

Abstract
The NF-κB signaling pathway is an attractive therapeutic target for cancer and chronic inflammatory diseases. In this study, we report the first strategy to achieve NF-κB inhibition with a peptide inhibitor loaded into perfluorocarbon nanoparticles with the use of a simple post-formulation mixing approach that utilizes an amphipathic cationic fusion peptide linker strategy for cargo insertion. A stable peptide-nanoparticle complex is formed (dissociation constant ~0.14 μM) and metered inhibition of both NF-κB signaling and downstream gene expression (ICAM-1) is demonstrated in leukemia/lymphoma cells. This post-formulation cargo loading strategy enables the use of a generic synthetic or biologic lipidic nanostructure for drug conjugation that permits flexible specification of types and doses of peptides and/or other materials as diagnostic or therapeutic agents for metered incorporation and cellular delivery.
Keywords: Nanoparticle, Peptide, Lipid, Membrane, Drug delivery, Inflammation
1. Introduction
NF-κB plays an essential role in the initiation and development of cancer and other chronic inflammatory diseases, by regulating a variety of genes that control inflammation, innate and adaptive immune response, cell cycle, and apoptosis [1–4]. As a mediator of oncogenic transformation, NF-κB is a well recognized target for anticancer treatment [5–7]. Also, NF-κB is required for proper immune cell function and, if depleted, can lead to inadequate surveillance against noxious agents and cancer [8]. Among signaling pathway inhibitors, peptide drugs represent a class of attractive therapeutic agents, because of their high specificity, affinity, activity, and low toxicity [9,10]. However, because of inadequate delivery approaches, rapid proteolytic inactivation and poor bioavailability have limited clinic applications of peptide drugs [10].
One such peptide candidate for NF-κB inhibition is the Nemo Binding Domain (NBD) inhibitory peptide that has been shown to control the signaling events that release NF-κB from the cytoplasmic compartment to translocate to the nucleus and stimulate inflammatory responses [11]. Although the NBD peptide has been fused to another cell penetrating peptide, antennapedia, and used to good effect in systemic applications such as muscular dystrophy [12], the potential lack of cellular specificity of a generalized cell penetrating peptide fusion construct begs the need for a more constrained delivery system. Therefore, we ultimately seek to develop a nanoparticle-based peptide therapeutic approach to the delivery of agents that would inhibit NF-κB but not result in complete suppression, instead eliciting a metered response that could be monitored and adjusted as needed depending on the condition being treated.
Previously, we have demonstrated that loading cationic amphipathic peptides such as the bee venom peptide melittin [13] into perfluorocarbon (PFC) nanoparticles, results in a 10-fold increase in the melittin circulation time while protecting the peptide from both destruction and off-target effects, which enables increased tumor cell delivery and dramatic inhibition of tumor growth [14]. The PFC nanoparticles, which comprise a hydrophobic perfluorocarbon (PFC) core surrounded by a lipid surfactant monolayer, have been used for molecular imaging and site-specific drug delivery in chronic inflammation diseases and cancer [12,15,16] making them a suitable platform for NBD peptide delivery in an effort to extend the circulating lifetime and enhancing therapeutic efficacy over the free peptide. In this work, we propose to utilize an amphipathic cationic peptide linker to incorporate therapeutic peptide into PFC nanoparticles for cellular delivery. This amphipathic cationic peptide linker is generated by a designed modification of melittin that eliminates melittin’s cytotoxicity but retains lipid membrane insertion function [17]. If successful, we anticipate that this cargo-peptide-nanoparticle system could serve as a paradigm for designing subsequent therapeutic peptide delivery approaches.
2. Materials and methods
2.1. Nanoparticle and Alexa Fluor 488 labeled peptide synthesis
Perfluorocarbon (PFC) nanoparticle emulsions were formulated using methods described previously [18]. Briefly, a lipid/surfactant co-mixture of 99 mol% egg lecithin and 1 mol% dipalmitoyl-phosphatidylethanolamine, DPPE (Avanti Polar Lipids, Piscataway, NJ) was dissolved in methanol:chloroform(1:3 in volume). Solvent was evaporated under reduced pressure to produce a lipid film, which was dried in a 50 °C vacuum oven overnight to obtain the surfactant. Then the surfactant (2.0%,w/v), perfluorooctyl bromide (PFOB) (Gateway Specialty Chemicals, St Peters, MO)(20%, w/v), and distilled, deionized water were blended and emulsified at 20,000 PSI for 4 min in an ice bath (S110 Microfluidics emulsifier, Microfluidics, Newton, MA). For fluorescent nanoparticles, Alexa Fluor 488 was incorporated into the surfactant layer. To incorporate NBD peptide (TALDWSWLQTE) or mutant NBD (mutNBD) peptide (TALDASALQTE) [11] into the PFC nanoparticles for protected delivery, we first fused NBD or mutNBD peptide onto the N-terminal of linker peptide (VLTTGLPALISWIKRKRQQ) [17] with two glycines added as a spacer. These combination peptides, “NBD-Linker” and “mutNBD-Linker”, were synthesized by GenScript Co (Piscataway, NJ). Conjugation of Alexa Flour 488 to the peptide NBD-Linker was carried out in solution. The TFP ester (2,3,5,6-tetrafluorophenyl) (Sigma, St. Louis, MO) of the dye was chosen for the labeling reaction since the TFP ester moiety is more stable in solution compared to the commonly used succinimidyl ester. The methodology for the labeling reaction is as follows: 5 mg of NBD-Linker was dissolved in 0.1 M of sodium bicarbonate buffer. The pH of the buffer was adjusted close to neutral (~7.3) to increase N-terminus selective labeling of the peptide. The required amount of the dye (1.84 mg, 1.5 equiv) was dissolved in 300 μL of DMF (N,N-Dimethylformamide). While stirring/vortexing the peptide solution, the dye solution was added slowly. The reaction was run in dark overnight. The completion of the reaction was monitored by analyzing an aliquot amount of the reaction mixture by reversed phase HPLC (C18 column, flow rate 0.7 ml/min) in 40–60% gradient of Acetonitrile (containing 0.075% TFA)/Water (containing 0.1% TFA). Based on HPLC, the reaction was ~87% complete. The labeled peptide was then purified on preparative HPLC (C18 column) following the same solvent gradient as mentioned above. The pure fractions were then lyophilized to obtain the final pure labeled peptide. Fluorescent labeling was further confirmed by FCS analysis.
