

Abstract
Autosomal-recessive cerebellar ataxias comprise a clinically and genetically heterogeneous group of neurodegenerative disorders. In contrast to their dominant counterparts, unraveling the molecular background of these ataxias has proven to be more complicated and the currently known mutations provide incomplete coverage for genotyping of patients. By combining SNP array-based linkage analysis and targeted resequencing of relevant sequences in the linkage interval with the use of next-generation sequencing technology, we identified a mutation in a gene and have shown its association with autosomal-recessive cerebellar ataxia. In a Dutch consanguineous family with three affected siblings a homozygous 12.5 Mb region on chromosome 3 was targeted by array-based sequence capture. Prioritization of all detected sequence variants led to four candidate genes, one of which contained a variant with a high base pair conservation score (phyloP score: 5.26). This variant was a leucine-to-arginine substitution in the DUF 590 domain of a 16K transmembrane protein, a putative calcium-activated chloride channel encoded by anoctamin 10 (ANO10). The analysis of ANO10 by Sanger sequencing revealed three additional mutations: a homozygous mutation (c.1150_1151del [p.Leu384fs]) in a Serbian family and a compound-heterozygous splice-site mutation (c.1476+1G>T) and a frameshift mutation (c.1604del [p.Leu535X]) in a French family. This illustrates the power of using initial homozygosity mapping with next-generation sequencing technology to identify genes involved in autosomal-recessive diseases. Moreover, identifying a putative calcium-dependent chloride channel involved in cerebellar ataxia adds another pathway to the list of pathophysiological mechanisms that may cause cerebellar ataxia.
Main Text
Autosomal-recessive cerebellar ataxias are a heterogeneous group of rare neurodegenerative disorders in which progressive spinocerebellar ataxia, due to involvement of the cerebellum, brainstem, and/or spinocerebellar long tracts, is the key feature. Clinically, patients are characterized by gait and balance impairment, upper limb coordination problems, and impairment of speech, swallowing, and eye movements. The overall prevalence is estimated to be around five to six per 100,000.1 Autosomal-recessive cerebellar ataxias are often associated with other neurological (e.g., polyneuropathy, spasticity) or nonneurological (e.g., cardiomyopathy, cataract) symptoms and can thus lead to complex phenotypes. Most autosomal-recessive ataxias have an early onset age, formerly defined as < 20 years of age,2 but some have been shown to begin much later. In contrast to the rapidly increasing number of genes involved in the autosomal-dominant cerebellar ataxias, the molecular background of the recessive cerebellar ataxias has been only partly elucidated. Many patients with a recessive ataxia are therefore still left without a molecular diagnosis.
Here, we describe the identification of a gene involved in autosomal-recessive cerebellar ataxia, with downbeat nystagmus and involvement of lower motor neurons as additional clinical features. The gene was identified in a Dutch remotely consanguineous family (Figure 1A). The current study was approved by the Medical Ethics Committee of the Radboud University Nijmegen Medical Centre. Written informed consent to participate in the study was obtained from the patients and their participating relatives (and all other patients described in this paper). All three affected siblings from this family displayed impaired coordination of limbs and gait with an onset between 20 and 35 years. Neurological investigation revealed a gaze-evoked downbeat nystagmus with hypermetric saccades, dysarthric speech, and brisk knee reflexes. Mild atrophy of the upper part of the lower limbs with fasciculations was observed in two of the three affected siblings. These two also had evidence of lower motor neuron involvement on electromyography (Table 1). In all three affected siblings, brain imaging displayed marked cerebellar atrophy with normal supratentorial structures (Figure S1, available online).
Figure 1.
