

Abstract
CK syndrome (CKS) is an X-linked recessive intellectual disability syndrome characterized by dysmorphism, cortical brain malformations, and an asthenic build. Through an X chromosome single-nucleotide variant scan in the first reported family, we identified linkage to a 5 Mb region on Xq28. Sequencing of this region detected a segregating 3 bp deletion (c.696_698del [p.Lys232del]) in exon 7 of NAD(P) dependent steroid dehydrogenase-like (NSDHL), a gene that encodes an enzyme in the cholesterol biosynthesis pathway. We also found that males with intellectual disability in another reported family with an NSDHL mutation (c.1098 dup [p.Arg367SerfsX33]) have CKS. These two mutations, which alter protein folding, show temperature-sensitive protein stability and complementation in Erg26-deficient yeast. As described for the allelic disorder CHILD syndrome, cells and cerebrospinal fluid from CKS patients have increased methyl sterol levels. We hypothesize that methyl sterol accumulation, not only cholesterol deficiency, causes CKS, given that cerebrospinal fluid cholesterol, plasma cholesterol, and plasma 24S-hydroxycholesterol levels are normal in males with CKS. In summary, CKS expands the spectrum of cholesterol-related disorders and insight into the role of cholesterol in human development.
Main Text
X-linked intellectual disability (XLID) disorders account for 16% of all intellectual disabilities in males.1 This high frequency arises in part because males, unlike females, have only one X chromosome. To date, 91 genes involved in XLID have been cloned with demonstrated causative mutations and another 35 XLID syndromes have been mapped.2 Despite this progress, ∼50% of affected families lack an identified causative mutation and thus remain undiagnosed.3
CK syndrome (CKS) is a recently described XLID disorder named after the first reported patient.4 It is characterized by mild to severe cognitive impairment, seizures beginning in infancy, microcephaly, cerebral cortical malformations, and a thin body habitus.4 Distinctive features include downslanting palpebral fissures, a high nasal bridge, a high arched palate, micrognathia, and disproportionate short stature without distinctive radiographic findings (Figure S1, available online). Affected males also have behavior problems, including aggression, attention deficit hyperactivity disorder, and irritability.4
Using DNA from the first described family,4 we performed an X chromosome single-nucleotide variant (SNV) scan of family members giving consent to the protocol (H07-02142), approved by the clinical research ethics board at the University of British Columbia. We identified linkage to Xq28 (Figure 1). Two-point linkage analysis was performed with MLink from the FASTLINK software package, version 4.0P,5 and the Allegro program, version 1.1b.6 Multipoint linkage analysis was performed with the Allegro program. The maximum two-point and multipoint LOD scores were, respectively, 1.43 (θ = 0) and 2.29 (θ = 0) with marker rs941400. Haplotype and microsatellite analysis narrowed the interval to 5 Mb, marker DXS1684 to the telomere (Figure 1). Analysis of the 36.5 NCBI build of the human genome sequence revealed 133 positional candidate genes, expressed sequence tags (ESTs), and noncoding RNAs annotated within this region.
Figure 1.

Family 1 Pedigree, Linkage Analysis, DNA Sequencing Results, and NSDHL Mutation
(A) A five-generation family segregating CKS in males.4 The propositus is indicated by an arrow. Affected males are indicated in black. Female carriers of the NSDHL mutation are indicated by a black dot in the center. Females I-2, II-2, and II-4 are assumed obligate carriers. X chromosome linkage analysis identified a critical disease locus on Xq28 (red bar). The maximum two-point (1.43, θ = 0) and multipoint (2.29, θ = 0) LOD scores were at marker rs941400 (highlighted).
(B) The SNVs (left) and indels (right) identified in each individual in short-read analysis. SNVs and indels affecting the coding sequence are in parentheses; those events unique to the individual are indicated in red. The single indel unique to the propositus V-3 was in NSDHL.
(C) Diagram of the NSDHL showing coding (orange) and noncoding (light blue) exons. The exons showing homology to the steroid dehydrogenases are underlined in red.
(D) Chromatograms showing the NSDHL c.696_698delGAA mutation segregating with CKS in family 1. The predicted protein translation is shown above chromatograms.
(E) Lysine 232 is not conserved across species but lies between well-conserved amino acids.
