

Abstract
Psoriasis, an immune-mediated, inflammatory disease of the skin and joints, provides an ideal system for expression quantitative trait locus (eQTL) analysis, because it has a strong genetic basis and disease-relevant tissue (skin) is readily accessible. To better understand the role of genetic variants regulating cutaneous gene expression, we identified 841 cis-acting eQTLs using RNA extracted from skin biopsies of 53 psoriatic individuals and 57 healthy controls. We found substantial overlap between cis-eQTLs of normal control, uninvolved psoriatic, and lesional psoriatic skin. Consistent with recent studies and with the idea that control of gene expression can mediate relationships between genetic variants and disease risk, we found that eQTL SNPs are more likely to be associated with psoriasis than are randomly selected SNPs. To explore the tissue specificity of these eQTLs and hence to quantify the benefits of studying eQTLs in different tissues, we developed a refined statistical method for estimating eQTL overlap and used it to compare skin eQTLs to a published panel of lymphoblastoid cell line (LCL) eQTLs. Our method accounts for the fact that most eQTL studies are likely to miss some true eQTLs as a result of power limitations and shows that ∼70% of cis-eQTLs in LCLs are shared with skin, as compared with the naive estimate of < 50% sharing. Our results provide a useful method for estimating the overlap between various eQTL studies and provide a catalog of cis-eQTLs in skin that can facilitate efforts to understand the functional impact of identified susceptibility variants on psoriasis and other skin traits.
Introduction
Transcriptional regulation of gene expression is essential for almost every process in a cell, and abnormal transcriptional regulation is likely to be involved in the etiology of many diseases. Advances in high-throughput gene expression profiling and genotyping technologies have recently enabled researchers to study the genetic variants that regulate gene expression at a genomic scale.1 Such genome-wide association studies (GWAS) of gene expression have identified thousands of genetic loci affecting the expression of specific transcripts. Each of these loci is called an expression quantitative trait locus (eQTL). The identification of eQTLs will enhance our understanding of global transcriptional regulation and regulatory variation. Furthermore, as GWAS of diseases identify many susceptibility variants with no known functional effects,2 eQTL studies might help clarify the function of many newly identified susceptibility variants.3
Studies of global gene expression were initially performed in model organisms (ranging from yeast4,5 to flies6 to mice7), but more recent studies have directly examined human cells.8–11 In humans, the vast majority of validated eQTLs map to within a few hundred kilobase pairs of the associated transcription unit. Loosely, these loci are termed cis-eQTLs. In contrast to cis-eQTLs, loci located far from the transcripts that they regulate have been much harder to confidently identify in humans and are, loosely, termed trans-eQTLs. One example of the utility of eQTL analysis is the GWAS for asthma (MIM 600807) reported by Moffatt and colleagues.12 The study showed that a set of noncoding genetic variants that is strongly associated with childhood asthma also regulates expression levels of ORMDL3 (MIM 610075), focusing attention on ORMDL3 as a target for additional functional studies. Most human studies have measured transcript abundance in blood cells (peripheral blood lymphocytes and immortalized lymphoblastoid cell lines [LCLs]); only a small number of studies have examined it in other tissues (e.g., liver13 or brain14). There is a controversy regarding whether associations observed in LCLs will translate to other tissues, and some recent studies have suggested that overlap between eQTL signals among tissues might be relatively small. Dimas et al. compared lists of significant eQTLs identified in three cell types (LCLs, fibroblasts, and T cells) and estimated that 69 to 80% of cis-eQTLs operate in a cell-type-specific manner.15 However, our analyses provide evidence that current eQTL studies are typically underpowered and that, as a result, directly comparing lists of significant eQTLs leads to underestimation of the overlap percentage. Indeed, our results show that overlap in eQTLs across tissues can be substantial.
In this study we report the mapping of eQTLs in skin tissue from psoriatic patients and healthy controls. Psoriasis (MIM 177900) is an immune-mediated, inflammatory, and hyperproliferative disease of the skin and joints. It provides an ideal system for eQTL mapping analysis, because psoriasis has a strong genetic basis and affected tissue (skin) is readily accessible.
The current work presents two major advances. First, it describes a large catalog of genetic variants influencing transcript levels in both normal and psoriatic skin. This catalog is based on gene expression and genotype data that we have collected from normal skin from healthy controls (normal skin), normal-appearing skin from psoriatic patients (uninvolved skin), and diseased skin from psoriatic patients (lesional skin). This catalog represents a genetic map of gene regulation in skin and provides a useful tool for examining the functional impact of genetic variants associated with psoriasis and other diseases.
Second, it describes a method for accurately estimating the overlap of eQTLs between tissues; we use this method to compare our eQTL catalog in skin with a previously described catalog for LCLs. An accurate estimate of overlap in eQTLs across tissues can help researchers to quantify the benefits of studying eQTLs in different tissues. We also note that a simple and naive estimate based on the overlap of discoveries will necessarily underestimate the true overlap because each eQTL study will detect only a subset of all eQTLs. Here, we describe a procedure that takes statistical power into account to provide a more accurate estimate of the percentage of overlapping eQTLs between two tissues.
These two aspects of our work shed light on transcriptional regulation by genetic variants in skin and provide an insight into the genetics of gene expression in different tissue types. Our work also describes a practical approach for estimating the overlap of signals from two experimental settings. This method has the potential to be applied to a wide range of biological studies.
Material and Methods
Mapping eQTLs in Skin
Subjects
We enrolled 58 psoriatic patients and 64 healthy controls in the study. Patients had to have at least one well-demarcated, erythematous, scaly psoriatic plaque that was not limited to the scalp. In those instances where there was only a single psoriatic plaque, the case was considered only if the plaque occupied more than 1% of the total body surface area. Study subjects did not use any (a) systemic antipsoriatic treatments for 2 wks prior to biopsy or (b) topical antipsoriatic treatments for 1 wk prior to biopsy. Informed consent was obtained from all subjects, under protocols approved by the institutional review board of the University of Michigan Medical School, and the study was conducted according to the principles of the Declaration of Helsinki. Subjects with failed gene expression profiling or failed genotyping were excluded from the analysis. The final analysis included 53 psoriatic patients and 57 healthy controls.
Genotype Data
Subjects (as a subset of a total of 1409 cases and 1436 controls) were genotyped by Perlegen Sciences with the use of four proprietary, high-density oligonucleotide arrays in partnership with the Genetic Association Information Network (GAIN). A series of quality control filters, which are described in detail in Nair et al.,16 were applied to the genotype data. In brief, we excluded markers with < 95% genotype call rates, with a minor allele frequency < 1%, with a Hardy-Weinberg equilibrium (HWE) p value < 10−6, with > 2 mismatches among 48 pairs of individuals that were genotyped twice, or with > 2 Mendelian inconsistencies among 27 trios; we also excluded samples with call rates < 95% and with outlier heterozygosities. In total, 438,670 autosomal SNPs were genotyped for 53 psoriatic patients and 57 controls. As previously described,17–19 we used information on patterns of haplotype variation in the HapMap CEU samples (release 21) to infer missing genotypes “in silico.” We analyzed only SNPs that were genotyped or could be imputed with relatively high confidence (estimated r2 between imputed SNP and true genotypes > 0.3, so that patterns of haplotype sharing between sampled individuals and HapMap samples consistently indicated a specific allele; we use this r2-based threshold, rather than one based on the posterior probability of each imputed genotype, because it naturally accommodates SNPs with different allele frequencies and because it is the same threshold used in many recent GWAS, including our psoriasis study16).
