandem with reduced response to neutral stimuli, which could explain the previous findings.24,25
These new findings generate novel research opportunities and raise questions about the comparability of imaging–genomic and basic 5HTT data on threat responding. Development further complicates interpretations: fMRI data suggest that the amygdala responds more strongly to threats in adolescents than adults, at least under some circumstances.26 Therefore, although there may be associations between the 5HTT genotype and responses to neutral stimuli in adults, in youth there may be associations between the 5HTT genotype and responses to threat stimuli. Work in rodents finds interrelated developmental differences in amygdala function, response to 5HTT manipulations, and threat responding.9 As a result, it is important to establish in humans the degree to which the SLC6A4 genotype specifically affects responding to neutral or emotional stimuli at various ages. Adding yet further complexity to this emerging set of findings, studies using positron emission tomography find no relation between the SLC6A4 genotype and in vivo measurements of 5HTT binding potential in adults.27 Although many explanations could produce such unexpected, negative findings, some suggest that they result from development: effects of genetic variation in 5HTT may shape behavior early in life in ways that are not reflected in adult measurements of 5HTT binding potential.
Other complexities concern the contexts under which amygdala hypersensitivity manifests. Early interpretations of imaging–genetics 5-HTT data treated amygdala hyperactivity as a relatively static phenomenon, correlated with genotype. However, amygdala activity is plastic, changing with the context of experimental tasks.28 Between-group differences in amygdala function related to anxiety or age only manifest in specific experimental contexts.26,29 One would expect a similar context dependency on genetic effects.
Ideally, fMRI experiments designed to elucidate associations between amygdala activity and genetic predictors of psychopathology should use particular tasks. These tasks should be selected based on their ability to elicit, in studies in patients and healthy subjects, between-group differences in task performance or autonomic physiology. This approach generates research that directly links many of the processes depicted in Figure 1. Unfortunately, most 5HTT imaging–genetics work uses tasks that do elicit strong amygdala responses in subjects, considered as a group, but do not possess such prior knowledge linking neural system function, behavior in the laboratory, and clinical features. Thus, clinical applicability is limited in most published work, because it is does not use tasks sensitive to psychopathology-related perturbations that manifest on the employed cognitive tasks. Consideration of experimental context appears particularly important in child psychiatry, given the powerful effects of context on children's behavior and brain function. As such, the experimental context ideally suited for eliciting between-group differences in children may contrast with that ideally suited for adults.
Only one pediatric fMRI study has examined the relation between the SLC6A4 genotype and amygdala function.30 Unlike prior studies in adults, this study used an experimental task where prior independent data demonstrated between-group differences in task performance relevant to mood and anxiety disorders.31 The study then mapped 5HTT-related associations with amygdala function under task conditions previously shown to elicit behavioral differences between controls and adolescents with familial or personal risk for mood and anxiety disorders.
Figure 2 shows data from two fMRI studies of amygdala function, i.e., the pediatric imaging–genetics study of the SLC6A4 genotype discussed above,30 and a study on adolescent anxiety and major depressive disorders.32 Three findings emerged from these studies. First, as expected, there was an association between enhanced amygdala response and the low-activity 5HTT S or LG alleles in 33 healthy adolescents (Figure 2a). This occurred specifically when adolescents rated levels of fear experienced while viewing fear faces. Second, like healthy adolescent carriers of the low-activity 5HTT alleles, adolescents with anxiety or major depressive disorders showed a greater amygdala response than did healthy adolescents while rating fear levels to fear faces (Figure 2b). Third, genetics and diagnosis interacted: in the 31 psychiatrically impaired adolescents, genetic associations with amygdala function exhibited an opposite trend in these patients as had manifested in healthy adolescents (Figure 2a). Thus, individual differences related to genes and to diagnosis may shape amygdala response to threats in youth.
FIGURE 2.