2.2. Incorporation of NBD-Linker or mutNBD-Linker into PFC nanoparticles
NBD peptide or mutNBD peptide incorporated nanoparticles were formulated by mixing PFC nanoparticles with known amount of NBD-Linker or mutNBD-Linker, which was dissolved in MilliQ H2O at 10 mM. 1–4 μl 1 mM or 1–15 μl 10 mM NBD-Linker was added to 30 μl of PFC nanoparticles with mixing. After incubation at 4 °C overnight, the mixture was centrifuged at 100g for 10 min to remove unincorporated peptides. The peptide in the supernatant was quantified by measuring intrinsic tryptophan fluorescence (described below) with a standard curve.
2.3. Size distribution and zeta potential of nanoparticles
The size distributions of the nanoparticles with or without cargo incorporated were analyzed by dynamic light scattering (Brookhaven Instruments Corp., Holtsville, NY). The size distribution was plotted by particle number. Zeta potential (ζ) values were determined with a Brookhaven Instruments PALS Zeta Potential Analyzer (Brookhaven Instruments Corp., Holtsville, NY). Data were acquired in the phase-analysis light scattering (PALS) mode following solution equilibration at 25°C.
2.4. Electron microscopy
NBD-Linker incorporated PFC nanoparticles were depicted by transmission electron microscopy. Procedures have been described in detail previously [19].
2.5. Giant Unilamellar Vesicles (GUV) preparation and confocal microscopy
GUVs were prepared by the electroformation method [20] from a lipid mixture containing 99.9 mol% of 1,2-dioleoyl-sn-glycero-3-phosphocholine, DOPC (Avanti Polar Lipids, Piscataway, NJ) and 0.1 mol% of fluorescent dye DiD (Invitrogen, Molecular Probes, Carlsbad, CA). Briefly, chloroform mixture of lipids and dye at 2 mg/ml total lipid concentration was dried on a surface of two parallel platinum electrodes resulting in a creation of a thin lipid film on each electrode. Next, platinum electrodes were immersed into a chamber containing 300 mM sucrose solution and connected to a power generator. Electroformation of GUVs attached to the platinum electrodes was performed at 2.3 V and 10 Hz for 1 h at room temperature followed by the detachment of GUVs from the platinum electrodes done at 2.3 V, 2 Hz for 30 min. For GUV observation, 50 μl of solution containing GUVs was transferred into a Lab-Tek observation chamber (Fisher Scientific, Pittsburgh, PA) containing 450 μl of 10 mM HEPES, pH 7.2, 100 mM KCl and 20 μM of Alexa Fluor 546 (Invitrogen, Molecular Probes, Carlsbad, CA). Alexa Fluor 546 dye was used to assess the permeabilization of GUVs in the presence of mellitin with time. Observation of GUVs and confocal microscopy was done on Zeiss LSM 510 microscope (Zeiss, Thornwood, NY).
2.6. Fluorescence correlation spectroscopy (FCS) [21]
FCS is a quantitative technique, which detects fluorescence intensity fluctuations as fluorescent molecules diffuse through a small observation volume (<1 fL). Statistical analysis of these fluorescence intensity fluctuations allows simultaneous determination of the number of fluorescent species in the system and their mobility.
In our case the observation volume is defined by the focal volume of a laser scanning microscope, while the statistical analysis of the intensity trace is calculated as follows:
| (1) |
where G is the autocorrelation function, F is the fluorescence intensity as a function of time, τ is the correlation time and the angular brackets refer to time averaging, while δF(t) = F(t) − 〈F(t)〉.
The correlation curve obtained from the experiment is fitted with a mathematical function which describes the fluorescence intensity fluctuations in the observation volume according to Brownian diffusion and it also accounts for the photophysical characteristics of the dye:
| (2) |
where N is the average number of fluorescent particles in the observation volume, T is the fraction of fluorophores in the triplet state, τT is the lifetime of the triplet state of the fluorophore, τD is the characteristic diffusion time of the fluorophore, ω0 the waist radius of the laser focus. The structural parameter S = ωz/ω0measures the aspect ratio of observation volume which is assumed to have Gaussian shape. The diffusion time τD is related to the diffusion coefficient D through the expression:
| (3) |
The waist of the focus ω0 was determined by fitting the autocorrelation curve obtained in the same experimental conditions using free Alexa Fluor 488 [22]. In the case of labeled peptide binding to the nanoparticles the correlation curves were analyzed using the two component model as was described previously [23].
2.7. Surface plasmon resonance
The kinetics of NBD-Linker incorporation into lipid monolayers of PFC nanoparticles was studied by surface plasmon resonance (SPR). SPR detects change in the reflective index of a surface (Biacore-X 100 and carboxymethylated dextran chip L1 from Biacore Inc, Piscataway, NJ). A uniform lipid monolayer on an L1 chip was created by injecting PFC nanoparticles (3 μl/min) for 30 min. Loosely deposited nanoparticles were removed by performing extra washing steps after immobilization to ensure a stable baseline. Complete coverage was confirmed by injecting the bovine serum albumin (1 mg/ml in PBS) at 15 μl/min for 2 min. Different peptides in selected concentrations were injected at a flow rate of 30 μl/min for 1 min. At the end of each experiment the chip was regenerated by two consecutive injections of 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate, CHAPS (50 μl, 100 μl/min). The data were analyzed with BiaEvaluation software (Biacore Inc, Piscataway, NJ). A two-state model, [17].