Gene Expression Data
Two biopsies (one lesional, one uninvolved skin; 6 mm each) were taken under local anesthesia from each psoriatic subject, whereas one 6 mm punch biopsy (normal skin) was taken from healthy controls. Lesional skin biopsies were taken from psoriasis plaques, and uninvolved skin biopsies were taken from the buttocks, at least 10 cm away from the nearest plaque. The normal-skin biopsies were also taken from the buttocks. RNA from each biopsy was isolated with the RNeasy kit (QIAGEN, Valencia, CA). Samples were run on Affymetrix U133 Plus 2.0 arrays for evaluating the expression of ∼54,000 probes, in accordance with the manufacturer's protocol. The raw data from 180 microarrays were processed via the Robust Multichip Average (RMA) method. We considered expression levels for all ∼54,000 probes on the array. To avoid outliers, we also applied a second inverse normal transformation step to residuals for each trait, after adjusting for sex and batch effects. Procedures for extracting RNA, controlling RNA quality, and preprocessing gene expression data are described in detail elsewhere.20 The microarray data have been deposited into NCBI Gene Expression Omnibus (GEO) under accession number GSE13355.
eQTL Mapping
We tested SNP-gene expression associations separately in normal skin (n = 57), in uninvolved skin (n = 53), and in lesional skin (n = 53). Given the small sample size in each analysis (< 60) and hence the relatively low statistical power, we tested only cis-associations between each transcript (i.e., probe) and those SNPs in its cis-candidate region (from 1Mb upstream of the transcription start site to 1Mb downstream of the transcription end site). Specifically, we used the score test in Merlin (fastassoc option) to test the association.21 For genotyped SNPs, the number of copies of one allele was modeled. For imputed SNPs, the dosage (i.e., the expected number of copies) of one allele was modeled. Unless noted otherwise, we used a p value threshold of 9 × 10−7 as the significance threshold to originally identify cis-eQTLs, which corresponds to a false discovery rate (FDR) of approximately 0.01 in each of the three skin types. For gene expression traits that were significantly associated with more than one cis-SNP, we considered only the most significant cis-SNP.
Measuring cis-eQTL Overlap among Three Skin Types
To test whether cis-associations identified in one skin type can be replicated in the other two skin types, we started with the significant SNP-gene pairs identified in one skin type and then evaluated evidence for association in the other two skin types, using a nominal p value threshold of 0.05. We did not use the genome-wide p value significance threshold of 9 × 10−7 for this analysis, which focuses on a small number of SNP-gene pairs. Ideally we would use the method we develop below (see next section) to estimate the cis-eQTL overlap among three skin types, but the small sample sizes of the three skin types prevented us from using the sample splitting strategy if we wanted to maintain adequate statistical power for the study.
Estimating the Overlap of cis-eQTL Signals in Lymphoblastoid Cell Lines and in Skin
Intuition
To understand our method, consider the following hypothetical situation. Suppose that in a study of LCLs from 100 individuals we detect 1000 eQTLs. Furthermore, suppose that in a second study of identically treated LCLs from 60 individuals we find evidence for 300 of these eQTLs and that in a study of skin samples from 60 individuals we find evidence for only 270 of these eQTLs. In this hypothetical setting, the raw eQTL overlap is only 270/1000 (or 27%) but the power-adjusted estimate of the overlap in eQTLs is 270/300 (or 90%). More details for our method follow.
Statistical Methods
The simplest approach to comparing eQTL lists between two experimental settings is to evaluate the overlap of lists of significant eQTLs compiled separately for each setting. Unfortunately, this method will underestimate the overlap percentage whenever either of the two studies is underpowered (in that case, many true eQTLs might be detected in one study but missing from the list of eQTLs detected in the second study). Here, we propose a method that takes into account the statistical power of the studies. Very briefly, our procedure starts by splitting the study/tissue with the larger sample size into two parts. One part identifies eQTLs in the tissue, and the second part provides unbiased estimators for the power to replicate eQTL signals. This estimated power is then used to adjust the observed raw overlap percentage and hence to obtain a power-adjusted estimate of the overlap in eQTL signals.
Estimating the Overlap Percentage
In our method, we assume that eQTL analyses are performed in two studies: in study 1 (here, the study using lymphoblastoid cell lines), we use a nominal p value cutoff of α1 to generate a list of significant eQTLs, which corresponds to an FDR of FDR1, whereas in study 2 (here, the study using skin tissue), we use a nominal p value cutoff of α2, corresponding to an FDR of FDR2. Let π be the percentage of eQTLs in study 1 that are also eQTLs in study 2; let πraw be the observed percentage of significant eQTLs in study 1 that are also significant in study 2. Because both eQTL lists are necessarily incomplete, πraw will result in an underestimate of π. Our aim is, thus, to arrive at a better estimator of the true overlap percentage π. To do this, we attempt to estimate a power-adjusted expected overlap in significant eQTLs, πadjusted.
To arrive at this power-adjusted expected overlap, we start with the list of significant eQTLs in study 1 and consider (see Figure 1 for a detailed decision diagram) a series of possibilities that might lead these eQTLs to replicate in study 2 (i.e., to be overlapping eQTLs): (a) a fraction (π) of true positive eQTLs in study 1 are also true eQTLs in study 2 and are expected to replicate in study 2 with a particular power; (b) a fraction (1− π) of true positive eQTLs in study1 will not be true eQTLs in study 2 but might “replicate” by chance, with the probability determined by the significance threshold α2, which is simply the false positive rate; (c) false positive eQTLs in study 1 might also replicate by chance, with the probability also determined by the significance threshold α2. We note that it is possible that a small fraction of false positive eQTLs in study 1 will represent true eQTLs in study 2, but for simplicity, we assume that this number will be approximately zero (see Supplemental Material and Methods, available online, for the complete decision diagram with all possibilities and the full description of the method that leads to the simplified version presented here). Therefore:
| (Equation 1) |
Figure 1.
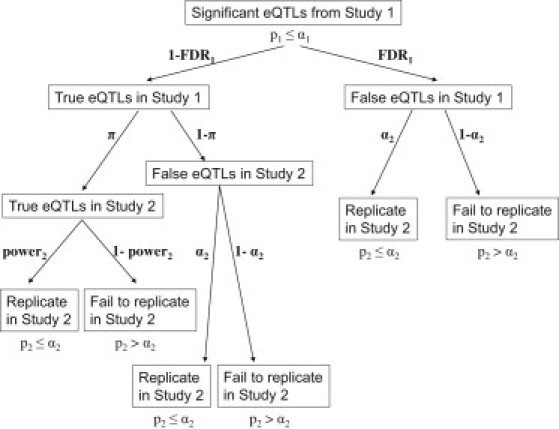
Simplified Diagram for Categorization of Significant eQTLs from Study1 into Groups for the Estimation of the Overlap Percentage
where power2 is the statistical power of study 2 to detect eQTLs that are true positives in both study 1 and study 2 (overlapping eQTLs). Algebraic manipulation of Equation 1 above gives:
Thus, we can estimate π as:
| (Equation 2) |
In Equation 2, is an observed quantity and α2 is the (arbitrary) p value threshold used in study 2. Given α1, FDR1 can be estimated via the Benjamini and Hochberg procedure.22 Therefore, to estimate π, the major work is to estimate power2.