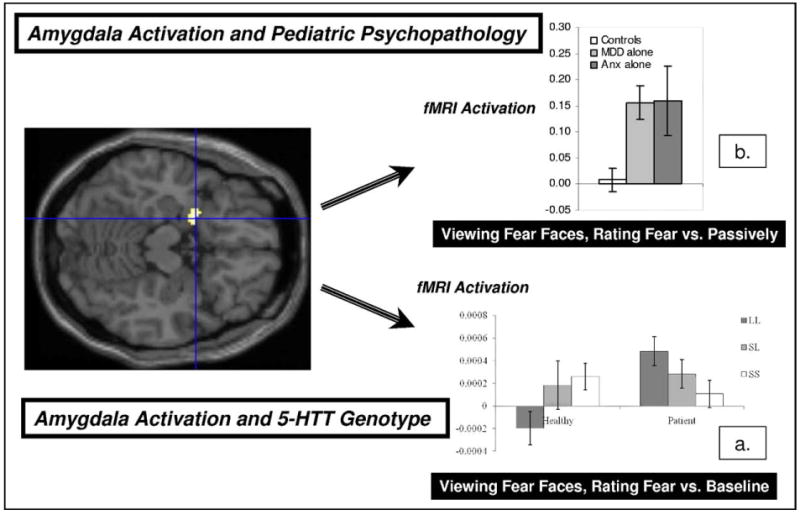
5-HTT imaging–genetics in adolescents. Note: Illustrated are findings from two studies on relations among amygdala function, 5-HTT genotype, and developmental psychopathology. Amygdala topography is displayed on the left-hand side, in an axial slice. (a) Data from an imaging–genetics study in adolescents.30 Levels of amygdala activation are shown for six groups, comprising three groups of healthy adolescents on the left-hand side and three groups of patients with anxiety disorders or major depressive disorder (MDD) on the right-hand side. Each of these three groups is formed based on the level of 5-HTT function (low [S, S], medium [S, L], high [L, L]) associated with the three genotypes, with LG and S carriers being classified together. These data are for an functional magnetic resonance imaging (fMRI) contrast of fear-face–viewing events, viewed while fear levels are rated compared with a low-level, null-event baseline. Amygdala activity in healthy subjects is highest for the SS/SLG low-activity-allele group, whereas amygdala activity in patients is highest for the high-activity LA/LA allele group. (b) Data from an imaging study focused on adolescent psychopathology,29 which includes the subjects shown in panel a. Levels of amygdala activation are shown in healthy adolescents, nonanxious adolescents with MDD, and euthymic adolescents with anxiety for a contrast of fear-face–viewing events, viewed while fear levels are rated compared with events where these same faces are passively viewed. Anx = anxiety.
These two studies may provide important clues on the modulating effects from psychopathology in imaging–genetics. Few prior fMRI studies in any age group assessed such modulation by simultaneously studying healthy and impaired subjects. However, given the small samples and existing complexities, these data should encourage more definitive studies in larger samples, studied under experimental conditions where links to psychopathology have been established through independent studies of information processing.
Monoamine Oxidase A Polymorphism
Monamine Oxidase and Behavior
Although MAO exists as two isoenzymes, this review focuses on MAO-A, the isoenzyme subjected to imaging–genetics research. The MAO-A gene, lying on the X chromosome, has functional variants differing in tandem repeats or single-nucleotide polymorphisms, which exhibit functional differences in vitro.33-37 Because MAO-A catabolizes the monoamines, dopamine, noradrenaline, and serotonin, MAO inhibition raises monoamine levels, which enhances activity in these neurotransmitter systems and modulates development. Effects on brain function from the specific functional MAO-A-gene variants are expected to influence behavior through genetic effects on monoamines, with reverberating effects on the brain acutely and chronically during brain development.