2.8. Circular dichroism spectroscopy
Jasco J-810 spectropolarimeter (Jasco Inc, Eastern, MD) was utilized for CD spectra measurements of free NBD-Linker and lipid bound NBD-Linker (lipid–peptide molar ratio was 10:1). Spectra were scanned in a 1 mm path length quartz cuvette in the far-UV range from 190 to 260 nm at a scan rate of 100 nm/min and all spectra were collected under argon. An average 20 scans was used for all spectra. Buffer used was 10 mM potassium phosphate buffer pH 7.0.
2.9. Tryptophan fluorescence spectroscopy of NBD-Linker-nanoparticles
The proximity of NBD-Linker peptides with respect to the core perfluorocarbon structures can be defined by measuring their intrinsic tryptophan fluorescence. NBD-Linker contains three tryptophan residues at position 5, 7, and 25, which are potentially quenchable by the bromine atoms in the core perfluoroctylbromine material if in close proximity [17]. Tryptophan fluorescence emission spectra (300 nm–500 nm) were measured after excitation at 280 nm in a fluorescent spectrofluorometer (Varian Inc, Palo Alto, CA).
2.10. Cell culture
F8 cells (from the Lee Ratner laboratory) were maintained in a humidified atmosphere of 95% air and 5% CO2 in the cell culture medium: RPMI, 10% FBS, 4 mM glutamine, and 100 μ penicillin/100 μg/ml strep (WashU Tissue Culture Support Center, St. Louis, MO).
2.11. Transcription factor assays and Akt expression
After F8 cells were treated with NBD-Linker or mutNBD-Linker loaded nanoparticles at various concentrations, indicated in the Fig. 4, for 8 h, cytoplasmic and nuclear proteins were extracted by using a nuclear extract kit (Active Motif, Carlsbad, CA) following the manufacturer’s instruction. Protein concentrations were determined by the BCA protein assay (Pierce, Rockford, IL). Transcription factor assays were perform by using TransAMTM NF-κB p65 Transcription Fact Assay Kit (Active Motif, Carlsbad, CA), according to the manufacturer’s instruction. Total Akt (tAkt) and phosphorylated Akt (pAkt) expression were evaluated by ELISA (cell signaling technology, Boston, MA), according to the manufacturer’s instruction.
Fig. 4.

Inhibition of NF-kB signaling pathway by NBD-Linker incorporated PFC nanoparticles. (A). NBD-Linker incorporated PFC nanoparticles inhibit NF-kB protein (P65) translocation into the nucleus in a dose-dependent fashion. Data presented as mean ± s.d. (n = 3). (B). At a concentration of 30 μM, NBD-Linker loaded nanoparticles reduce P65 nuclear translocation by half, but mutNBD-Linker loaded nanoparticles do not inhibit P65 nuclear translocation. (C). Expression of NF-kB dependent gene (ICAM-1) was inhibited by NBD-Linker incorporated PFC nanoparticle treatment. The histograms were from one of six sets of independent experiments. Black and grey curves represent ICAM-1 expression with and without treatment for 9.5 h, respectively. (D). Bar graph of mean fluorescence intensity from ICAM 1 stained F8 cells without treatment and with treatment of either NBD-Linker or mutNBD-Linker nanoparticles at concentration of 30 μM. ICAM-1 expression is not significantly affected by mutNBD-Linker nanoparticles. (E–F). NBD-Linker incorporated PFC nanoparticles do not affect Akt signaling, an signaling pathway upstream of NF-κB. Total Akt (tAkt) (E) and phosphorylated Akt (pAkt) (F) levels do not differ significantly between F8 cells without or with treatment at selected concentrations. Data presented as mean ± STD (n = 6).
2.12. Flow cytometry analysis and antibodies
After F8 cells were treated with NBD-Linker loaded nanoparticles (30 μM) for 9.5 h, cells were incubated with FITC Hamster Anti-Mouse CD54 (BD Pharmingen, San Jose, CA) and staining buffer (HBSS, 2% FBS, and 1 mM EDTA) for 60 min on ice and then washed before analyzed on flow cytometry (CyAnTM ADP with Summit™ Software, Dako, Carpinteria, CA).
3. Results
3.1. Physical characterization of peptide-nanoparticle constructs
To generate NBD peptide loaded nanoparticles, we produced two discrete components: base PFC nanoparticles as delivery vehicles; and therapeutic NBD peptides fused onto the peptide linker. Base PFC nanoparticles, consisting of hydrophobic core surrounded by a lipid monolayer, are created according to our standard methods of formulation [18]. The NBD peptide is conjugated onto the N-terminal of the peptide linker with two glycines added as a spacer. The sequence of the new peptide (NBD-Linker) is shown in Fig. 1A. Controlled incorporation of NBD-Linker into PFC nanoparticles is achieved by mixing selected amounts of NBD-Linker with PFC nanoparticles. Free (unloaded) NBD-Linker was removed by gentle centrifugation (10 min @100g) and measured by tryptophan fluorescence to determine the amount of peptide that was retained on the nanoparticle. By varying the amount of NBD-Linker added to the nanoparticles, copies of NBD-Linker loaded into each nanoparticle ranged between 220 and 24,900. The mean diameters of PFC nanoparticles without and with various amount of peptide loading are equivalent (Fig. 1B). The morphology and size of nanoparticles loaded with NBD-Linker was also visualized by transmission electron microscopy with lipid membrane staining (Fig. 1C).
Fig. 1.