In theory, power2 is determined by the effect sizes of overlapping eQTLs in study 2, the sample size of study 2, and the type I error rate of study 2 (α2). To estimate power2 we calculate power2raw, the statistical power of a study on tissue 1 with the same sample size as study 2 and significance level α2 to detect significant study 1 eQTLs. Because significant study 1 eQTLs include both identified true study 1 eQTLs and false positives, we use the following formula to adjust for false positives when estimating power2 (the formula is obtained with the use of a decision tree idea similar to the one in Figure 1; see Supplemental Material and Methods for the derivation):
| (Equation 3) |
Note that a simple estimate of power2raw based on the observed effect sizes of each eQTL in study 1 would be biased because of the winner's curse.23,24 To avoid the bias, we estimate power2raw by using a sample splitting strategy: we split study1 into mutually exclusive and independent sets: study1A and study1B. Study 1A is used to identify significant eQTLs in study 1, whereas study 1B is used to provide unbiased estimates for effect sizes of these eQTLs. Given the sample size of study 2, we can then estimate power2raw on the basis of the effect-size estimates. The sample splitting strategy can be further simplified if splitting is done such that study 1B has the same number of subjects and the same data structure (i.e., the same pattern of related and unrelated individuals) as study 2. In this setting, the proportion of signals identified in study 1A that are also significant in study 1B is power2raw. The results presented in this paper use this simplified sample-splitting strategy.
Our approach assumes that the distribution of effect sizes is similar for overlapping and nonoverlapping eQTLs in study 1. In addition, it assumes that the distribution of effect sizes for overlapping eQTLs is similar between the two studies. Violation of these assumptions could lead to an underestimate of power2 (for example, if overlapping eQTLs typically have larger effect sizes than the nonoverlapping ones) or to an overestimate of power2 (for example, if overlapping eQTLs typically have smaller effect sizes in study 2 than in study 1, where they were originally detected). In the Discussion, we describe results from empirical data that support these assumptions.
Returning to the example in the “Intuition” section, consider a setting in which 1000 eQTLs are detected with a 1% FDR in study 1. If 90% of the true eQTLs in study 1 are also true eQTLs in study 2, and if we set α2 = 0.05 and assume the power of study 2 is 30%, then we expect to see 273 significant eQTLs in study 2 (using the formula from Equation 1). So = 27.3%, which is approximately one-third of the true overlap percentage 90%. However, if we apply Equation 2 with power2 = 0.3, α2 = 0.05, and FDR1 = 0.01, we get = 90.0%.
Estimating the Variance of the Overlap Percentage
We use the jackknife resampling technique to estimate the variance of . We randomly remove one subject from study 1B and one subject from study 2 to obtain new estimators for and power2 and hence a new estimator for . We repeat this procedure and obtain multiple estimators for and then estimate the variance of as:
Data for Lymphoblastoid Cell Lines
Genotype and expression data for LCLs were originally published in Dixon et al.9 for a set of 183 families (340 subjects total). Affymetrix U133 Plus 2.0 arrays were used for gene expression profiling, and Sentrix HumanHap300 Genotyping BeadChips (Illumina, San Diego, CA) were used for SNP genotyping. We then used MACH17–19 to impute all HapMap SNPs, with phased HapMap CEU sample haplotypes as templates. We split the 183 families randomly into two sets, where set 1A contained 126 families and set 1B contained one randomly selected individual from each of the remaining 57 families (so that the sample size matched our study of normal skin).
Applying the Method to Estimating the Overlap of cis-eQTL Signals in LCLs and in Skin
We first performed eQTL analysis in study 1A. As we did in the skin study, we tested only associations between each transcript and those SNPs within 1 Mb of the transcript. We used a range of nominal p value thresholds, which corresponded to FDRs (FDR1) of 0.001, 0.0005, and 0.0001. To avoid multiple counting of the same cis-eQTL signal, we focus on the most significant SNP-transcript pair for each transcript. For the study in skin (study 2), we focused our analysis on the data from the 57 healthy controls (the data from 53 patients were also analyzed). We used a range of α2 values (p value thresholds) in study1B and study2: 0.05, 0.001, and 0.0005.
Studying Other Features of Skin cis-eQTLs
Relationship of Skin eQTL SNPs to Association Signals in Psoriasis GWAS
We compiled a list of 9462 SNPs associated with levels of at least one transcript in normal, uninvolved, or lesional skin with p < 9 × 10−7 (corresponding to the FDR at 0.01). From this list we selected 389 independent skin eQTL SNPs. We thinned the eQTL list using linkage disequilibrium (LD) while favoring SNPs with stronger cis-association p values. Specifically, we used an r2 threshold of 0.2 so that each of the 9462 eQTL SNPs is either in the pruned list of 389 or has a proxy SNP with r2 > 0.2 in the pruned list. Using a quantile-quantile (Q-Q) plot, we compared the distribution of psoriasis association p values for these 389 eQTL SNPs against the null expectation. Disease-association p values were derived from a meta-analysis of two psoriasis GWAS, the GAIN psoriasis GWAS16 and the Kiel psoriasis study,25 and then all HapMap SNPs were imputed in the same way as mentioned above. To further compare these 389 SNPs with remaining GWAS SNPs, we removed all skin eQTL SNPs from the GWAS SNP set and then randomly picked 389 of the remaining SNPs 5000 times. This allowed us to derive confidence intervals (CIs) for the p value distribution of non-eQTL SNPs. Because we were interested in testing whether eQTL SNPs could reveal new psoriasis-susceptibility loci, we removed from both skin eQTL and non-eQTL SNP lists those SNPs that were within 1 Mb of the seven replicated loci from our recently published GWAS (i.e., HLA-C [MIM 142840], IL12B [MIM 161561], TNIP1 [MIM 607714], IL13 [MIM 147683], TNFAIP3 [MIM 191163], IL23A [MIM 605580] / STAT2 [MIM 600556], and IL23R [MIM 607562]).
Gene Ontology Enrichment Analysis of Genes Associated with cis-eQTLs
We searched for Gene Ontology (GO) terms that were significantly enriched in each list of genes associated with eQTLs in the three skin types. This GO category-enrichment analysis was performed with the publicly available software DAVID.
Results
Mapping cis-eQTLs in Skin
As described previously,20 we found that expression profiles for lesional skin were markedly different from those of normal and uninvolved skin. Using principal component analysis (PCA) (Figure S2), we achieved near-perfect separation of lesional skin from normal and uninvolved skin, whereas the latter two skin types were intermixed. Here, we do not focus on a comparison of expression levels between the tissues,20 but instead report on the cis-eQTLs in the different skin types.