More than 50 years of research implicates MAO-A inhibition in behavioral and mood regulation. Interest grew after the discovery of a Dutch family in which male carriers of an MAO-A gene deletion exhibited a complex psychiatric phenotype with intellectual disability and impulsive aggression.34 A similar phenotype emerged in the MAO-A knockout mouse.33
These initial genetic findings implicated developmental factors, based on the early emergence of aggressive behavior in the above-noted Dutch family and based on demonstrable effects of MAO-A inhibition on brain development in the knockout mouse. This work generated early interest in gene–behavior associations. However, as with the 5HTT gene, particular interest emerged in examining how the environment interacts with the MAO-A gene. Although gene-association studies implicated MAO-A in various human behaviors,35-37 perhaps the most consistent findings emerged for aggressive behavior, where early maltreatment predicted high risk in male carriers of low-expression MAO-A alleles.36 Evidence seems to support this gene-by-environment interaction more unequivocally than for the 5HTT gene.37
Recent attempts to extend this work focused on research with girls and intermediate phenotypes. For example, a recent study found that prenatal nicotine exposure, a pharmacological/environmental risk factor for aggression, interacted with the MAO-A gene to predict adolescent antisocial behavior in unique ways in boys and girls.38 Boys exposed to prenatal nicotine showed the highest risk for antisocial behavior in the presence of low-activity MAO-A alleles, whereas girls showed the opposite pattern. This study also demonstrated gene-by-environment interactions for an alternative phenotype, face-emotion labeling, where data exist concerning the underlying mediating neural circuitry. As such, the work naturally raises questions about mediating neural circuitry, as can be addressed through genetic–imaging. Moreover, the findings raise particular questions about how events occurring early in development might modify neural circuit ontogeny through interactions with genes.
Imaging, Genetics, and Monamine Oxidase A
Questions on neural mediators have been addressed through imaging–genetics studies. Indeed, considerable cognitive neuroscience work generates paradigms well suited for explorations of the underlying neural circuitry of gene–behavior associations manifest in work with MAO-A. So far, imaging–genetics MAO-A studies only involve adults, where they report evidence of associations between genotype and prefrontal function. The largest set of studies1,39 examined MAO-A gene effects on social–emotional function, emotional memory, and inhibition. Predicted frontal and subcortical brain regions showed activation patterns that differed between individuals possessing the low- and high-expressing MAO-A variants, with findings being moderated by sex and temperament. Further imaging–genetics work on temperament and the MAO-A gene could bring a developmental focus to imaging–genetics in this area, as already is emerging in work on the 5HTT gene.
Studying Phenotypes and their Relation to Genes
Selecting Phenotypes
Although one set of studies begins by selecting genes, another set begins by selecting a phenotype. In this second approach, the phenotype is then related to brain structure or function before being related to genetic variation. In these studies, selection of the phenotype is a starting point and is a crucial element of the research design. Indeed, recent work focuses considerable attention on alternative phenotypes.
The most straightforward approach relies on diagnoses, but it has disadvantages, because diagnosis is unlikely to map onto dysfunction in specific brain circuits. Moreover, this categorical approach possesses less statistical power than approaches using dimensions. Thus, alternative phenotyping approaches have characterized patients with bipolar disorder (BD) according to age of onset; those with autism by severity of language impairment; and those with attention-deficity/hyperactivity disorder (ADHD) or BD by level of impulsivity.40-43 This work can be readily extended through imaging–genetics. For example, a genetic fMRI study on autism and language impairment could link neural activation during a language-based task to relevant polymorphisms.
Another approach relies on behaviors measured with laboratory-based paradigms, as in the above-noted work on the MAO-A gene, prenatal cigarette exposure, and face-emotion processing. When such behaviors are heritable, state independent, and associated with illness, they are called endophenotypes.44 Examples include delayed motor inhibition in ADHD,45 deficits in prepulse inhibition or anti-saccades in schizophrenia,46 and deficits in face-emotion labeling or sustained attention in BD.47,48 Compared with clinical phenotypes, such endophenotypes are more easily linked to brain function, particularly when they examine behaviors that also can be modeled in experimental organisms.
Phenotypes, Genetics, and Child Psychiatry
Few imaging–genetics studies in child psychiatry begin by selecting a phenotype; indeed, the number of such studies in psychiatry generally is small. Considerable more imaging–genetics work is needed that selects multiple clinical groups and nonimpaired subjects and then examines relations in each group between genes and brain imaging measurements. In this section, we provide examples in ADHD, autism, and BD. In each instance, investigators used diagnosis as the inclusion criterion and then examined associations between genotype and structural or functional imaging measurements. As more studies use approaches to link distinct diagnoses to measurements of brain function and then genes, future reviews on child psychiatry imaging–genetics will be able to summarize the manner in which gene–brain relations vary by clinical groupings.