Characterization of NF-κB inhibiting PFC nanoparticles generated with the use of a linker peptide. (A). Sequence of the NBD peptide conjugated on the N-terminal of the linker peptide (Italic) with two Glycines. (B). Mean hydrodynamic diameter and zeta potential of nanoparticles with or without incorporation of NBD-Linker, respectively. (C). Transmission electron micrograph of PFC nanoparticles incorporated with NBD-Linker. Scale bar represents 250 nm (D). A schematic illustration of PFC nanoparticle with enlarged NBD-Linkers incorporated in the lipid monolayer.
Zeta potential represents the surface charge status of nanoparticles. The native PFC nanoparticles exhibit negative zeta potential at −13.74 ± 0.7 mV (Fig. 1B) due to the negative electron density of the phosphate head groups of the lipid monolayer. The NBD-Linker construct carries 3 net positive charges. Thus, we anticipate that the zeta potential of PFC nanoparticles would shift to more positive values after the peptides are loaded. As shown in Fig. 1B, the more peptide that is loaded, the more positive is the zeta potential on the NBD-Linker loaded PFC nanoparticles. At a loading of 24,900 copies of NBD-Linker peptides per nanoparticle, the NBD-Linker loaded PFC nanoparticles exhibit a zeta potential of + 30.18 ± 0.52 mV. The zeta potential change confirms the incorporation of NBD-Linker into PFC nanoparticles. The schematic illustration of the structure of the PFC nanoparticle with NBD-Linker incorporation in its lipid membrane is depicted in Fig. 1D.
3.2. NBD-Linker incorporation into the lipid membrane
To visually depict the lipid membrane incorporation of NBD-Linker into generic lipidic structures, we conjugated Alexa Fluor 488 Dye onto the NBD-Linker and mixed them with Giant Unilamellar Vesicles (GUV), which encapsulate a liquid core with a lipid bilayer. The fluorescence from the Alexa Fluor 488 Dye conjugated NBD-Linker (green rings in Fig. 2B) and that from lipophilic-stained (DiD dye) GUV (red rings in Fig. 2A) were co-localized (yellow rings in Fig. 2C). These results illustrate the integration of NBD-Linker into the lipid membrane of GUV.
Fig. 2.

Visualization of NBD-Linker incorporation into the lipid membrane. A–C. Confocal microscope images show NBD-Linker incorporated onto the lipid membrane of Giant Unilamellar Vesicles (GUV). (A). Confocal image of GUV with membrane labeled with lipophilic dye DiD (red rings). (B). Confocal image of Alexa Fluor 488 labeled NBD-Linker (green rings). (C). Co-localization of Alexa Fluor 488 labeled NBD-Linker and the lipid membrane of GUV (yellow rings). (D). Schematic of the FCS observation volume formed by the focused laser beam (~1 fL). (E). Normalized autocorrelation curves for Alexa Fluor 488, Alexa Fluor 488 labeled NBD-Linker (NBD-Linker-488), nanoparticles incorporated with labeled peptide (NP–NBD–Linker-488). (F). Diffusion time of Alexa Fluor 488, Alexa Fluor 488 labeled NBD-Linker (NBD-Linker-488), nanoparticles incorporated with labeled peptide (NP–NBD–Linker-488), and nanoparticles formulated with Alexa Fluor 488 conjugated lipids. Data presented as mean ± STD (n = 3).
To further illustrate the incorporation of the NBD-Linker into the lipid monolayer of nanoparticles, we employed Fluorescence Correlation Spectroscopy (FCS), which is a single-molecule sensitive fluorescence technique permitting high-accuracy determination of the diffusion coefficients of fluorescently labeled particles in solution [23–25]. The diffusion coefficient of a particle characterizes the mobility of a particle in solution, and it is inversely proportional to the characteristic diffusion timeτD, which is directly measured by FCS. τD represents the average time the particle spends in the FCS detection volume (illustrated as green oval in Fig. 2D) created by a focused laser beam. The longer the FCS diffusion time of a particle, the bulkier it is. By fitting the autocorrelation curve obtained in the experiments, two diffusion time components were detected by FCS analysis from the mixture of Alexa Fluor 488 labeled NDB-Linker and PFC nanoparticles. One component exhibited the same diffusion time as the Alexa Fluor 488 conjugated NBD-Linker, and the other component manifested a comparable diffusion time to that of PFC nanoparticles containing Alexa Fluor 488 conjugated lipid. Normalized autocorrelation curves and the fittings of Alexa Fluor 488, Alexa Fluor 488 labeled NDB-Linker, and Alexa Fluor 488 labeled NDB-Linker plus PFC nanoparticles are shown in Fig. 2E. In addition, analysis of the FCS diffusion times shows that the diffusion time of Alexa Fluor 488 conjugated NBD-Linker (67.2 ± 1.8 μs) is about two times slower than the diffusion time of the Alexa Fluor 488 (35.8 ± 1.4 μs). After achieving Alexa Fluor 488 labeled NDB-Linker incorporation, the resultant fluorescent nanoparticles exhibited strikingly longer diffusion times (1300 ± 100 μs), which is comparable to the diffusion time of the Alexa Fluor 488 lipid labeled nanoparticle (1000 ± 100 μs), as shown in Fig. 2F.
3.3. Mechanism of NBD-Linker and PFC interaction
To investigate the release rate of the NBD-Linker from the PFC nanoparticles, surface plasma resonance experiments were performed with a Biacore X 100, which allows quantitative analysis of interaction between peptide and lipid membrane [26]. Consistent with our previous study [19], immobilization of PFC nanoparticles onto the L1 sensor chip surface results in maximal response of 4000 RU, which implies complete coverage of the senor surface by nanoparticles. NBD-Linker peptide at selected concentrations was injected at 30 μl/min for 1 min into the detection flow cell, which contains a volume of 0.06 μl. After injection, a 1-h wash with running buffer followed. The kinetics of interaction between NBD-Linker and immobilized PFC nanoparticles are illustrated by the sensorgrams shown in Fig. 3A. We consistently recorded two populations of peptides in the peptide–lipid interactions. One population is loosely attached to the PFC nanoparticles and is quickly washed away. The other population is tightly incorporated into the PFC nanoparticles, and remains associated with PFC nanoparticles after 1 h washing. The dissociation constant of the entire process is 0.14 μM. Also, the more NBD-Linker is injected, the more NBD-Linker remains with PFC nanoparticles (Fig. 3B).