Using a nominal p value threshold of 9 × 10−7 (corresponding to an FDR for cis-associations of ∼0.01 for each of the three skin types), we identified 331, 275, and 235 independent cis-associations in normal, uninvolved, and lesional skin, respectively. We have created a publicly available database containing the catalogs of cis-eQTLs for each of the three skin types, which will allow researchers to interrogate their specific SNPs or genes of interest. Figure 2 gives two examples of cis-association between gene transcripts and their nearby SNPs: ERAP2 (MIM 609497) shows clear cis-association in normal skin, peaking at rs2910686, and similar signals in both uninvolved and lesional skin; RPS26 (MIM 603701) has one of the most significant cis-associations in uninvolved and lesional skin, peaking at rs11171739. Although the signal is less significant in normal skin, the same overall pattern of association is observed.
Figure 2.
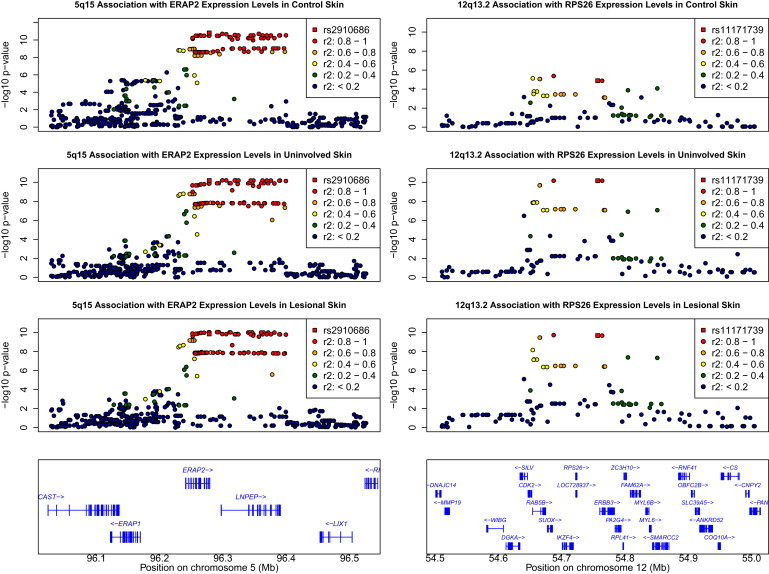
Regional Plots for Evidence of cis-Association between SNPs and ERAP2 or RPS26
The most significant SNPs (the most significant SNP associated with ERAP2 in normal skin on the left panel; the most significant SNP associated with RPS26 in uninvolved skin on the right panel) are highlighted with a square. The other SNPs are drawn as circles and color coded according to the degree of linkage disequilibrium with the most significant SNP.
We then measured the overlap of cis-eQTLs among the three skin types by testing how many significant cis-eQTLs in one skin type were replicated in other two skin types at a nominal p value threshold of 0.05. The results are shown in Figure 3: 95.1%, 96.7%, and 98.7% of the significant cis-eQTLs in normal, uninvolved, and lesional skin, respectively, were detected in the other two skin types. Furthermore, we observed only two cis-eQTLs in each set that were observed only in that skin type, consistent with an FDR of 0.01 (i.e., in a set of 200 signals, we expect to see two false positives). These results, consistent with the similar cis-association patterns observed in the three skin types (Figure 2), indicate that nearly all cis-eQTLs currently identified are shared by normal, uninvolved, and lesional skin. Therefore, the dramatic physiological changes that are apparent in psoriatic skin appear to have little impact on the identity of cis-eQTLs in skin.
Figure 3.
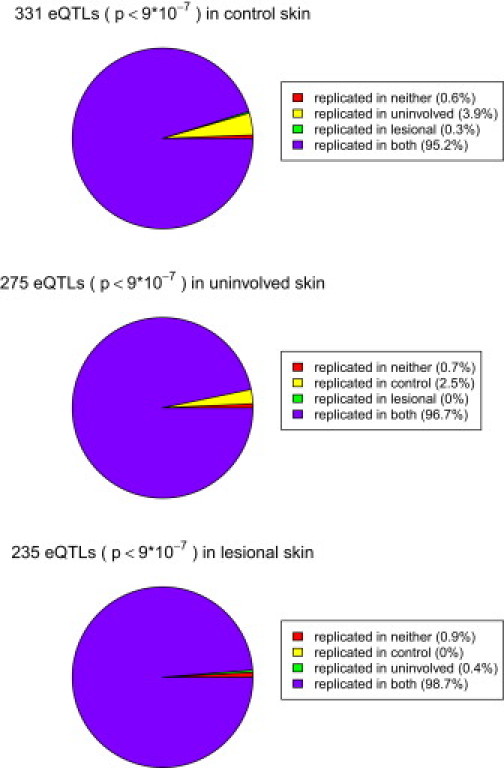
The Sharing of cis-eQTLs in Normal, Uninvolved, and Lesional Skin with the Other Two Types of Skin
The p value threshold for discovery is 9 × 10−7, and the p value threshold for replication is 0.05.
Estimating the Overlap of cis-eQTL Signals in Lymphoblastoid Cell Lines and in Skin
We have developed a more accurate method for estimating the eQTL overlap between two tissues. Using our method, we estimated the percentage of true eQTLs in LCLs that are also true eQTLs in normal skin. As our method requires an approximation in the formula, we controlled the FDR in study 1 relatively tightly (i.e., controlling FDR1 at 0.001, 0.0005, and 0.0001). We allowed α2 to take a range of different values (0.05, 0.001, and 0.0005) and then estimated the overlap percentage for all combinations of FDR1 and α2. As summarized in Table 1, the different FDR1 and α2 thresholds give relatively consistent estimates for the percentage of overlapping eQTLs between tissues: around 70% of the true cis-eQTLs in LCLs are estimated to be present in normal skin. The naive estimator suggests overlap percentages ranging from 30% to 50% depending on the statistical thresholds used in the analysis. As an example, if we set FDR1 = 0.0005 and α2 = 0.001, the observed overlap percentage () was 0.316 and the power was estimated at power2 = 0.462. Using Equation 2, we estimated the true overlap percentage to be 68.3% (95% CI from jackknife resampling: 66.4%–70.2%). We also estimated the overlap of cis-eQTLs between LCLs and uninvolved skin, as well as between LCLs and lesional skin. These additional comparisons produced similar estimates of ∼70% shared cis-eQTLs (Table 1). These results suggest that a majority of cis-eQTLs are shared between skin and LCLs.
Table 1.