In child psychiatry, the largest number of genetic–imaging studies focus on ADHD, where four structural and one functional study currently exist.49 Each study first linked ADHD to imaging measurements, which in turn were then linked to dopamine genes. This is a reasonable starting point for such work, given dopamine's role in ADHD pathophysiology and treatment. One study used a longitudinal approach to show that, in the dopamine D4 receptor gene, the dopamine D4 receptor seven-repeat allele was associated with thinner prefrontal and parietal cortices and better outcome in patients with ADHD.50 There were no associations with polymorphisms in the dopamine D1 receptor or dopamine transporter (DAT1) genes.
Durston et al.51,52 conducted functional genetic–imaging research on ADHD, focusing on the DAT1 gene in youth with ADHD, their unaffected siblings, and controls. The investigators tested two hypotheses: that during a cognitive control task, the impact of the DAT1 genotype on striatal and cerebellar activation would mirror that of methylphenidate; and that genotype would have a differential impact on activation in those with a familial risk for ADHD versus low-risk controls. They found that, across groups, striatal and cerebellar activation varied as a function of genotype, although genotype had a more significant impact on activation in subjects at risk for ADHD than in low-risk individuals. These data suggest that striatal dysfunction mediates genetic risk for ADHD, conferred at least partly by DAT1 genotype.
Fewer studies examine autism than ADHD. Nevertheless, considerable imaging work more broadly examines brain size. Wassink et al.53 drew on this literature and studies on 5HT by examining associations between cortical gray matter and SLC6A4 genotype. They found an association between the 5HTT short allele and increased cortical gray matter, particularly in the frontal lobes.
In BD, several candidate behavioral endophenotypes have been suggested, including deficits in face-emotion labeling. Coupling this line of research with studies on amygdala response to face-emotion tasks in BD, Liu et al.54 used a genome-wide association scan (GWAS) to examine genetic associations with amygdala activation in healthy youth and those with BD. Few studies have coupled GWAS with fMRI;55 such a strategy obviates candidate genes but, given the multiple statistical tests employed, ideally should involve a large sample. Liu et al. found an impact of polymorphisms in the gene DOK5 (rs2023454), a substrate of TrkB/C receptors involved in neurotrophin signaling, on amygdala activation while subjects rated their emotional response to neutral faces. The impact of genotype was nonsignificantly greater in patients than in controls. Given the small sample of this study, these results should be considered preliminary.
This work on GWAS addresses limitations in candidate-gene association studies, such as those reviewed above for the 5HTT and MAO-A gene. Compared with such studies, GWAS examines relatively broad features of the genome. Moreover, as with GWAS, molecular geneticists currently use other rapidly developing techniques to probe functions manifesting across the entire genome as opposed to within particular genes. This includes work on copy number variants, noncoding ribonucleic acid variation, DNA methylation, epistasis, and other related features.56 As it continues to expand, such work is likely to be highly relevant to imaging–genetics research in child psychiatry, because brain development might be shaped by the effects of these broadly influential genetic processes. Nevertheless, as of this writing, no imaging–genetics research in child psychiatry adopts these genetic approaches.
Imaging genetics has emerged as a technique that combines genetics and imaging to generate insights on pathophysiology. The main advantage afforded by the technique pertains to its reliance on noninvasive methods to assess processes in humans that are examined using more invasive methods in rodents and nonhuman primates. This provides scientists with heretofore unseen opportunities to directly link genes to neural systems from a mechanistic point of view. These opportunities then can be translated to generate insights pertinent to mental illness biomarkers.
The review discusses a range of imaging–genetic approaches and related findings as generated with diverse techniques. Nevertheless, the review focuses in most depth on one particular set of examples emerging from imaging–genetics research on 5HT and its relation to amygdala function. This focus reflects the large body of imaging–genetics and basic work devoted to related topics, coupled with the fact that the early, limited applications of child psychiatry imaging–genetics consider these issues in most depth. The complexities of findings in this area demonstrate the need for a deep, precisely articulated, cross-species perspective, whereby scientists work in tandem with multiple species to model gene–brain–behavior associations at particular points in development. Such an approach necessitates a focus on measurements, such as fear conditioning or attention orienting, which exhibit cross-species comparability in terms of the underlying circuitry and its relation to behaviors that can be quantified in laboratory-based studies in diverse species.