By long-range PCR we amplified 1,535,643 bp of genomic sequence containing 111,381 bp of coding sequence and used Illumina reversible terminator-based sequencing to sequence the amplicons. Sufficient sequence coverage for unambiguously identifying variants was obtained for 85.3%, 80.6%, and 87.5% of the coding sequence in individuals V-3, III-4, and IV-11, respectively (Figure S2). Analysis identified a total of 6200 SNVs and 581 indels (Figure 1). Of the SNVs, 5106 were not found in four reference genomes.7–10 Capillary resequencing of 44,925 bp confirmed 86% of the SNV and indel observations. Of the 1347 SNVs and one indel unique to the proband, one SNV and one indel met the following criteria: (1) is absent from dbSNP, (2) is confirmed by capillary sequencing, (3) changes an amino acid change, and (4) segregates with CKS. The SNV was a mutation in F8 (c.1064G>A [p.Arg355Gln]), which encodes the blood coagulation factor 8 associated with hemophilia A. However, this mutation was considered clinically irrelevant because these males do not have bleeding problems. The indel was in exon 7 of NAD(P) dependent steroid dehydrogenase-like (NSDHL [MIM 300275]) (NM 015922.1:c.696_698del [p.Lys232del]) (Figure 1). The NSDHL mutation was not observed in 150 North American control chromosomes or in the 357 genomes evaluated for indels as part of the 1000 Genomes Project. We did not observe NSDHL mutations among 79 males (58 syndromic and 21 nonsyndromic) with intellectual disability (Table S1). During the course of our studies, however, Tarpey et al.11 reported that 1 of 208 families with X-linked intellectual disability had an NSDHL mutation (c.1098dup [p.Arg367SerfsX33, reported as p.R367fsX31 by Tarpey et al.11]) (Figure 2). Careful clinical evaluation of this family by F.L.R. showed that the p.Arg367SerfsX33 mutation, which extends the protein past the native stop codon and into the 3′ untranslated region (Figure 2), also causes CKS in this family (Figure 3).
Figure 2.
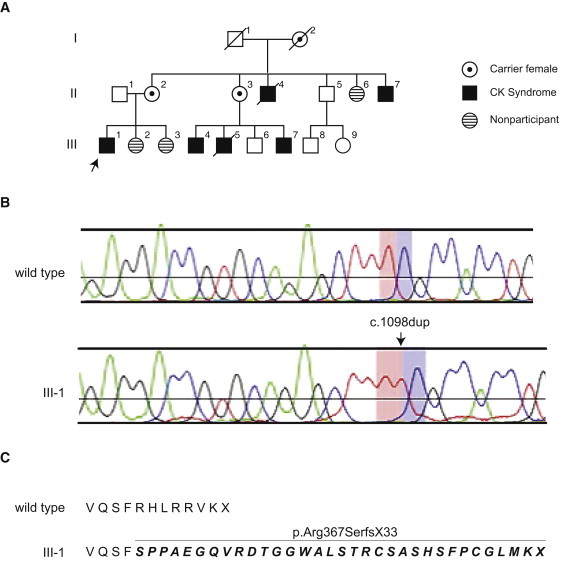
Family 2 Pedigree and NSDHL Mutation
(A) A three-generation family identified by Tarpey et al.11 segregating CKS4 in males. The propositus is indicated by an arrow. Affected males are indicated in black. Female carriers of the NSDHL mutation are indicated by a black dot in the center. X chromosome linkage analysis identified a critical disease locus on Xq28 with a LOD score of 1.06.
(B) Chromatograms showing the NSDHL c.1098dup mutation segregating with CKS.
(C) The predicted protein translation showing that the frameshift extends the protein past the native stop codon and into the 3′ untranslated region.
Figure 3.

Males Affected with CKS
(A) Affected males from family 1 (V-3 and IV-8) hemizygous for the c.696_698del (p.Lys232del) NSDHL mutation and from family 2 (III-1, III-4, III-7, and II-7) hemizygous for the c.1098dup (p.Arg367SerfsX33) NSDHL mutation.
(B) Summary of clinical features in males with CKS from families 1 and 2.
The NSDHL enzyme, which localizes to the surface of the endoplasmic reticulum and lipid droplets, is a C4 demethylase involved in postsqualene cholesterol biosynthesis.12–14 Because CKS males and their mothers had normal plasma cholesterol, steroid hormone levels, and lipoprotein profiles (Table 1), we cultured lymphoblastoid cells expressing p.Lys232del or p.Arg367SerfsX33 NSHDL in cholesterol-poor medium and measured sterols as described.15 Although of lesser severity, the sterol aberrations were similar to those reported for the allelic disorder congenital hemidysplasia with ichthyosiform nevus and limb defects syndrome (CHILD [MIM 308050]) (Figure 4) (R.I.K., unpublished data) and in mice with Nsdhl mutations.16 The aberrations include accumulation of 4-methyl sterol intermediates, 4,4-dimethyl sterol intermediates, lathosterol, and desmosterol.16
Table 1.