Estimating the Overlap of cis-eQTLs between LCLs and the Three Types of Skin with the Use of Different Significance Thresholds
|
Different Thresholds |
57 Normal Skin |
53 Uninvolved Skin |
53 Lesional Skin |
|||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| FDR1 | α2 | power2 |
|
power2 |
|
power2 |
|
|||||||||
| 0.001 | 0.05 | 0.520 | 0.771 | 0.652 (0.642, 0.662) | 0.495 | 0.764 | 0.623 (0.612, 0.634) | 0.499 | 0.764 | 0.629 (0.618, 0.640) | ||||||
| 0.001 | 0.001 | 0.296 | 0.429 | 0.689 (0.669, 0.709) | 0.278 | 0.402 | 0.691 (0.671, 0.711) | 0.266 | 0.402 | 0.661 (0.641, 0.681) | ||||||
| 0.001 | 0.0005 | 0.264 | 0.367 | 0.719 (0.692, 0.746) | 0.252 | 0.345 | 0.730 (0.703, 0.757) | 0.239 | 0.345 | 0.692 (0.667, 0.717) | ||||||
| 0.0005 | 0.05 | 0.538 | 0.805 | 0.646 (0.636, 0.656) | 0.514 | 0.798 | 0.620 (0.609, 0.631) | 0.519 | 0.798 | 0.627 (0.616, 0.638) | ||||||
| 0.0005 | 0.001 | 0.316 | 0.462 | 0.683 (0.664, 0.702) | 0.297 | 0.434 | 0.684 (0.665, 0.703) | 0.285 | 0.434 | 0.656 (0.636, 0.676) | ||||||
| 0.0005 | 0.0005 | 0.284 | 0.398 | 0.713 (0.686, 0.740) | 0.271 | 0.374 | 0.724 (0.698, 0.750) | 0.256 | 0.374 | 0.684 (0.660, 0.708) | ||||||
| 0.0001 | 0.05 | 0.587 | 0.856 | 0.666 (0.656, 0.676) | 0.564 | 0.849 | 0.643 (0.632, 0.654) | 0.552 | 0.849 | 0.628 (0.617, 0.639) | ||||||
| 0.0001 | 0.001 | 0.364 | 0.534 | 0.681 (0.663, 0.699) | 0.342 | 0.506 | 0.675 (0.657, 0.693) | 0.332 | 0.506 | 0.655 (0.637, 0.673) | ||||||
| 0.0001 | 0.0005 | 0.330 | 0.471 | 0.700 (0.675, 0.725) | 0.315 | 0.442 | 0.712 (0.688, 0.736) | 0.301 | 0.442 | 0.681 (0.659, 0.703) | ||||||
We used permutations to further evaluate the performance of our method under the null hypothesis of no overlap. Specifically, we generated 20 permuted data sets by shuffling expression phenotypes (independently of genotype) for the skin eQTL data. In each of these permuted data sets, the expected true overlap is zero, and the estimated overlap obtained with our method is also very close to zero (Table S1).
Unaccounted-for population substructure could generate false eQTLs or mask the signal of true eQTLs, adversely affecting estimates of eQTL overlap between tissues. In our data, the genomic control value was 1.009 for the skin data set. Dixon et al.9 previously reported their genomic control value as 1.01 in the LCL data set.
Studying other Features of Skin cis-eQTLs
Relationship of Skin eQTL SNPs to Association Signals in Psoriasis GWAS
Out of a total of 9462 SNPs that passed the eQTL significance threshold of 9 × 10−7 in normal, uninvolved, or lesional skin (FDR = 0.01), we identified 389 independent skin eQTLs (r2 < 0.2) and examined their potential importance in the context of psoriasis and other complex genetic disorders that have been subjected to GWAS. First, using the meta-analysis results for two psoriasis GWAS, we compared the distribution of disease-association p values for SNPs that define eQTLs and those that do not. For this comparison, we exclude SNPs within 1 Mb of regions known to be associated with psoriasis, so as to more directly evaluate the ability of eQTLs to suggest new loci. Figure 4 shows the Q-Q plot for the 389 independent eQTL SNPs in skin, with CIs estimated by sampling the same number of non-eQTL SNPs. The Q-Q plot clearly shows a trend for eQTL SNPs to be more strongly associated with psoriasis than non-eQTL SNPs, an observation that is consistent with other recent studies.26 Furthermore, the majority of eQTL SNPs exceed the 75% CI obtained by sampling non-eQTL SNPs, and six of the top eight ranked eQTL SNPs exceed the 95% CI determined by sampling non-eQTL SNPs. Table S2 presents the most significant eQTLs identified in normal, uninvolved, and lesional skin. Table 2 lists the top eight eQTL SNPs from the Q-Q plot, along with their cis-association and psoriasis GWAS results. Although the overlap between eQTL signals and psoriasis associations is intriguing, we recognize that further follow-up genotyping will be required to confirm these signals. Still, examination of the genes in this list (FUT2 [MIM 182100], RPS26, ERAP1 [MIM 606832], and ERAP2) suggests several plausible biological connections, which are detailed in the Discussion.
Figure 4.
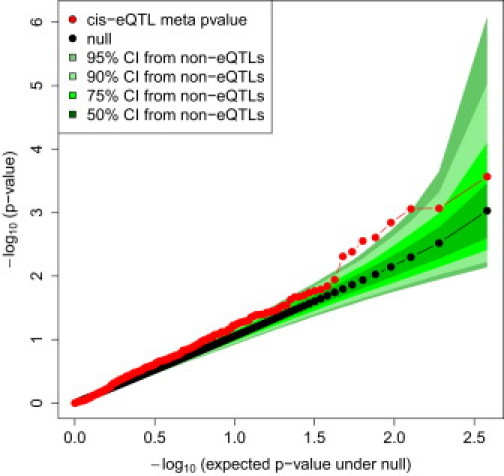
Quantile-Quantile Plot of Psoriasis GWAS p Values for 389 Independent eQTL SNPs in Skin, with Confidence Intervals Defined by Non-eQTL SNPs
Table 2.
cis-Association and Psoriasis Association Meta-Analysis Results for the Eight Independent Skin eQTL SNPs with the Most Significant Psoriasis Association
| Marker |
cis-Association |
Psoriasis GWAS |
||||||
|---|---|---|---|---|---|---|---|---|
| cis-Associated Gene |
cis-Association p Value |
Alleles Risk/Nonrisk | Risk Allele Frequency | OR | Meta p Valuea | |||
| Normal | Uninvolved | Lesional | ||||||
| rs492602 | FUT2 | 1.5 × 10−4 | 1.9 × 10−6 | 1.0 × 10−9 | G/A | 0.482 | 1.169 | 2.7 × 10−4 |
| rs12039309 | TMEM77 | 3.2 × 10−7 | 5.3 × 10−6 | 2.7 × 10−5 | G/T | 0.271 | 1.212 | 8.5 × 10−4 |
| rs11171739 | RPS26 | 1.3 × 10−5 | 6.8 × 10−11 | 2.1 × 10−10 | C/T | 0.418 | 1.174 | 8.7 × 10−4 |
| rs13008446 | LOC348751 | 1.9 × 10−6 | 1.7 × 10−4 | 1.3 × 10−7 | A/G | 0.655 | 1.191 | 1.4 × 10−3 |
| rs8082268 | C17orf45 | 1.0 × 10−7 | 6.7 × 10−7 | 4.4 × 10−5 | C/T | 0.651 | 1.188 | 2.4 × 10−3 |
| rs2910686 | ERAP2 | 5.6 × 10−11 | 8.5 × 10−11 | 6.1 × 10−11 | C/T | 0.427 | 1.144 | 2.7 × 10−3 |
| rs139910 | TNRC6B | 2.6 × 10−7 | 5.0 × 10−4 | 3.3 × 10−2 | C/G | 0.744 | 1.117 | 4.1 × 10−3 |
| rs503612 | ENDOD1 | 2.9 × 10−9 | 9.6 × 10−9 | 6.9 × 10−9 | C/A | 0.564 | 1.148 | 4.8 × 10−3 |
The p value from the psoriasis GWAS meta-analysis (GAIN study + Kiel study). For all eight SNPs, the effect directions are consistent in the two studies of the meta-analysis.