Fig. 3.

Mechanism of NBD-Linker incorporation into the lipid membrane of PFC nanoparticles. (A). Incorporation of NBD-Linker into the PFC nanoparticles. Sensorgram, acquired by BIAcore X100, depicts the kinetics of the NBD-Linker incorporation into the PFC nanoparticles, which are immobilized on the surface of an L1 sensor chip. The NBD-Linker concentrations were 0.8, 1, 2, 5, and 10 μM (B). Stable incorporation of NBD-Linker incorporation onto PFC nanoparticles at various loading concentrations. (C). Secondary structural change of NBD-Linker after lipid insertion, which was measured by circular dichroism spectroscopy. Free NBD-Linker (light grey) presented unordered structure; while lipid bounded NBD-Linker (black) adopted α-helical structure. (D). Relative location of Tryptophans of NBD-Linker in PFC nanoparticles. Fluorescence emission spectra demonstrating both quenching and blue shift of endogenous tryptophan fluorescence of NBD-Linker. NBD-Linker concentration was 40 μM. The lipid:peptide ratio was 10.
By measuring the circular dichroism (CD) spectra of free NBD-Linker and PFC nanoparticle incorporated NBD-Linker, the results show that NBD-Linker peptide retains α-helical secondary structure after the incorporation into the PFC nanoparticles. In Fig. 3C, the CD spectrum of free peptide presents a strong negative peak near 200 nm and another negative band near 220 nm, which suggests the random coil unordered form. Conversely, the CD spectrum of the incorporated peptide exhibits two minimum at 222 and 208 nm and a maximum between 190 and 195 nm, which demonstrates the α-helical secondary structure.
To define the relative location (or proximity) of the NBD-Linker with respect to the core perfluorocarbon structures, we created PFC nanoparticles with a perfluorooctyl bromide (PFOB) core, the bromine atoms of which would be expected to quench the fluorescent tryptophan atoms in the NBD-Linker by Förster resonance energy transfer (FRET), if they are sufficiently close. It is also known that when tryptophan residue inserted into the lipids without quencher, its endogenous fluorescence emission spectrum exhibits a blue shift due to the hydrophobic environment [27]. The tryptophan emission spectra of free NBD-Linker and the NBD-Linker mixed with PFOB nanoparticles are measured and shown in Fig. 3D. The results demonstrated that the addition of the nanoparticles resulted in both quenching and blue shift (leftward peak) of the tryptophan emission spectra from the incorporated linker peptides, with the residual tryptophan signal emanating from the remaining free surrounding peptide.
3.4. Metered NF-κB signaling modulation
F8 cells were treated with PFC nanoparticles loaded with selected amounts of the NBD-Linkers to evaluate the dose dependence response. After treatment, nuclear proteins were extracted from the F8 cells to determine the amount of NF-κB in the nucleus. The results demonstrated that NBD-Linker incorporated PFC nanoparticles inhibited the nuclear translocation of the P65, NF-κB protein. Furthermore, the higher the dose, the less of P65 is measured in the extracted nuclear protein (Fig. 4A). To further confirm that the inhibition effect is induced by NBD, we generated mutant NBD (mutNBD, see sequence in Materials and methods) loaded nanoparticles by using the peptide linker. Nanoparticles loaded with mutNBD did not affect P65 nuclear translocation (Fig. 4B). To check functional inhibition of gene transcription and translation, we examined responses of ICAM-1 level, which is expressed on the surface of the F8 cells in response to NF-κB mediation [28]. The results indicate clearly that the ICAM-1 expression is suppressed by the treatment with NBD-Linker incorporated PFC nanoparticles (Fig. 4C). The mean fluorescence intensity from the F8 cells without treatment or treated with mutNBD-Linker loaded nanoparticles (162.83 ± 9.99 or 146.31 ± 3.62415, respectively) is more than 2-fold stronger than that from the F8 with the treatment of NBD-Linker loaded nanoparticles (73.29 ± 3.56) (Fig. 4D). The Akt signaling pathway is upstream of NF-κB. We show that NBD incorporated nanoparticles do not affect either total Akt (tAkt) or phosphorylated Akt (pAkt) level in F8 cells (Fig. 4E, F).
4. Discussion
In this study, we reported a new peptide-nanoparticle delivery system for therapeutic peptide drug delivery by using PFC nanoparticles, which are dual functional vehicles for noninvasive imaging and site-specific drug delivery [15,16,18,29]. Our results showed that the PFC nanoparticle size and integrity are not affected by NBD-Linker loading, while the surface charge of the NBD-Linker loaded PFC nanoparticles is shifted to the more positive range. Furthermore, we employed fluorescence probe to visualize the lipid loading of NBD-Linker. Scanning confocal microscopy confirmed colocalization of NBD-Linker with the lipid membrane of the GUV and demonstrated that the lipid membrane barrier was intact after the incorporation of the peptide drug. Moreover, FCS results demonstrate that the mobility of the nanoparticles is not affected by the loading of the NBD-Linker peptides. These results verified incorporation of NBD-Linker into the lipid membrane of the delivery vehicle, and the integrity of the PFC nanoparticles retained after the peptide incorporation.