Serum Cholesterol, Lipoprotein, and Sterol Profiles for Members in Family 1 and Family 2
|
p.Lys232del NSDHL Positive |
p.Arg367SerfsX33 NSDHL Positive |
p.Lys232del NSDHL Negative |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| III-2 | III-5 | III-8 | IV-2 | IV-6 | IV-7 | IV-8 | V-1a | V-2 | V-3 | II-2 | III-1 | IV-10 | IV-11 | |
| Sex | F | F | F | F | M | F | M | F | F | M | F | M | F | M |
| Age (yrs) | 67 | 53 | 65 | 43 | 11 | 16 | 22 | 23 | 21 | 19 | 41 | 21 | 42 | 44 |
| X inactivation ratiob | 69:31 | 89:11 | 86:14 | 71:29 | NR | NR | NR | 70:30 | 90:10 | NR | NR | NR | NR | NR |
| Cholesterol (mg/dL) | 183.7 | 384.4 | 230.9 | 181.0 | 164.3 | 225.1 | 228.5 | 266.8 | 169.4 | 138.8 | 176.7 | 182.5 | 238.2 | 176 |
| 24S (ng/ml) | 48.5 | 31.1 | NR | 43.6 | NR | 80.8 | 70.9 | 62.1 | 65.6 | 46.4 | NR | NR | NR | 40.6 |
|
Lipoproteins | ||||||||||||||
| LDL (mg/dL) | 107.5 | 266.1 | 129.9 | 95.9 | 99.0 | 150.4 | 163.6 | 139.2 | 99.8 | 86.2 | 106 | 115 | 147.3 | 77.7 |
| HDL (mg/dL) | 49.1 | 43.7 | 46.4 | 75.0 | 56.1 | 45.6 | 42.5 | 81.2 | 56.1 | 29.8 | 47.9 | 22.4 | 70.4 | 63.4 |
| Steroids | ||||||||||||||
| Estradiol (pg/ml) | 27.2 | NR | 24.2 | 217.9 | 13.3 | 97.0 | 34.1 | NR | 249.8 | 20.4 | NR | NR | 38.7 | NR |
| Testosterone (ng/ml) | 0.7 | < 0.2 | 0.60 | 0.4 | < 0.2 | 0.8 | 6.8 | 1.6 | 0.4 | 3.8 | NR | NR | 0.69 | NR |
| DHEAS (ug/dl) | 106.8 | 29.5 | 77.4 | 117.9 | 70.0 | 333.6 | 187.9 | 62.6 | 143.7 | 138.8 | NR | NR | NR | NR |
| Cortisol (ug/dl) | NR | 10.7 | 11.9 | 14.1 | 9.7 | 10.8 | 17.9 | 24.3 | 14.6 | NR | NR | NR | NR | NR |
| Progesterone (ng/ml) | NR | <0.2 | NR | 21.4 | NR | 4.1 | NR | 109.1 | 1.89 | NR | NR | NR | NR | NR |
Abbreviations are as follows: 24S, 24S-hydroxycholesterol; LDL, low density lipoprotein; HDL, high density lipoprotein; DHEAS, 5-Dehydroepiandrosterone sulfate; F, female; M, male; NR, not reported or not checked.
Normal value ranges are as follows:
Cholesterol: adult male (110.2–220.4); adult female (162.4–201.8); pediatric male (125.7–230); pediatric female (106.3–216.6).
24S: males and females ages 11–70 years (30.1–105.9).
LDL: adult male (58–116); adult female (58–131.5); pediatric male and female (< 110).
HDL: adult and pediatric male and female (> 34.8).
Estradiol: adult male (15–45); adult female (30–450); adult postmenopausal female (< 59.9); prepubertal (< 10.9).
Testosterone: adult male (2.8–8.8); adult female (0.1–0.8); prepubertal (< 0.2).
DHEAS: adult female (33.2–431); prepurbertal (7.4–66.3).
Cortisol: morning levels; adult male/female (5–25).
Progesterone: adult postmenopausal females (< 1); adult preovulatory females (< 1); adult midcycle females (5–20); adult females in third trimester of pregnancy (48.4–425).
Individual V-1 was 32 wks pregnant at the time of blood work. Reference ranges for total cholesterol and lipoproteins are based on those reported by Piechota and Statszewski.40 All values were within the normal range for pregnancy in the third trimester.
X inactivation ratio for an additional p.Lys232del NSDHL-positive female (IV-9) is 58:42; for a p.Lys232del NSDHL-negative female (V-4), the ratio is 36:64.
Figure 4.

NSDHL Mutations Associated with CKS
(A) Comparative sterol profiles of a CKS male (V-3) of family 1 and an unaffected male. The sterol abnormalities have the same pattern as those observed in CHILD. Sterols were measured in lysates from lymphoblastoid cells cultured in cholesterol poor medium for 3 days. K232del = Lys232del. Error bars represent one standard deviation.
(B) Comparative sterol profiles of a CKS male (III-1) of family 2 and an unaffected male. Again, the sterol abnormalities have the same pattern as those observed in CHILD. Sterols were measured in lysates from lymphoblastoid cells cultured in cholesterol-poor medium for 3 days. R367SfsX33 = Arg367SerfsX33. Error bars represent one standard deviation.
(C) Predicted tertiary structure of wild-type NSDHL (blue) and p.K232del NSDHL (ochre). The protein structures are superimposed to highlight differences. The region between G229 and T239 (white box) is enlarged below; K232 is shown on the wild-type protein in the enlargement.
(D) Immunoblot showing NSDHL expression in unaffected male (Cnt) and p.K232del primary skin fibroblasts (left) and in unaffected male (Cnt) and pR367SfsX33 lymphoblastoid cells (right).
(E) Quantitative real-time PCR measurement of NSDHL mRNA steady-state levels in unaffected male (Cnt) and p.K232del primary skin fibroblasts (left) and in unaffected male (Cnt) and p.R367SfsX33 lymphoblastoid cells (right). Error bars represent one standard deviation for three biological replicates.