We also studied this list of skin eQTL SNPs in the context of other complex genetic diseases that have been subjected to GWAS. Among 1482 significant (p < 10−5) SNP associations from 321 published GWAS curated by the National Human Genome Research Institute, we found 14 skin eQTL SNPs (Table S3), which are associated with 19 disease traits, whereas we expected to see only five overlapping SNPs by chance, with the 95% CI being 2–10 for overlapping SNPs.
Enrichment of eQTLs for Genes Involved in MHC Class I Antigen Presentation
Using DAVID,27,28 we searched for biological processes enriched in eQTL-associated transcripts from lesional, uninvolved, and normal skin, as well as LCLs. This analysis revealed significant enrichment for eQTLs regulating genes involved in the processing and presentation of endogenous peptide antigens via MHC class I in lesional skin (Table S4). We also observed a similar but nonsignificant trend in uninvolved skin, normal skin, and LCLs. The skin eQTL-associated genes observed to be enriched in this GO category included ERAP1 (endoplasmic reticulum aminopeptidase 1; also known as ARTS-1), TAP2 (transporter, ATP-binding cassette, major histocompatibility complex, 2 [MIM 170261]), ERAP2 (endoplasmic reticulum aminopeptidase 2; also known as LRAP), and TAPBPL (TAP binding protein-like [MIM 607081]). These genes are intimately involved in the transport (TAP2, TAPBLPL) and processing (ERAP1, ERAP2) of peptides within the endoplasmic reticulum for subsequent presentation on the surface of cells within the antigen-binding groove of MHC I class molecules.29 These results provide further evidence for the genetic control of the expression of genes involved in MHC class I antigen presentation in the skin.
Localization of cis-eQTLs With Respect To the Transcription Start Site of the Transcripts They Putatively Regulate
We studied the localization of the most significant eQTL for each cis-association in normal, uninvolved, and lesional skin with respect to the transcription start site of the gene it putatively regulates. The most significant cis-eQTLs localize closely (most of them within 100 kb) and roughly symmetrically around the transcription start site (Figure S3). This localization pattern in the skin confirms previous observations in LCLs.9,11,30 Although many mechanisms control mRNA levels, this observed localization pattern suggests that many, if not all, eQTLs play a role in the regulation of transcription.
Discussion
We report an eQTL map of human skin and identify eQTLs in normal skin, uninvolved skin from psoriatic individuals, and lesional psoriatic skin. Our results thus provide a useful resource for studying the regulation of gene expression in skin. Our analysis shows that the vast majority of strong cis-eQTLs are shared in the three skin tissue types, which indicates that the physiology of the disease does not change the identity of those strong cis-eQTLs. This finding does not preclude a role for cis-eQTLs in psoriasis or other skin diseases. First, although it appears that the same set of transcripts are cis-regulated in all three skin types, differences in genotype frequencies for the regulatory SNP between cases and controls can result in differences in expression levels for the transcripts they regulate between psoriatic and normal skin. Second, it is possible that the same transcripts can have different downstream effects in normal and diseased skin tissues.
We have developed a refined method for estimating eQTL overlap between two tissues. Our method can provide a more accurate estimator for the eQTL overlap percentage whenever either of the two studies is underpowered. Our multistep procedure first generates a list of potential eQTLs and then uses unbiased estimates for eQTL effect sizes to estimate the expected number of replicating eQTLs for a specific sample size. The proportion of overlapping eQTLs can then be interpreted in this context. Our method can be useful in a variety of settings where estimation of the overlap of two signal lists is needed. For example, in theory the method can be applied to estimate the overlap of areas of the brain activated by two different stimuli in an fMRI (functional magnetic resonance imaging) experiment.
Using our method, we have estimated that around 70% of the significant cis-eQTLs in LCLs are also observed in skin, a value that greatly exceeds the raw overlap of 30%–50% obtained with the use of a naive estimator. If overlapping eQTLs typically had larger effect sizes than nonoverlapping eQTLs, we might expect these to replicate across tissues more often than expected for an “average” eQTL. To empirically examine this possibility, we divided the set of eQTLs identified in the lymphocyte data into two equally sized groups: a “large effect size” group with the largest effect eQTLs and a “small effect size” group with the remainder. As shown in Table S5 (and described in Supplemental Material and Methods), we found very similar estimates of eQTL overlap between tissues for “large effect” and “small effect” eQTLs. We also compared eQTL effect sizes between LCLs and skin for overlapping loci. Our results show that the effect sizes are similar for the two groups (Figure S4), providing further reassurance of the validity of our method.
We also compared the LCL cis-eQTLs in our analysis with cis-eQTLs identified in fibroblasts and T cells generated by Dimas and colleagues.15 Even though the raw overlap percentages were rather low, after we adjusted for the power of the study (because sample sizes of LCLs, fibroblasts, and T cells are the same in Dimas et al.,15 the power of the study can be estimated by using the results from LCLs in that same study), we estimated that 65%–70% of significant LCL cis-eQTLs were also present in fibroblasts and T cells (Table 3 and Supplemental Material and Methods). This finding is consistent with our results comparing LCLs and skin. The same methods we have described here to compare eQTL sets between tissues could be used to compare eQTL sets between many different groups, including comparisons of eQTL lists between populations, sexes, and cases and controls.
Table 3.
|
Overlap with LCLs (Dixon et al.) |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
α2 = 0.01 |
α2 = 0.005 |
α2 = 0.001 |
||||||||||
| Dimas et al. | ||||||||||||
| FDR1 = 0.001 | ||||||||||||
| LCLs | 0.368 | 1.000a | 0.312 | 1.000a | 0.241 | 1.000a | ||||||
| Fibroblasts | 0.237 | 0.633 | 0.202 | 0.643 | 0.151 | 0.624 | ||||||
| T cells | 0.257 | 0.690 | 0.230 | 0.732 | 0.167 | 0.691 | ||||||
| FDR1 = 0.005 | ||||||||||||
| LCLs | 0.326 | 1.000a | 0.272 | 1.000a | 0.208 | 1.000a | ||||||
| Fibroblasts | 0.210 | 0.632 | 0.177 | 0.644 | 0.131 | 0.630 | ||||||
| T cells | 0.235 | 0.712 | 0.204 | 0.744 | 0.143 | 0.687 | ||||||
Assumed.
Our method provides an estimate of the fraction of eQTLs shared between two tissues, but this is only one of many questions of interest when comparing the impact of genetic variation on gene regulation across tissues. For example, it would be desirable to evaluate whether eQTLs maintain their relative importance across tissues: perhaps, even if eQTLs are generally shared between tissues, their relative impact on gene expression will vary. Additionally, one might be interested in examining how these proportions vary for specific categories of genes. For example, one might propose that eQTLs regulating the expression of genes involved in repair might be relatively conserved across tissues, whereas those involved in more tissue-specific processes (such as those involved in the specialization of different skin cell types) might be more often tissue specific. Whereas the first question can be addressed with more detailed statistical models, the second question can be addressed by focusing the analysis on subsets of genes of related function. Both analyses will benefit from large sample sizes and larger eQTL lists.