After confirming NBD-Linker incorporation into the lipid membrane of PFC nanoparticles, we studied the mechanism of peptide drug loading by investigating peptide lipid interaction. With the use of SPR to study the kinetics of peptide drug loading, we found that the NBD-Linker interacts with lipid membrane by both electrostatic and hydrophobic interaction, which is consistent with previous report [30]. One population, which interacts with PFC nanoparticles through electrostatic interaction, is loosely attached to the PFC nanoparticles and is quickly washed away. The other population, which interacts with PFC nanoparticles through hydrophobic interaction, is tightly incorporated into the PFC nanoparticles, and remains associated with PFC nanoparticles after 1 h washing. Considering that the volume of the testing chamber is 0.06 μl, and the chamber was washed with 3600 μl of running buffer at constant flow rate, these results suggest that the NDB-Linker incorporation is quite stable. Consistently, as the amphipathic peptides integrate into the lipid membrane, they undergo conformational change and assume an α-helical secondary structure. The hydrophobic interaction between α-helical segment of the peptide and the surrounding lipid induce a negative free energy change, which maintains system stability [30]. This α-helical formation of membrane incorporated NBD-Linker was confirmed by the CD spectra.
Previously, we demonstrated that the tryptophan fluorescence of melittin is quenched after melittin incorporated into the nanoparticles with PFOB core, which indicates that the tryptophan residue of melittin is located at the tail region of the lipid monolayer and very close to the PFOB core of the nanoparticles [19]. After NBD-Linker incorporated into the lipid membrane of PFOB nanoparticles, the tryptophan fluorescence spectra exhibited both quenching and blue shift. These results suggested that some of the tryptophan residues on the NBD-Linker are located close to the core of the PFOB nanoparticles, while others settle in the lipid membrane away from the hydrophobic core. As illustrated in Fig. 5, the NBD-Linker has three tryptophan residues. After NBD-Linker peptides incorporated into the lipid membrane and formed the α-helic secondary structure, two of the tryptophan residues face the same direction, which could be in proximity to the core of the PFC nanoparticle, while the third one could face the opposite direction in the lipid membrane but away from the core and not quenched by the bromine.
Fig. 5.
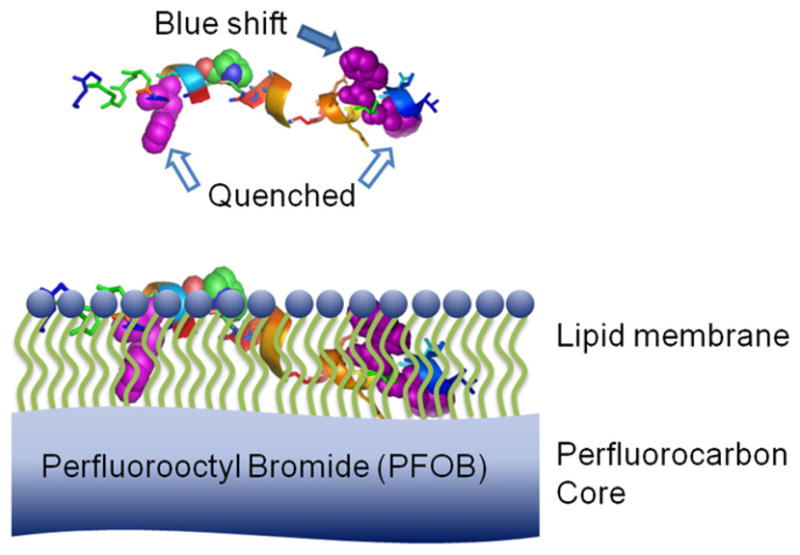
A schematic of relative location of NBD-Linker incorporated into lipid membrane of PFC nanoparticles. Top: NBD-Linker in α-helical structure. Three tryptophans are highlighted with purple spheres and pointed by arrows. Bottom: NBD-Linker incorporated into the lipid monolayer of PFOB nanoparticle. Two tryptophans (white arrows), are close to the PFOB core. The fluorescence of these two trypophans was quenched by PFOB; while the third Tryptophan in the lipid membrane was away from the PFOB core, which contributed to the blue shift of the tryptophan emission spectra.
Next, the functional activity of the PFC nanoparticles loaded with NBD-Linker was assessed by investigating NF-κB activation, namely NF-κB nuclear translocation and the expression of the NF-κB dependent genes. To evaluate the therapeutic effect of this NF-κB modulating nanoparticle, we employed a leukemia/lymphoma cell line (F8), which manifests constitutive NF-κB activation [31]. This cell line is derived from a large granular lymphocytic (LGL) leukemia transgenic mice model [32], which faithfully represents the leukemia/lymphoma induced by Tax promoter expression, resulting in morphology and phenotype of F8 cells in vitro that are indistinguishable from those observed in vivo [28,32]. Tax, a transactivator protein, is encoded in the genome of HTLV-1, and is critical in promoting transformation of the cells [10,33]. In F8 cells with Tax expression, NF-κB is activated constitutively [34].
Under physiological conditions, NF-κB is sequestered in the cytoplasm as an inactive complex with the inhibitory protein IκB. Under pathological conditions, such as infection, IκB is phosphorylated by the activated IKK complex. This phosphorylation results in unbiquitination and degradation of IκB. Consequently, NF-κB is free to translocate to the nucleus where it initiates the expression of NF-κB dependent genes. The IKK complex consists of two catalytic subunits subunits, IKKα/IKKβ, and one regulatory subunit, IKKγ (also known as NEMO) [35–37]. By interrupting the interaction between IKKγ and the NEMO Binding Domain (NBD) on the carboxyl-terminal of IKKα/IKKβ, the activation of IKK complex can be inhibited, which in turn prohibits the subsequent phosphorylation of IκB and activation of NF-κB. Thus, the NBD peptide inhibits NF-κB activation by preventing subunit interactions in the IKK complex, while mutNBD peptide is not capable of doing so [11].