The reviewed findings demonstrate the early stage of imaging–genetics research in child psychiatry. Given this early stage, it is likely to be many years before imaging–genetics findings directly shape views of diagnosis or classification. Nevertheless, other work in translational medicine suggests that imaging–genetics research could influence clinical care in the not-so-distant future. Specifically, much like work on basic mechanisms of pathophysiology for other illnesses, imaging–genetics research in child psychiatry may generate novel mental illness treatments relatively soon.
Footnotes
Disclosure: Drs. Pine, Ernst, and Leibenluft report no biomedical financial interests or potential conflicts of interest.
References
- 1.Meyer-Lindenberg A, Weinberger DR. Intermediate phenotypes and genetic mechanisms of psychiatric disorders. Nat Rev Neurosci. 2006;7(10):818–827. doi: 10.1038/nrn1993. [DOI] [PubMed] [Google Scholar]
- 2.Hyman SE. Can neuroscience be integrated into the DSM-V? Nat Rev Neurosci. 2007;8(9):725–732. doi: 10.1038/nrn2218. [DOI] [PubMed] [Google Scholar]
- 3.Couzin-Frankel J. Genetics. The promise of a cure: 20 years and counting. Science. 2009;324(5934):1504–1507. doi: 10.1126/science.324_1504. [DOI] [PubMed] [Google Scholar]
- 4.Woolf SH. The meaning of translational research and why it matters. JAMA. 2008;299(2):211–213. doi: 10.1001/jama.2007.26. [DOI] [PubMed] [Google Scholar]
- 5.Pine DS, Helfinstein SM, Bar-Haim Y, Nelson E, Fox NA. Challenges in developing novel treatments for childhood disorders: lessons from research on anxiety. Neuropsychopharmacology. 2009;34(1):213–228. doi: 10.1038/npp.2008.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Reiss AL. Childhood developmental disorders: an academic and clinical convergence point for psychiatry, neurology, psychology and pediatrics. J Child Psychol Psychiatry. 2009;50(1-2):87–98. doi: 10.1111/j.1469-7610.2008.02046.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gothelf D, Furfaro JA, Penniman LC, Glover GH, Reiss AL. The contribution of novel brain imaging techniques to understanding the neurobiology of mental retardation and developmental disabilities. Ment Retard Dev Disabil Res Rev. 2005;11(4):331–339. doi: 10.1002/mrdd.20089. [DOI] [PubMed] [Google Scholar]
- 8.Meyer-Lindenberg A, Hariri AR, Munoz KE, et al. Neural correlates of genetically abnormal social cognition in Williams syndrome. Nat Neurosci. 2005;8(8):991–993. doi: 10.1038/nn1494. [DOI] [PubMed] [Google Scholar]
- 9.Gross C, Hen R. The developmental origins of anxiety. Nat Rev Neurosci. 2004;5(7):545–552. doi: 10.1038/nrn1429. [DOI] [PubMed] [Google Scholar]
- 10.Green AE, Munafo MR, Deyoung CG, Fossella JA, Fan J, Gray JR. Using genetic data in cognitive neuroscience: from growing pains to genuine insights. Nat Rev Neurosci. 2008;9:710–720. doi: 10.1038/nrn2461. [DOI] [PubMed] [Google Scholar]
- 11.Arnold PD, Macmaster FP, Richter MA, et al. Glutamate receptor gene (GRIN2B) associated with reduced anterior cingulate glutamatergic concentration in pediatric obsessive-compulsive disorder. Psychiatry Res. 2009;172(2):136–139. doi: 10.1016/j.pscychresns.2009.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Arnold PD, MacMaster FP, Hanna GL, et al. Glutamate system genes associated with ventral prefrontal and thalamic volume in pediatric obsessive-compulsive disorder. Brain Imaging Behav. 2009;3:64–76. doi: 10.1007/s11682-008-9050-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grant P, Lougee L, Hirschtritt M, Swedo SE. An open-label trial of riluzole, a glutamate antagonist, in children with treatment-resistant obsessive-compulsive disorder. J Child Adolesc Psychopharmacol. 2007;17(6):761–767. doi: 10.1089/cap.2007.0021. [DOI] [PubMed] [Google Scholar]