Results from the eQTL mapping in skin are also useful in interpreting genetic susceptibility loci identified by GWAS of multiple complex traits, including skin diseases. We examined whether eQTL SNPs were more likely to be associated with psoriasis. This is analogous to other analyses that might focus on SNPs that are likely to be functional because, for example, they encode nonsynonymous SNPs. This focused analysis of eQTL SNPs identified SNPs near FUT2 (rs492602), RPS26 (rs11171739), ERAP1 (rs7063), and ERAP2 (rs2910686) loci as attractive candidates for further analysis in psoriasis and other autoimmune diseases. Other lines of evidence support the idea that these genes might be important for psoriasis. For example, FUT2 encodes a fucosyltransferase involved in the synthesis of blood-group antigens,31 which are also involved in the fucosylation of cell-surface proteins on epithelia.32 rs492602, the peak eQTL SNP in the region, also corresponds to the peak psoriasis association signal in the region. RPS26 is linked to antigen processing and presentation and T cell-mediated immunity. eQTL SNPs near RPS26 (e.g., rs2292239) have been associated with type I diabetes,13 though their relevance as a direct disease determinant has been questioned.33 In our data, the same SNPs show suggestive association with psoriasis (p value = 0.01) and with RPS26 transcript levels in both uninvolved and lesional skin (p value < 10−9 in both tissues). We also observed highly significant eQTL associations for ERAP1 and ERAP2. The products of these genes are intimately involved in the process of trimming peptides in preparation for loading into MHC class I molecules;34 variants near ERAP1 are associated with ankylosing spondylitis (MIM 106300),35 another autoimmune disorder. As this paper went to press, association between ERAP1 and psoriasis was reported in a large independent sample.36 Psoriasis, psoriatic arthritis (MIM 607507), and ankylosing spondylitis are the only major autoimmune diseases that are primarily associated with MHC class I, and ERAP1 is involved in MHC class I antigen processing.
In summary, we have provided a catalog of genetic variants influencing transcript levels in skin and developed a method for estimating eQTL overlap between two tissues. In the future, larger eQTL studies will enable us to study both cis- and trans-eQTLs and hence to provide a comprehensive profile for the genetics of gene expression in skin. With the efforts from our group and others, more data are being collected from different tissues, and researchers will soon be able to comprehensively study the tissue specificity of eQTLs and to have a better understanding of common and tissue-specific transcription-regulation mechanisms. In addition, newly developed technologies (e.g., exon microarrays and next-generation RNA sequencing for high-throughput gene expression profiling) will not only provide us with higher quality data, but will also enable us to study the regulation of different isoforms of the same transcript and to provide a more sophisticated picture of the genetic regulation of transcription.
Acknowledgments
The authors thank the psoriasis patients and healthy controls who participated in this study. This research was supported by grants AR042742, AR050511, AR054966, HG002651, and MH084698 from the National Institutes of Health and by the Ann Arbor Veterans Affairs Hospital. The authors thank Antigone Dimas and Emmanouil Dermitzakis for kindly sharing their eQTL results from fibroblasts, LCLs, and T cells. Genotyping of the German study panel was supported by the German Ministry of Education and Research (BMBF) through the National Genome Research Network (NGFN), the Popgen Biobank, and it received infrastructure support through the Deutsche Forschungsgemeinschaft (DFG) excellence cluster “Inflammation at Interfaces.”
Contributor Information
Jun Ding, Email: junding@umich.edu.
Gonçalo R. Abecasis, Email: goncalo@umich.edu.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
A catalog of published genome-wide association studies curated by the National Human Genome Research Institute, http://www.genome.gov/gwastudies/
Database for Annotation, Visualization and Integrated Discovery (DAVID), http://david.abcc.ncifcrf.gov/
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
University of Michigan Center for Statistical Genetics skin eQTL database, http://www.sph.umich.edu/csg/junding/eQTL
References
- 1.Rockman M.V., Kruglyak L. Genetics of global gene expression. Nat. Rev. Genet. 2006;7:862–872. doi: 10.1038/nrg1964. [DOI] [PubMed] [Google Scholar]
- 2.McCarthy M.I., Abecasis G.R., Cardon L.R., Goldstein D.B., Little J., Ioannidis J.P., Hirschhorn J.N. Genome-wide association studies for complex traits: consensus, uncertainty and challenges. Nat. Rev. Genet. 2008;9:356–369. doi: 10.1038/nrg2344. [DOI] [PubMed] [Google Scholar]
- 3.Cookson W., Liang L., Abecasis G., Moffatt M., Lathrop M. Mapping complex disease traits with global gene expression. Nat. Rev. Genet. 2009;10:184–194. doi: 10.1038/nrg2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brem R.B., Yvert G., Clinton R., Kruglyak L. Genetic dissection of transcriptional regulation in budding yeast. Science. 2002;296:752–755. doi: 10.1126/science.1069516. [DOI] [PubMed] [Google Scholar]
- 5.Yvert G., Brem R.B., Whittle J., Akey J.M., Foss E., Smith E.N., Mackelprang R., Kruglyak L. Trans-acting regulatory variation in Saccharomyces cerevisiae and the role of transcription factors. Nat. Genet. 2003;35:57–64. doi: 10.1038/ng1222. [DOI] [PubMed] [Google Scholar]
- 6.Wayne M.L., McIntyre L.M. Combining mapping and arraying: An approach to candidate gene identification. Proc. Natl. Acad. Sci. USA. 2002;99:14903–14906. doi: 10.1073/pnas.222549199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chesler E.J., Lu L., Shou S., Qu Y., Gu J., Wang J., Hsu H.C., Mountz J.D., Baldwin N.E., Langston M.A. Complex trait analysis of gene expression uncovers polygenic and pleiotropic networks that modulate nervous system function. Nat. Genet. 2005;37:233–242. doi: 10.1038/ng1518. [DOI] [PubMed] [Google Scholar]
- 8.Morley M., Molony C.M., Weber T.M., Devlin J.L., Ewens K.G., Spielman R.S., Cheung V.G. Genetic analysis of genome-wide variation in human gene expression. Nature. 2004;430:743–747. doi: 10.1038/nature02797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dixon A.L., Liang L., Moffatt M.F., Chen W., Heath S., Wong K.C., Taylor J., Burnett E., Gut I., Farrall M. A genome-wide association study of global gene expression. Nat. Genet. 2007;39:1202–1207. doi: 10.1038/ng2109. [DOI] [PubMed] [Google Scholar]
- 10.Göring H.H., Curran J.E., Johnson M.P., Dyer T.D., Charlesworth J., Cole S.A., Jowett J.B., Abraham L.J., Rainwater D.L., Comuzzie A.G. Discovery of expression QTLs using large-scale transcriptional profiling in human lymphocytes. Nat. Genet. 2007;39:1208–1216. doi: 10.1038/ng2119. [DOI] [PubMed] [Google Scholar]