(F) Immunoblot detection of endogenous NSDHL and V5-tagged NSDHL in HEK293T-Rex cells transfected with tetracycline-inducible expression constructs. After selection of stable cell lines, expression of the respective NSDHL was induced with tetracycline. For determining whether mutant proteins were degraded by the proteosome, protein levels were measured before and after inhibition of the proteosome with MG132.
(G) Indirect immunofluorescent subcellular localization of V5-tagged NSDHL (green) in HEK293T-REx cells after tetracycline induction and proteosome inhibition with MG132. Anti-Calnexin (Cal, red) was used to identify the endoplasmic reticulum. Cells were counterstained with DAPI (Scale bar represents 10 μm).
(H) Complementation at 30°C in yeast deficient for Erg26, the NSDHL ortholog, by wt or mutant NSDHL (p.K232del or p.R367SfsX33). The expression plasmid without an insert was used as the negative control (Neg Cnt). Immunoblot detection of wt and mutant (p.K232del or p.R367SfsX33) human NSDHL in Erg26-deficient yeast grown at 30°C or 37°C. 3-phosphoglycerate kinase (PGK) is shown as the loading control.
NSDHL mutations associated with CHILD are presumed to eliminate or greatly decrease NSDHL function because they include nonsense, frameshift, and deletion mutations.17 To test this, we assessed NSDHL expression in fibroblasts cultured from the affected skin of CHILD patients. Consistent with the nonsense mutations causing either nonsense-mediated mRNA decay or rapid degradation of a truncated protein, the cultures were a mosaic of cells with and without NSDHL expression (Figure S3).
Using the Swiss-Model server18 to predict the tertiary structure of NSDHL, we found that p.Lys232del disrupts a β-pleated sheet (Figure 4). By immunoblotting, the steady-state level of NSDHL in patient cells expressing either p.Lys232del or p.Arg367SerfsX33 NSDHL was markedly reduced despite comparable mRNA levels as measured by qRT-PCR (Figure 4). Deletion of the analogous amino acid Glu221 from mouse Nsdhl confirmed a stabilizing role for this amino acid when the protein was expressed in HEK293 cells (Figure S4). Also, immunoblotting for p.Lys232del and p.Arg367SerfsX33 NSDHL expressed in HEK293 cells detected low or undetectable steady-state levels unless the proteosome was inhibited with MG132 (Figure 4).
The p.Lys232del and p.Arg367SerfsX33 NSDHL had a distribution similar to that of wild-type NSDHL and partially colocalized with the endoplasmic reticulum protein calnexin (Figure 4). To test whether the mutant protein retained enzymatic activity, we assessed complementation in S. cerevisiae deficient for the NSDHL ortholog Erg26.19 The appropriate cDNAs were cloned into the pAG-416-GPD-DEST vector and inserted as single copies into the yeast strain SGD200, which is deficient for Erg26.13 Interestingly, both p.Lys232del and p.Arg367SerfsX33 NSDHL complemented at 30°C (Figure 4). Immunoblotting detected protein levels comparable to those of wild-type NSDHL at 30°C but detected little mutant protein when the yeast were grown at 37°C (Figure 4). Therefore, at a permissive temperature of 30°C, the mutant NSDHL proteins are able to correctly fold and function at a level comparable to wild-type, whereas at the restrictive temperature of 37°C, abnormal folding of the mutant proteins results in protein degradation. Given that NSDHL mutations associated with CHILD syndrome and the Nsdhl loss-of-function alleles found in Bpa mice do not show complementation at the permissive temperature,19 we conclude that the p.Lys232del and p.Arg367SerfsX33 mutations are temperature-sensitive hypomorphic alleles of NSDHL. From this, we postulate that these hypomorphic alleles retain sufficient function to allow survival of males and to mitigate the severe features of CHILD syndrome, particularly in cooler tissues such as skin.
Because the developing brain synthesizes cholesterol de novo,20 we used in situ hybridization and immunohistochemistry to assess NSDHL expression and NSDHL localization, respectively, in the mouse and human brain. The mouse and human tissues were obtained in accordance with protocols approved by the University of British Columbia's ethical review board and institutional policies. In both species, cortical neurons and glia express NSDHL throughout development (Figure S5). Therefore, we hypothesized that deficiency of NSDHL could cause the cortical brain malformations observed in males with CKS (Figure 5).4 Indeed, histopathological studies of embryonic day 10.5 (E10.5) forebrains from male mice with a Bpa8H loss-of-function allele of Nsdhl19 showed a thin and disorganized cortex and, as measured by TUNEL and BrdU incorporation, significantly increased numbers of apoptotic cells as well as increased cellular proliferation (Figure 5). This paradoxical observation can be explained by the toxic and proliferative effects of methylsterols (L.E.K. and R.I.K., unpublished data).
Figure 5.