If the activation of the NF-κB signaling pathway is inhibited, less NF-κB will translocate into the nucleus. For NF-κB signaling, P65 is the resultant transcription factor controlling gene expression in classical NF-κB activation. Thus, we compared the nuclear P65 between F8 cells without and with the inhibitory treatment at different dosages. The observed dose-dependent suppression of the nuclear levels of constitutively activated NF-κB is precisely the desired outcome. At the same time, this NF-κB inhibitory nanoparticle did not affect the upstream signaling pathway, e.g. Akt signaling. These results suggest a specific inhibitory effect of NBD incorporated nanoparticles on NF-κB activation, and validate this peptide-nanoparticle as a promising delivery system.
5. Conclusion
In summary, we have formulated, characterized, and evaluated the function of a nanoparticle that inhibits NF-κB activation by using a flexible cargo linker peptide. One of the challenges in formulating therapeutic peptides into the delivery vehicles is the preservation of the activity of the peptide throughout the formulation procedures. The peptide linker strategy proposed here enables the addition of therapeutic peptide into the nanoparticles after their formulation. Therefore, therapeutic peptide could be loaded on the delivery nanoparticles in a simple mixing step without encountering harsh particle formulation steps, and the activity of the therapeutic peptide remains intact, which should facilitate sterile preparation of such compounds. This peptide linker strategy in conjunction with lipidic nanodelivery vehicles, either wholly synthetic or native, exemplifies a promising method of delivering small peptides for signaling pathway modulation.
Acknowledgments
The authors would like to thank Ralph Fuhrhop for the base PFC nanoparticle formulation. This work was supported by the National Institutes of Health Grants (HL073646 and CA119342 to Samuel A. Wickline; CA10073, CA94056, and CA63417 to Lee Ratner), and Leukemia & Lymphoma Society grant (LLS6067-10 to Lee Ratner).
Appendix
Figures with essential color discrimination. Figures 1,2 & 5 in this article are difficult to interpret in black and white. The full color images can be found in the on-line version, at doi:10.1016/j. biomaterials.2010.08.080.
References
- 1.Karin M, Cao Y, Greten FR, Li ZW. NF-kappaB in cancer: from innocent bystander to major culprit. Nat Rev Cancer. 2002;2:301–10. doi: 10.1038/nrc780. [DOI] [PubMed] [Google Scholar]
- 2.Karin M. Nuclear factor-kappaB in cancer development and progression. Nature. 2006;441:431–6. doi: 10.1038/nature04870. [DOI] [PubMed] [Google Scholar]
- 3.Pasparakis M. Regulation of tissue homeostasis by NF-kappaB signalling: implications for inflammatory diseases. Nat Rev Immunol. 2009;9:778–88. doi: 10.1038/nri2655. [DOI] [PubMed] [Google Scholar]
- 4.Bhoj VG, Chen ZJ. Ubiquitylation in innate and adaptive immunity. Nature. 2009;458:430–7. doi: 10.1038/nature07959. [DOI] [PubMed] [Google Scholar]
- 5.Garg A, Aggarwal BB. Nuclear transcription factor-kappaB as a target for cancer drug development. Leukemia. 2002;16:1053–68. doi: 10.1038/sj.leu.2402482. [DOI] [PubMed] [Google Scholar]
- 6.Baud V, Karin M. Is NF-kappaB a good target for cancer therapy? hopes and pitfalls. Nat Rev Drug Discov. 2009;8:33–40. doi: 10.1038/nrd2781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lopez-Guerra M, Colomer D. NF-kappaB as a therapeutic target in chronic lymphocytic leukemia. Expert Opin Ther Targets. 2010;14:275–88. doi: 10.1517/14728221003598930. [DOI] [PubMed] [Google Scholar]
- 8.Smale ST. Selective transcription in response to an inflammatory stimulus. Cell. 2010;140:833–44. doi: 10.1016/j.cell.2010.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bellmann-Sickert K, Beck-Sickinger AG. Peptide drugs to target G protein-coupled receptors. Trends Pharmacol Sci. doi: 10.1016/j.tips.2010.06.003. In Press. [DOI] [PubMed] [Google Scholar]
- 10.Bidwell GL, 3rd, Raucher D. Therapeutic peptides for cancer therapy. Part I -peptide inhibitors of signal transduction cascades. Expert Opin Drug Deliv. 2009;6:1033–47. doi: 10.1517/17425240903143745. [DOI] [PubMed] [Google Scholar]
- 11.May MJ, D’Acquisto F, Madge LA, Glockner J, Pober JS, Ghosh S. Selective inhibition of NF-kappaB activation by a peptide that blocks the interaction of NEMO with the IkappaB kinase complex. Science. 2000;289:1550–4. doi: 10.1126/science.289.5484.1550. [DOI] [PubMed] [Google Scholar]
- 12.Acharyya S, Villalta SA, Bakkar N, Bupha-Intr T, Janssen PM, Carathers M, et al. Interplay of IKK/NF-kappaB signaling in macrophages and myofibers promotes muscle degeneration in Duchenne muscular dystrophy. J Clin Invest. 2007;117:889–901. doi: 10.1172/JCI30556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weissmann G, Hirschhorn R, Krakauer K. Effect of melittin upon cellular and lysosomal membranes. Biochem Pharmacol. 1969;18:1771–5. doi: 10.1016/0006-2952(69)90167-1. [DOI] [PubMed] [Google Scholar]