- 11.Stranger B.E., Nica A.C., Forrest M.S., Dimas A., Bird C.P., Beazley C., Ingle C.E., Dunning M., Flicek P., Koller D. Population genomics of human gene expression. Nat. Genet. 2007;39:1217–1224. doi: 10.1038/ng2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moffatt M.F., Kabesch M., Liang L., Dixon A.L., Strachan D., Heath S., Depner M., von Berg A., Bufe A., Rietschel E. Genetic variants regulating ORMDL3 expression contribute to the risk of childhood asthma. Nature. 2007;448:470–473. doi: 10.1038/nature06014. [DOI] [PubMed] [Google Scholar]
- 13.Schadt E.E., Molony C., Chudin E., Hao K., Yang X., Lum P.Y., Kasarskis A., Zhang B., Wang S., Suver C. Mapping the genetic architecture of gene expression in human liver. PLoS Biol. 2008;6:e107. doi: 10.1371/journal.pbio.0060107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Myers A.J., Gibbs J.R., Webster J.A., Rohrer K., Zhao A., Marlowe L., Kaleem M., Leung D., Bryden L., Nath P. A survey of genetic human cortical gene expression. Nat. Genet. 2007;39:1494–1499. doi: 10.1038/ng.2007.16. [DOI] [PubMed] [Google Scholar]
- 15.Dimas A.S., Deutsch S., Stranger B.E., Montgomery S.B., Borel C., Attar-Cohen H., Ingle C., Beazley C., Gutierrez Arcelus M., Sekowska M. Common regulatory variation impacts gene expression in a cell type-dependent manner. Science. 2009;325:1246–1250. doi: 10.1126/science.1174148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nair R.P., Duffin K.C., Helms C., Ding J., Stuart P.E., Goldgar D., Gudjonsson J.E., Li Y., Tejasvi T., Feng B.J., Collaborative Association Study of Psoriasis Genome-wide scan reveals association of psoriasis with IL-23 and NF-kappaB pathways. Nat. Genet. 2009;41:199–204. doi: 10.1038/ng.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Scott L.J., Mohlke K.L., Bonnycastle L.L., Willer C.J., Li Y., Duren W.L., Erdos M.R., Stringham H.M., Chines P.S., Jackson A.U. A genome-wide association study of type 2 diabetes in Finns detects multiple susceptibility variants. Science. 2007;316:1341–1345. doi: 10.1126/science.1142382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li Y., Willer C., Sanna S., Abecasis G. Genotype imputation. Annu. Rev. Genomics Hum. Genet. 2009;10:387–406. doi: 10.1146/annurev.genom.9.081307.164242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li Y., Willer C.J., Ding J., Scheet P., Abecasis G.R. MaCH: using sequence and genotype data to estimate haplotypes and unobserved genotypes. Genet. Epidemiol. 2010 doi: 10.1002/gepi.20533. Published online November 5, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gudjonsson J.E., Ding J., Li X., Nair R.P., Tejasvi T., Qin Z.S., Ghosh D., Aphale A., Gumucio D.L., Voorhees J.J. Global gene expression analysis reveals evidence for decreased lipid biosynthesis and increased innate immunity in uninvolved psoriatic skin. J. Invest. Dermatol. 2009;129:2795–2804. doi: 10.1038/jid.2009.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen W.M., Abecasis G.R. Family-based association tests for genomewide association scans. Am. J. Hum. Genet. 2007;81:913–926. doi: 10.1086/521580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Benjamini Y., Hochberg Y. Controlling the false positive discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc., B. 1995;57:289–300. [Google Scholar]
- 23.Göring H.H., Terwilliger J.D., Blangero J. Large upward bias in estimation of locus-specific effects from genomewide scans. Am. J. Hum. Genet. 2001;69:1357–1369. doi: 10.1086/324471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xiao R., Boehnke M. Quantifying and correcting for the winner's curse in genetic association studies. Genet. Epidemiol. 2009;33:453–462. doi: 10.1002/gepi.20398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ellinghaus E., Ellinghaus D., Stuart P.E., Nair R.P., Debrus S., Raelson J.V., Belouchi M., Fournier H., Reinhard C., Ding J. Genome-wide association study identifies a psoriasis susceptibility locus at TRAF3IP2. Nat. Genet. 2010;42:991–995. doi: 10.1038/ng.689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nicolae D.L., Gamazon E., Zhang W., Duan S., Dolan M.E., Cox N.J. Trait-associated SNPs are more likely to be eQTLs: annotation to enhance discovery from GWAS. PLoS Genet. 2010;6:e1000888. doi: 10.1371/journal.pgen.1000888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dennis G., Jr., Sherman B.T., Hosack D.A., Yang J., Gao W., Lane H.C., Lempicki R.A. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003;4:3. [PubMed] [Google Scholar]
- 28.Huang W., Sherman B.T., Lempicki R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 29.Peaper D.R., Cresswell P. Regulation of MHC class I assembly and peptide binding. Annu. Rev. Cell Dev. Biol. 2008;24:343–368. doi: 10.1146/annurev.cellbio.24.110707.175347. [DOI] [PubMed] [Google Scholar]
- 30.Veyrieras J.B., Kudaravalli S., Kim S.Y., Dermitzakis E.T., Gilad Y., Stephens M., Pritchard J.K. High-resolution mapping of expression-QTLs yields insight into human gene regulation. PLoS Genet. 2008;4:e1000214. doi: 10.1371/journal.pgen.1000214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kelly R.J., Rouquier S., Giorgi D., Lennon G.G., Lowe J.B. Sequence and expression of a candidate for the human Secretor blood group alpha(1,2)fucosyltransferase gene (FUT2). Homozygosity for an enzyme-inactivating nonsense mutation commonly correlates with the non-secretor phenotype. J. Biol. Chem. 1995;270:4640–4649. doi: 10.1074/jbc.270.9.4640. [DOI] [PubMed] [Google Scholar]
- 32.Borén T., Falk P., Roth K.A., Larson G., Normark S. Attachment of Helicobacter pylori to human gastric epithelium mediated by blood group antigens. Science. 1993;262:1892–1895. doi: 10.1126/science.8018146. [DOI] [PubMed] [Google Scholar]
- 33.Plagnol V., Smyth D.J., Todd J.A., Clayton D.G. Statistical independence of the colocalized association signals for type 1 diabetes and RPS26 gene expression on chromosome 12q13. Biostatistics. 2009;10:327–334. doi: 10.1093/biostatistics/kxn039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Saveanu L., Carroll O., Lindo V., Del Val M., Lopez D., Lepelletier Y., Greer F., Schomburg L., Fruci D., Niedermann G., van Endert P.M. Concerted peptide trimming by human ERAP1 and ERAP2 aminopeptidase complexes in the endoplasmic reticulum. Nat. Immunol. 2005;6:689–697. doi: 10.1038/ni1208. [DOI] [PubMed] [Google Scholar]
- 35.Burton P.R., Clayton D.G., Cardon L.R., Craddock N., Deloukas P., Duncanson A., Kwiatkowski D.P., McCarthy M.I., Ouwehand W.H., Samani N.J., Wellcome Trust Case Control Consortium. Australo-Anglo-American Spondylitis Consortium (TASC) Biologics in RA Genetics and Genomics Study Syndicate (BRAGGS) Steering Committee. Breast Cancer Susceptibility Collaboration (UK) Association scan of 14,500 nonsynonymous SNPs in four diseases identifies autoimmunity variants. Nat. Genet. 2007;39:1329–1337. doi: 10.1038/ng.2007.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Strange A., Capon F., Spencer C.C., Knight J., Weale M.E., Allen M.H., Barton A., Band G., Bellenguez C., Bergboer J.G. A genome-wide association study identifies new psoriasis susceptibility loci and an interaction between HLA-C and ERAP1. Nat. Genet. 2010;42:985–990. doi: 10.1038/ng.694. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.