Mutant NSDHL Localization and Functionality
(A) Neuropathology of E10.5 male wild-type (wt) (i−iv) and Nsdhl mutant (Bpa8H, v–viii,) mice. (i and v) Whole mounts of the embryos (scale bar represents 1mm). (ii and vi) Serial 7 μm horizontal sections through the forebrain at the level of the lens and optic cup stained with hematoxylin and eosin (scale bar represents 50 μm); the black box represents the area of higher magnification shown in panels iii–iv and vii–viii. Sections of forebrain stained for proliferation by BrdU incorporation (iii and vii) or for apoptosis by TUNEL-labeling (iv and viii) (scale bar represents 20 μm). The tissue was counterstained with hematoxylin.
(B) Quantification of dorsal anterior forebrain thickness, proliferation, and apoptosis in wt and Bpa8H male forebrains. The left graph shows the relative thickness of the dorsal anterior forebrain (FB) measured in four adjacent sections (wt, n = 6; Bpa8H, n = 3; p = 0.32). The middle graph shows the percentage of BrdU-positive cells (wt, n = 6; Bpa8H, n = 3; p = 0.16). The right graph shows the percentage of TUNEL-positive (apoptotic) cells (wt, n = 6; Bpa8H, n = 3; p = 0.48). In all graphs, the error bars represent one standard deviation.
(C) MRI scan of the proband V-3, age 6 yrs, showing a simplified gyral pattern in the frontal and parietal cortex (arrows).
From these observations, we hypothesized that accumulation of methylsterols, not cholesterol deficiency alone, causes CKS. Three patient observations support this: (1) as measured by isotope dilution liquid chromatography-tandem mass spectrometry,21 postnatal plasma 24S-hydroxycholesterol levels, a measure of brain cholesterol turnover,22 did not differ from controls for absolute 24S-hydroxycholesterol levels or 24S-hydroxycholesterol: cholesterol ratios (Table 1); (2) the cerebrospinal fluid (CSF) cholesterol level of one affected male was normal, whereas his CSF methylsterol levels were elevated (data not shown); and (3) the phenotype and neuropathology of males with CKS are distinctly different than that observed in humans or mice with deficiency of sterol delta-7-reductase,23,24 the last step in the synthesis of cholesterol.25
Accumulation of substrate and consequent toxicity, with or without cholesterol deficiency, also explains the diversity of phenotypes observed with defects of cholesterol biosynthesis. These include Greenberg dysplasia (MIM 215140), mevalonic aciduria (MIM 610377), X-linked dominant chondrodysplasia punctata (CDPX2 [MIM 302960]), lathosterolosis (MIM 607330) and desmosterolosis (MIM 602398), as well as Smith-Lemli-Opitz syndrome (SLOS [MIM 270400]), CHILD syndrome, and CKS.26,27 Similarly, in Insig double-knockout mice, the accumulation of cholesterol precursors in the presence of normal cholesterol levels causes phenotypes ranging from facial clefting28 to hair-growth defects,29 and consistent with this, the pathology is ameliorated or reversed by blocking the pathway with HMG-CoA reductase inhibitors.28,29
Study of SLOS also implicates the accumulation of enzymatic substrates, not cholesterol deficiency alone, as the cause of disease.26,30 First, cultured fibroblasts with mutations predicted to have no DHCR7 activity can synthesize cholesterol at rates that can be as high as 50% of all sterols; this suggests that cells have alternate pathways for synthesizing cholesterol.30 Second, the oxidized derivatives of 7-dehydrocholesterol retard growth of cultured rat embryos, are toxic to cultured cells, and induce gene-expression changes similar to those observed in cells deficient for 7-dehydrocholesterol reductase activity.26,31 Understanding the role of these substrates in human biology is thus crucial to treating these disorders and understanding the role of cholesterol in human behavior.32,33
Alternatively, the anomalies of CKS might be attributable, at least in part, to deficient hedgehog signaling as has been suggested in SLOS30 and in studies of Nsdhl-deficient mouse placentas.34 Autoprocessing of the hedgehog protein requires cholesterol as a cofactor and covalent adduct.35 Also, cells defective in cholesterol biosynthesis have a defective response to Sonic hedgehog signaling because of reduced Smoothened activity.36,37 In contrast to SLOS or mutations of Sonic hedgehog, however, CKS individuals do not have polydactyly, syndactyly, genital anomalies, or, as judged by MRI, a rostral-caudal gradient of neuropathology, the forme fruste of holoprosencephaly. Thus, deficient hedgehog signaling does not fully explain the pathology of CKS and again suggests a pathology arising primarily from accumulation of methylsterols.
Interestingly, the pathology of CKS is also distinct from that of CHILD syndrome. This disorder, which affects females, is characterized by normal intellect, unilateral ichthyosiform skin lesions typically involving only the right side of the body, alopecia, ipsilateral limb defects with epiphyseal stippling, and occasional internal malformations.38 Mutations of NSDHL causing CHILD syndrome are presumed to be lethal to males on the basis of the skewing of the sex ratio and mouse models.16,17 Mouse models have skewing of X inactivation as adults (Figure S6).39 Also, we found that fibroblast cultures from affected skin of three CHILD patients had X inactivation ratios of 77:23, 96:4, and 92:8 (Figure S6). In the mouse model, the development of skewing is progressive, suggesting that the pathology of CHILD syndrome arises from cell death.39 In contrast, mothers carrying an NSDHL mutation causing CKS have X inactivation ratios ranging from 90:10 to 58:42 (Table 1), a range common in the general population; this provides additional in vivo support that the NSDHL mutations observed with CKS are hypomorphic.