- 14.Soman NR, Baldwin SL, Hu G, Marsh JN, Lanza GM, Heuser JE, et al. Molecularly targeted nanocarriers deliver the cytolytic peptide melittin specifically to tumor cells in mice, reducing tumor growth. J Clin Invest. 2009;119:2830–42. doi: 10.1172/JCI38842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Caruthers SD, Cyrus T, Winter PM, Wickline SA, Lanza GM. Anti-angiogenic perfluorocarbon nanoparticles for diagnosis and treatment of atherosclerosis. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2009;1:311–23. doi: 10.1002/wnan.9. [DOI] [PubMed] [Google Scholar]
- 16.Kaneda MM, Caruthers S, Lanza GM, Wickline SA. Perfluorocarbon nanoemulsions for quantitative molecular imaging and targeted therapeutics. Ann Biomed Eng. 2009;37:1922–33. doi: 10.1007/s10439-009-9643-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pan H, Myerson JW, Ivashyna O, Soman NR, Marsh JN, Hood JL, et al. Lipid membrane editing with peptide cargo linkers in cells and synthetic nanostructures. Faseb J. 2010;24:2928–37. doi: 10.1096/fj.09-153130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lanza GM, Winter PM, Caruthers SD, Hughes MS, Cyrus T, Marsh JN, et al. Nanomedicine opportunities for cardiovascular disease with perfluorocarbon nanoparticles. Nanomedicine (Lond) 2006;1:321–9. doi: 10.2217/17435889.1.3.321. [DOI] [PubMed] [Google Scholar]
- 19.Soman NR, Lanza GM, Heuser JM, Schlesinger PH, Wickline SA. Synthesis and characterization of stable fluorocarbon nanostructures as drug delivery vehicles for cytolytic peptides. Nano Lett. 2008;8:1131–6. doi: 10.1021/nl073290r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Angelova MI, Dimitrov SD. Liposome electroformation. Faraday Discuss Chem Soc. 1986;81:303–11. [Google Scholar]
- 21.Rigler R, Elson ES, editors. Fluorescence correlation spectroscopy: theory and applications. Berlin: Springer; 2001. [Google Scholar]
- 22.Petrasek Z, Schwille P. Precise measurement of diffusion coefficients using scanning fluorescence correlation spectroscopy. Biophys J. 2008;94:1437–48. doi: 10.1529/biophysj.107.108811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rhoades E, Ramlall TF, Webb WW, Eliezer D. Quantification of alpha-synuclein binding to lipid vesicles using fluorescence correlation spectroscopy. Biophys J. 2006;90:4692–700. doi: 10.1529/biophysj.105.079251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bacia K, Schwille P. Fluorescence correlation spectroscopy. Methods Mol Biol. 2007;398:73–84. doi: 10.1007/978-1-59745-513-8_7. [DOI] [PubMed] [Google Scholar]
- 25.Hess ST, Huang S, Heikal AA, Webb WW. Biological and chemical applications of fluorescence correlation spectroscopy: a review. Biochemistry. 2002;41:697–705. doi: 10.1021/bi0118512. [DOI] [PubMed] [Google Scholar]
- 26.Torreri P, Ceccarini M, Macioce P, Petrucci TC. Biomolecular interactions by surface plasmon resonance technology. Ann Ist Super Sanita. 2005;41:437–41. [PubMed] [Google Scholar]
- 27.Lakowicz JR. Principles of fluorescence spectroscopy. 2. New York: Kluwer Academic/Plenum Press; 1999. [Google Scholar]
- 28.Grossman WJ, Ratner L. Cytokine expression and tumorigenicity of large granular lymphocytic leukemia cells from mice transgenic for the tax gene of human T-cell leukemia virus type I. Blood. 1997;90:783–94. [PubMed] [Google Scholar]
- 29.Winter PM, Cai K, Caruthers SD, Wickline SA, Lanza GM. Emerging nanomedicine opportunities with perfluorocarbon nanoparticles. Expert Rev Med Devices. 2007;4:137–45. doi: 10.1586/17434440.4.2.137. [DOI] [PubMed] [Google Scholar]
- 30.Klocek G, Schulthess T, Shai Y, Seelig J. Thermodynamics of melittin binding to lipid bilayers. Aggregation and pore formation. Biochemistry. 2009;48:2586–96. doi: 10.1021/bi802127h. [DOI] [PubMed] [Google Scholar]
- 31.Bernal-Mizrachi L, Lovly CM, Ratner L. The role of NF-{kappa}B-1 and NF-{kappa}B-2-mediated resistance to apoptosis in lymphomas. Proc Natl Acad Sci U S A. 2006;103:9220–5. doi: 10.1073/pnas.0507809103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Grossman WJ, Kimata JT, Wong FH, Zutter M, Ley TJ, Ratner L. Development of leukemia in mice transgenic for the tax gene of human T-cell leukemia virus type I. Proc Natl Acad Sci U S A. 1995;92:1057–61. doi: 10.1073/pnas.92.4.1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Boxus M, Twizere JC, Legros S, Dewulf JF, Kettmann R, Willems L. The HTLV-1 Tax interactome. Retrovirology. 2008;5:76–99. doi: 10.1186/1742-4690-5-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sun SC, Ballard DW. Persistent activation of NF-kappaB by the tax transforming protein of HTLV-1: hijacking cellular IkappaB kinases. Oncogene. 1999;18:6948–58. doi: 10.1038/sj.onc.1203220. [DOI] [PubMed] [Google Scholar]
- 35.Karin M. The beginning of the end: IkappaB kinase (IKK) and NF-kappaB activation. J Biol Chem. 1999;274:27339–42. doi: 10.1074/jbc.274.39.27339. [DOI] [PubMed] [Google Scholar]
- 36.Rothwarf DM, Zandi E, Natoli G, Karin M. IKK-gamma is an essential regulatory subunit of the IkappaB kinase complex. Nature. 1998;395:297–300. doi: 10.1038/26261. [DOI] [PubMed] [Google Scholar]
- 37.Yamaoka S, Courtois G, Bessia C, Whiteside ST, Weil R, Agou F, et al. Complementation cloning of NEMO, a component of the IkappaB kinase complex essential for NF-kappaB activation. Cell. 1998;93:1231–40. doi: 10.1016/s0092-8674(00)81466-x. [DOI] [PubMed] [Google Scholar]