- 14.Lau JY, Goldman D, Buzas B, et al. BDNF gene polymorphisms (Val66Met) predicts amygdala and anterior hippocampus responses to emotional faces in anxious and depressed adolescents. Neuroimage. doi: 10.1016/j.neuroimage.2009.11.026. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Munafo MR, Freimer NB, Ng W, et al. 5-HTTLPR genotype and anxiety-related personality traits: a meta-analysis and new data. Am J Med Genet B Neuropsychiatr Genet. 2009;150B(2):271–281. doi: 10.1002/ajmg.b.30808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pezawas L, Meyer-Lindenberg A, Drabant EM, et al. 5-HTTLPR polymorphism impacts human cingulate-amygdala interactions: a genetic susceptibility mechanism for depression. Nat Neurosci. 2005;8(6):828–834. doi: 10.1038/nn1463. [DOI] [PubMed] [Google Scholar]
- 17.Munafo MR, Brown SM, Hariri AR. Serotonin transporter (5-HTTLPR) genotype and amygdala activation: a meta-analysis. Biol Psychiatry. 2008;63(9):852–857. doi: 10.1016/j.biopsych.2007.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Risch N, Herrell R, Lehner T, et al. Interaction between the serotonin transporter gene (5-HTTLPR), stressful life events, and risk of depression: a meta-analysis. JAMA. 2009;301(23):2462–2471. doi: 10.1001/jama.2009.878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Flint J, Mackay TF. Genetic architecture of quantitative traits in mice, flies, and humans. Genome Res. 2009;19(5):723–733. doi: 10.1101/gr.086660.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Flint J, Munafo MR. The endophenotype concept in psychiatric genetics. Psychol Med. 2007;37(2):163–180. doi: 10.1017/S0033291706008750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Flint J, Shifman S. Animal models of psychiatric disease. Curr Opin Genet Dev. 2008;18(3):235–240. doi: 10.1016/j.gde.2008.07.002. [DOI] [PubMed] [Google Scholar]
- 22.Abbott A. Psychiatric genetics: the brains of the family. Nature. 2008;454(7201):154–157. doi: 10.1038/454154a. [DOI] [PubMed] [Google Scholar]
- 23.Munafo MR, Stothart G, Flint J. Bias in genetic association studies and impact factor. Mol Psychiatry. 2009;14(2):119–120. doi: 10.1038/mp.2008.77. [DOI] [PubMed] [Google Scholar]
- 24.Canli T. Toward a neurogenetic theory of neuroticism. Ann N Y Acad Sci. 2008;1129:153–174. doi: 10.1196/annals.1417.022. [DOI] [PubMed] [Google Scholar]
- 25.Canli T, Omura K, Haas BW, Fallgatter A, Constable RT, Lesch KP. Beyond affect: a role for genetic variation of the serotonin transporter in neural activation during a cognitive attention task. Proc Natl Acad Sci U S A. 2005;102(34):12224–12229. doi: 10.1073/pnas.0503880102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guyer AE, Monk CS, McClure-Tone EB, et al. A developmental examination of amygdala response to facial expressions. J Cogn Neurosci. 2008;20(9):1565–1582. doi: 10.1162/jocn.2008.20114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Parsey RV, Hastings RS, Oquendo MA, et al. Effect of a triallelic functional polymorphism of the serotonin-transporter-linked promoter region on expression of serotonin transporter in the human brain. Am J Psychiatry. 2006;163(1):48–51. doi: 10.1176/appi.ajp.163.1.48. [DOI] [PubMed] [Google Scholar]