In summary, CKS expands the phenotypes associated with NSDHL mutations. In CHILD syndrome17 and in the bare patches and striated Nsdhl mutant mice,16 there is male lethality and tissue deficiency among carrier females. In contrast, males with CKS survive, and their mothers have no physical abnormalities.4 This diversity of phenotypes arising from dysfunction of NSDHL is likely the consequence of variations in flux through the cholesterol biosynthesis pathway (Figure 6). Our findings provide an entry point for further dissection of the role of cholesterol synthesis intermediates in human development.
Figure 6.

Model of How NSDHL Mutations Cause a Spectrum of Disease
Unimpeded cholesterol production allows normal neurodevelopment. In contrast, reduced flow or flux through the pathway because of a hypomorphic NSDHL enzyme leads to dose-dependent effects of cholesterol deficiency and/or sterol precursor buildup in the brain. This results in neurodevelopmental malformations (CK syndrome). As the block becomes more extreme, further loss of functional NSDHL leads to severe developmental aberrations and cell death, resulting in male lethality and CHILD syndrome.
Acknowledgments
The authors thank Daniel Goldowitz, Jan M. Friedman, Ken Inoue, David Cooke, Martin Bard, and Rosemarie Rupps for critical review of this manuscript. We thank Colin Ross for genotyping support, Daniel Goldowitz for mouse tissues, and the family for their collaboration. This work was supported in part by a British Columbia Children's Foundation Telethon Award (C.D.S.), a Scottish Rite Foundation Award (C.D.S.), a Child & Family Research Institute Establishment Award (C.F.B.), the BC Clinical Genomics Network of the Michael Smith Foundation for Health Research (C.F.B.), and the Réseau de Médecine Génétique Appliquée of Québec (J.L.M. and G.A.R.). C.F.B., S.J.M.J., and M.A.M. are scholars of the Michael Smith Foundation for Health Research.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes Project, http://www.1000genomes.org/
The Greenwood Genetic Center, XLMR update, http://www.ggc.org/xlmr.htm
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
The Swiss Model Server, http://swissmodel.expasy.org/SWISS-MODEL.html
Accession Numbers
The dbSNP accession numbers for the sequence variants reported in this paper are ss263199175, ss263199176, and ss263199177.
References
- 1.Stevenson R.E., Schwartz C.E. X-linked intellectual disability: unique vulnerability of the male genome. Dev Disabil Res Rev. 2009;15:361–368. doi: 10.1002/ddrr.81. [DOI] [PubMed] [Google Scholar]
- 2.Greenwood Genetic Center (GGC). XLMR Update - July 2010. Available at http://www.ggc.org/xlmr.htm.
- 3.Gécz J., Shoubridge C., Corbett M. The genetic landscape of intellectual disability arising from chromosome X. Trends Genet. 2009;25:308–316. doi: 10.1016/j.tig.2009.05.002. [DOI] [PubMed] [Google Scholar]
- 4.du Souich C., Chou A., Yin J., Oh T., Nelson T.N., Hurlburt J., Arbour L., Friedlander R., McGillivray B.C., Tyshchenko N. Characterization of a new X-linked mental retardation syndrome with microcephaly, cortical malformation, and thin habitus. Am. J. Med. Genet. A. 2009;149A:2469–2478. doi: 10.1002/ajmg.a.33071. [DOI] [PubMed] [Google Scholar]
- 5.Schäffer A.A., Gupta S.K., Shriram K., Cottingham R.W., Jr. Avoiding recomputation in linkage analysis. Hum. Hered. 1994;44:225–237. doi: 10.1159/000154222. [DOI] [PubMed] [Google Scholar]
- 6.Gudbjartsson D.F., Jonasson K., Frigge M.L., Kong A. Allegro, a new computer program for multipoint linkage analysis. Nat. Genet. 2000;25:12–13. doi: 10.1038/75514. [DOI] [PubMed] [Google Scholar]
- 7.Bentley D.R., Balasubramanian S., Swerdlow H.P., Smith G.P., Milton J., Brown C.G., Hall K.P., Evers D.J., Barnes C.L., Bignell H.R. Accurate whole human genome sequencing using reversible terminator chemistry. Nature. 2008;456:53–59. doi: 10.1038/nature07517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Levy S., Sutton G., Ng P.C., Feuk L., Halpern A.L., Walenz B.P., Axelrod N., Huang J., Kirkness E.F., Denisov G. The diploid genome sequence of an individual human. PLoS Biol. 2007;5:e254. doi: 10.1371/journal.pbio.0050254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wheeler D.A., Srinivasan M., Egholm M., Shen Y., Chen L., McGuire A., He W., Chen Y.J., Makhijani V., Roth G.T. The complete genome of an individual by massively parallel DNA sequencing. Nature. 2008;452:872–876. doi: 10.1038/nature06884. [DOI] [PubMed] [Google Scholar]