- 28.Mitchell DG, Nakic M, Fridberg D, Kamel N, Pine DS, Blair RJ. The impact of processing load on emotion. Neuroimage. 2007;34(3):1299–1309. doi: 10.1016/j.neuroimage.2006.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Beesdo K, Lau JY, Guyer AE, et al. Common and distinct amygdala-function perturbations in depressed vs anxious adolescents. Arch Gen Psychiatry. 2009;66(3):275–285. doi: 10.1001/archgenpsychiatry.2008.545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lau JY, Goldman D, Buzas B, et al. Amygdala function and 5-HTT gene variants in adolescent anxiety and major depressive disorder. Biol Psychiatry. 2009;65(4):349–355. doi: 10.1016/j.biopsych.2008.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pine DS, Klein RG, Roberson-Nay R, et al. Face emotion processing and risk for panic disorder in youth. J Am Acad Child Adolesc Psychiatry. 2005;44:664–672. doi: 10.1097/01.chi.0000162580.92029.f4. [DOI] [PubMed] [Google Scholar]
- 32.Beesdo K, Lau J, McClure-Tone EB, et al. Common and specific amygdala-function perturbations in depressed versus anxious adolescents. Arch Gen Psychiatry. 2009;66:275–285. doi: 10.1001/archgenpsychiatry.2008.545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim JJ, Shih JC, Chen K, et al. Selective enhancement of emotional, but not motor, learning in monoamine oxidase A-deficient mice. Proc Natl Acad Sci U S A. 1997;94(11):5929–5933. doi: 10.1073/pnas.94.11.5929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brunner HG, Nelen M, Breakefield XO, Ropers HH, van Oost BA. Abnormal behavior associated with a point mutation in the structural gene for monoamine oxidase A. Science. 1993;262(5133):578–580. doi: 10.1126/science.8211186. [DOI] [PubMed] [Google Scholar]
- 35.Buckholtz JW, Meyer-Lindenberg A. MAOA and the neurogenetic architecture of human aggression. Trends Neurosci. 2008;31(3):120–129. doi: 10.1016/j.tins.2007.12.006. [DOI] [PubMed] [Google Scholar]
- 36.Caspi A, Moffitt TE. Gene-environment interactions in psychiatry: joining forces with neuroscience. Nat Rev Neurosci. 2006;7(7):583–590. doi: 10.1038/nrn1925. [DOI] [PubMed] [Google Scholar]
- 37.Taylor A, Kim-Cohen J. Meta-analysis of gene-environment interactions in developmental psychopathology. Dev Psychopathol. 2007;19(4):1029–1037. doi: 10.1017/S095457940700051X. [DOI] [PubMed] [Google Scholar]
- 38.Wakschlag LS, Kistner EO, Pine DS, et al. Interaction of prenatal exposure to cigarettes and MAOA genotype in pathways to youth antisocial behavior. Mol Psychiatry. 2009 doi: 10.1038/mp.2009.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Buckholtz JW, Callicott JH, Kolachana B, et al. Genetic variation in MAOA modulates ventromedial prefrontal circuitry mediating individual differences in human personality. Mol Psychiatry. 2008;13(3):313–324. doi: 10.1038/sj.mp.4002020. [DOI] [PubMed] [Google Scholar]
- 40.Reif A, Jacob CP, Rujescu D, et al. Influence of functional variant of neuronal nitric oxide synthase on impulsive behaviors in humans. Arch Gen Psychiatry. 2009;66(1):41–50. doi: 10.1001/archgenpsychiatry.2008.510. [DOI] [PubMed] [Google Scholar]
- 41.Schulze TG, Hedeker D, Zandi P, Rietschel M, McMahon FJ. What is familial about familial bipolar disorder? Resemblance among relatives across a broad spectrum of phenotypic characteristics. Arch Gen Psychiatry. 2006;63(12):1368–1376. doi: 10.1001/archpsyc.63.12.1368. [DOI] [PubMed] [Google Scholar]
- 42.Bradford Y, Haines J, Hutcheson H, et al. Incorporating language phenotypes strengthens evidence of linkage to autism. Am J Med Genet. 2001;105(6):539–547. [PubMed] [Google Scholar]