- 10.Wang J., Wang W., Li R., Li Y., Tian G., Goodman L., Fan W., Zhang J., Li J., Zhang J. The diploid genome sequence of an Asian individual. Nature. 2008;456:60–65. doi: 10.1038/nature07484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tarpey P.S., Smith R., Pleasance E., Whibley A., Edkins S., Hardy C., O'Meara S., Latimer C., Dicks E., Menzies A. A systematic, large-scale resequencing screen of X-chromosome coding exons in mental retardation. Nat. Genet. 2009;41:535–543. doi: 10.1038/ng.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Caldas H., Herman G.E. NSDHL, an enzyme involved in cholesterol biosynthesis, traffics through the Golgi and accumulates on ER membranes and on the surface of lipid droplets. Hum. Mol. Genet. 2003;12:2981–2991. doi: 10.1093/hmg/ddg321. [DOI] [PubMed] [Google Scholar]
- 13.Gachotte D., Barbuch R., Gaylor J., Nickel E., Bard M. Characterization of the Saccharomyces cerevisiae ERG26 gene encoding the C-3 sterol dehydrogenase (C-4 decarboxylase) involved in sterol biosynthesis. Proc. Natl. Acad. Sci. USA. 1998;95:13794–13799. doi: 10.1073/pnas.95.23.13794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mo C., Valachovic M., Randall S.K., Nickels J.T., Bard M. Protein-protein interactions among C-4 demethylation enzymes involved in yeast sterol biosynthesis. Proc. Natl. Acad. Sci. USA. 2002;99:9739–9744. doi: 10.1073/pnas.112202799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kelley R.I. Diagnosis of Smith-Lemli-Opitz syndrome by gas chromatography/mass spectrometry of 7-dehydrocholesterol in plasma, amniotic fluid and cultured skin fibroblasts. Clin. Chim. Acta. 1995;236:45–58. doi: 10.1016/0009-8981(95)06038-4. [DOI] [PubMed] [Google Scholar]
- 16.Liu X.Y., Dangel A.W., Kelley R.I., Zhao W., Denny P., Botcherby M., Cattanach B., Peters J., Hunsicker P.R., Mallon A.M. The gene mutated in bare patches and striated mice encodes a novel 3beta-hydroxysteroid dehydrogenase. Nat. Genet. 1999;22:182–187. doi: 10.1038/9700. [DOI] [PubMed] [Google Scholar]
- 17.Bornholdt D., König A., Happle R., Leveleki L., Bittar M., Danarti R., Vahlquist A., Tilgen W., Reinhold U., Poiares Baptista A. Mutational spectrum of NSDHL in CHILD syndrome. J. Med. Genet. 2005;42:e17. doi: 10.1136/jmg.2004.024448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bordoli L., Kiefer F., Arnold K., Benkert P., Battey J., Schwede T. Protein structure homology modeling using SWISS-MODEL workspace. Nat. Protoc. 2009;4:1–13. doi: 10.1038/nprot.2008.197. [DOI] [PubMed] [Google Scholar]
- 19.Lucas M.E., Ma Q., Cunningham D., Peters J., Cattanach B., Bard M., Elmore B.K., Herman G.E. Identification of two novel mutations in the murine Nsdhl sterol dehydrogenase gene and development of a functional complementation assay in yeast. Mol. Genet. Metab. 2003;80:227–233. doi: 10.1016/s1096-7192(03)00137-9. [DOI] [PubMed] [Google Scholar]
- 20.Dietschy J.M., Turley S.D. Thematic review series: brain Lipids. Cholesterol metabolism in the central nervous system during early development and in the mature animal. J. Lipid Res. 2004;45:1375–1397. doi: 10.1194/jlr.R400004-JLR200. [DOI] [PubMed] [Google Scholar]
- 21.DeBarber A.E., Lütjohann D., Merkens L., Steiner R.D. Liquid chromatography-tandem mass spectrometry determination of plasma 24S-hydroxycholesterol with chromatographic separation of 25-hydroxycholesterol. Anal. Biochem. 2008;381:151–153. doi: 10.1016/j.ab.2008.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lütjohann D., von Bergmann K. 24S-hydroxycholesterol: a marker of brain cholesterol metabolism. Pharmacopsychiatry. 2003;36(Suppl 2):S102–S106. doi: 10.1055/s-2003-43053. [DOI] [PubMed] [Google Scholar]
- 23.Wassif C.A., Zhu P., Kratz L., Krakowiak P.A., Battaile K.P., Weight F.F., Grinberg A., Steiner R.D., Nwokoro N.A., Kelley R.I. Biochemical, phenotypic and neurophysiological characterization of a genetic mouse model of RSH/Smith—Lemli—Opitz syndrome. Hum. Mol. Genet. 2001;10:555–564. doi: 10.1093/hmg/10.6.555. [DOI] [PubMed] [Google Scholar]