- 43.Buxbaum JD, Silverman JM, Smith CJ, et al. Evidence for a susceptibility gene for autism on chromosome 2 and for genetic heterogeneity. Am J Hum Genet. 2001;68(6):1514–1520. doi: 10.1086/320588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gottesman II, Gould TD. The endophenotype concept in psychiatry: etymology and strategic intentions. Am J Psychiatry. 2003;160(4):636–645. doi: 10.1176/appi.ajp.160.4.636. [DOI] [PubMed] [Google Scholar]
- 45.Goos LM, Crosbie J, Payne S, Schachar R. Validation and extension of the endophenotype model in ADHD patterns of inheritance in a family study of inhibitory control. Am J Psychiatry. 2009;166(6):711–717. doi: 10.1176/appi.ajp.2009.08040621. [DOI] [PubMed] [Google Scholar]
- 46.Turetsky BI, Calkins ME, Light GA, Olincy A, Radant AD, Swerdlow NR. Neurophysiological endophenotypes of schizophrenia: the viability of selected candidate measures. Schizophr Bull. 2007;33(1):69–94. doi: 10.1093/schbul/sbl060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Brotman MA, Rooney MH, Skup M, Pine DS, Leibenluft E. Increased intrasubject variability in response time in youths with bipolar disorder and at-risk family members. J Am Acad Child Adolesc Psychiatry. 2009;48(6):628–635. doi: 10.1097/CHI.0b013e3181a27527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brotman MA, Guyer AE, Lawson ES, et al. Facial emotion labeling deficits in children and adolescents at risk for bipolar disorder. Am J Psychiatry. 2008;165(3):385–389. doi: 10.1176/appi.ajp.2007.06122050. [DOI] [PubMed] [Google Scholar]
- 49.Durston S, de Zeeuw P, Staal WG. Imaging genetics in ADHD: a focus on cognitive control. Neurosci Biobehav Rev. 2009;33(5):674–689. doi: 10.1016/j.neubiorev.2008.08.009. [DOI] [PubMed] [Google Scholar]
- 50.Shaw P, Lerch J, Greenstein D, et al. Longitudinal mapping of cortical thickness and clinical outcome in children and adolescents with attention-deficit/hyperactivity disorder. Arch Gen Psychiatry. 2006;63(5):540–549. doi: 10.1001/archpsyc.63.5.540. [DOI] [PubMed] [Google Scholar]
- 51.Durston S, Mulder M, Casey BJ, Ziermans T, van Engeland H. Activation in ventral prefrontal cortex is sensitive to genetic vulnerability for attention-deficit hyperactivity disorder. Biol Psychiatry. 2006;60(10):1062–1070. doi: 10.1016/j.biopsych.2005.12.020. [DOI] [PubMed] [Google Scholar]
- 52.Durston S, Fossella JA, Mulder MJ, et al. Dopamine transporter genotype conveys familial risk of attention-deficit/hyperactivity disorder through striatal activation. J Am Acad Child Adolesc Psychiatry. 2008;47(1):61–67. doi: 10.1097/chi.0b013e31815a5f17. [DOI] [PubMed] [Google Scholar]
- 53.Wassink TH, Hazlett HC, Epping EA, et al. Cerebral cortical gray matter overgrowth and functional variation of the serotonin transporter gene in autism. Arch Gen Psychiatry. 2007;64(6):709–717. doi: 10.1001/archpsyc.64.6.709. [DOI] [PubMed] [Google Scholar]
- 54.Liu X, Akula N, Skup M, Brotman MA, Leibenluft E, McMahon FJ. A genome-wide association study of amygdala activation in youths with and without bipolar disorder. J Am Acad Child Adolesc Psychiatry. doi: 10.1097/00004583-201001000-00007. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Potkin SG, Turner JA, Fallon JA, et al. Gene discovery through imaging genetics: identification of two novel genes associated with schizophrenia. Mol Psychiatry. 2009;14(4):416–428. doi: 10.1038/mp.2008.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Plomin R, Davis OS. The future of genetics in psychology and psychiatry: microarrays, genome-wide association, and non-coding RNA. J Child Psychol Psychiatry. 2009;50(1-2):63–71. doi: 10.1111/j.1469-7610.2008.01978.x. [DOI] [PMC free article] [PubMed] [Google Scholar]