- 24.Kelley R.I., Hennekam R.C.M. Smith-Lemli-Opitz Syndrome. In: Valle D., Beaudet A.L., Vogelstein B., Kinzler K.W., Antonarakis S.E., Ballabio A., editors. The Online Metabolic & Molecular Bases of Inherited Disease. McGraw-Hill; New York: 2001. [Google Scholar]
- 25.Kandutsch A.A., Russell A.E. Preputial gland tumor sterols. 3. A metabolic pathway from lanosterol to cholesterol. J. Biol. Chem. 1960;235:2256–2261. [PubMed] [Google Scholar]
- 26.Korade Z., Xu L., Shelton R., Porter N.A. Biological activities of 7-dehydrocholesterol-derived oxysterols: implications for Smith-Lemli-Opitz syndrome. J. Lipid Res. 2010;51:3259–3269. doi: 10.1194/jlr.M009365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fliesler S.J. Retinal degeneration in a rat model of smith-lemli-opitz syndrome: thinking beyond cholesterol deficiency. Adv. Exp. Med. Biol. 2010;664:481–489. doi: 10.1007/978-1-4419-1399-9_55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Engelking L.J., Evers B.M., Richardson J.A., Goldstein J.L., Brown M.S., Liang G. Severe facial clefting in Insig-deficient mouse embryos caused by sterol accumulation and reversed by lovastatin. J. Clin. Invest. 2006;116:2356–2365. doi: 10.1172/JCI28988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Evers B.M., Farooqi M.S., Shelton J.M., Richardson J.A., Goldstein J.L., Brown M.S., Liang G. Hair growth defects in Insig-deficient mice caused by cholesterol precursor accumulation and reversed by simvastatin. J. Invest. Dermatol. 2010;130:1237–1248. doi: 10.1038/jid.2009.442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kelley R.I., Hennekam R.C. The Smith-Lemli-Opitz syndrome. J. Med. Genet. 2000;37:321–335. doi: 10.1136/jmg.37.5.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gaoua W., Chevy F., Roux C., Wolf C. Oxidized derivatives of 7-dehydrocholesterol induce growth retardation in cultured rat embryos: a model for antenatal growth retardation in the Smith-Lemli-Opitz syndrome. J. Lipid Res. 1999;40:456–463. [PubMed] [Google Scholar]
- 32.Golomb B.A., Stattin H., Mednick S. Low cholesterol and violent crime. J. Psychiatr. Res. 2000;34:301–309. doi: 10.1016/s0022-3956(00)00024-8. [DOI] [PubMed] [Google Scholar]
- 33.Lalovic A., Merkens L., Russell L., Arsenault-Lapierre G., Nowaczyk M.J., Porter F.D., Steiner R.D., Turecki G. Cholesterol metabolism and suicidality in Smith-Lemli-Opitz syndrome carriers. Am. J. Psychiatry. 2004;161:2123–2126. doi: 10.1176/appi.ajp.161.11.2123. [DOI] [PubMed] [Google Scholar]
- 34.Jiang F., Herman G.E. Analysis of Nsdhl-deficient embryos reveals a role for Hedgehog signaling in early placental development. Hum. Mol. Genet. 2006;15:3293–3305. doi: 10.1093/hmg/ddl405. [DOI] [PubMed] [Google Scholar]
- 35.Chiang C., Litingtung Y., Lee E., Young K.E., Corden J.L., Westphal H., Beachy P.A. Cyclopia and defective axial patterning in mice lacking Sonic hedgehog gene function. Nature. 1996;383:407–413. doi: 10.1038/383407a0. [DOI] [PubMed] [Google Scholar]
- 36.Cooper M.K., Porter J.A., Young K.E., Beachy P.A. Teratogen-mediated inhibition of target tissue response to Shh signaling. Science. 1998;280:1603–1607. doi: 10.1126/science.280.5369.1603. [DOI] [PubMed] [Google Scholar]
- 37.Cooper M.K., Wassif C.A., Krakowiak P.A., Taipale J., Gong R., Kelley R.I., Porter F.D., Beachy P.A. A defective response to Hedgehog signaling in disorders of cholesterol biosynthesis. Nat. Genet. 2003;33:508–513. doi: 10.1038/ng1134. [DOI] [PubMed] [Google Scholar]
- 38.Happle R., Koch H., Lenz W. The CHILD syndrome. Congenital hemidysplasia with ichthyosiform erythroderma and limb defects. Eur. J. Pediatr. 1980;134:27–33. doi: 10.1007/BF00442399. [DOI] [PubMed] [Google Scholar]
- 39.Cunningham D., Spychala K., McLarren K.W., Garza L.A., Boerkoel C.F., Herman G.E. Developmental expression pattern of the cholesterogenic enzyme NSDHL and negative selection of NSDHL-deficient cells in the heterozygous Bpa(1H)/+ mouse. Mol. Genet. Metab. 2009;98:356–366. doi: 10.1016/j.ymgme.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Piechota W., Staszewski A. Reference ranges of lipids and apolipoproteins in pregnancy. Eur. J. Obstet. Gynecol. Reprod. Biol. 1992;45:27–35. doi: 10.1016/0028-2243(92)90190-a. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.