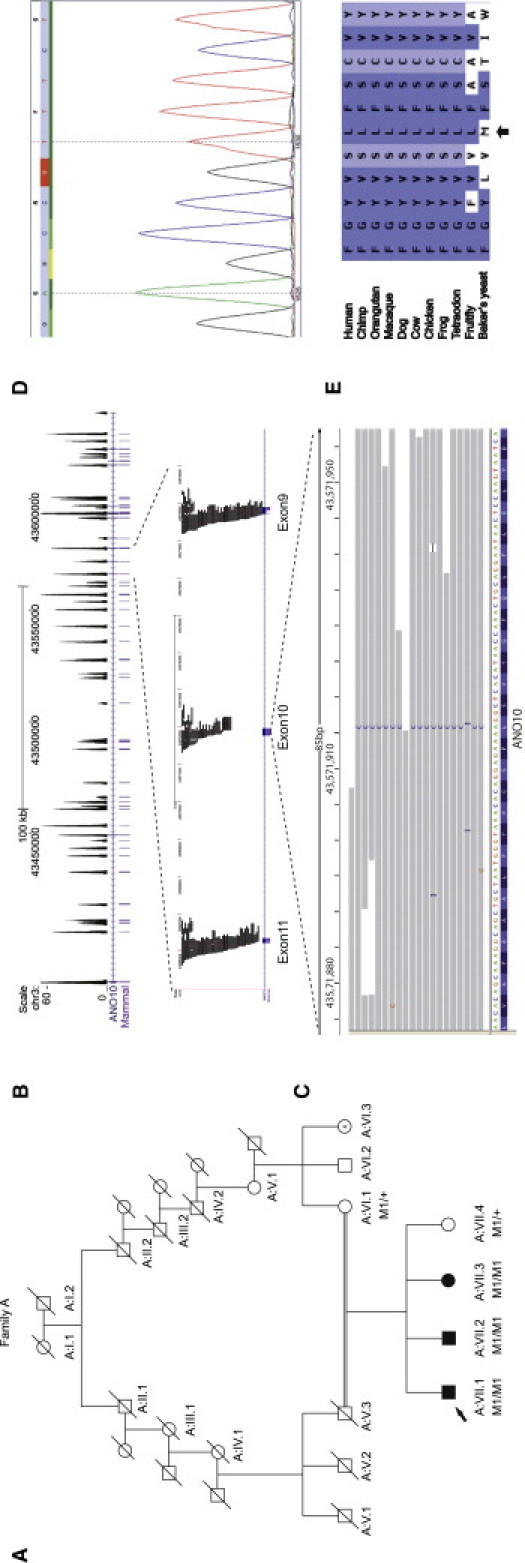
Identifcation of ANO10 Mutation in the Dutch Family A with Autosomal-Recessive Cerebellar Ataxia
(A) Pedigree for the Dutch family A together with the segregation of the mutation identified in this family. M1/M1 indicates homozygous carriers of the p.Leu510Arg mutation, and M1/+ indicates heterozygous carriers. The proband is indicated by an arrow. Unfortunately, DNA was not available for individual A:V.3.
(B) Coverage histogram of ANO10. Upper part: Sequence-read histograms uploaded to the UCSC Genome Browser display the sequencing depth of all exons of ANO10. Tracks displayed: scale, chromosomal position, read depth histogram per bp (between 0 and 60-fold coverage; higher than 60-fold coverage is not displayed), RefSeq gene track, highly conserved elements (PhastCons Placental Mammal Conserved Elements, 28-way Multiz Alignment). Lower part: Next-generation sequencing-read coverage of ANO10 exons 8–10. Tracks displayed: scale, chromosomal position, individual 454 sequencing reads, RefSeq gene track.
(C) Mutation visualization in the IGV browser. Individual reads overlapping with the mutation are displayed. Seventeen of eighteen reads show the homozygous mutation at genomic position chr3:43571913 (the reads are mapped and displayed on the + strand).
(D) Verification by Sanger sequencing of missense mutation c.1529T>G (p.Leu510Arg) in ANO10 (accession number NM_018075.3).
(E) Sequence comparison of ANO10 in different species. The amino acid that is mutated in the Dutch family is depicted, indicated by an arrow. The amino acid is highly conserved throughout different species, including the fruit fly.
Table 1.
Clinical Features of Patients with Recessive Cerebellar Ataxia and Mutations in ANO10
| Patient | A:VII.1 | A:VII.2 | A:VII.3 | B:II.3 | B:II.4 | B:II.5 | C:II.1 | C:II.3 |
|---|---|---|---|---|---|---|---|---|
| Age | 50 | 48 | 47 | 42 | 39 | 35 | 60 | 55 |
| Sex | M | M | F | F | F | M | F | F |
| Age at onset (yrs) | 25 | 20 | 32 | 15 | 15 | 13 | 45 | 25 |
| Age at assessment | 50 | 48 | 47 | 42 | 39 | 35 | 57 | 49 |
| Mental retardation | no | no | no | yes (mild) | yes (moderate) | no | no | no |
| Ocular pursuit | downbeat nystagmus | downbeat nystagmus | downbeat nystagmus | horizontal and vertical nystagmus | horizontal and vertical nystagmus | horizontal nystagmus | saccadic pursuit, nystagmus | multidirectional nystagmus |
| Saccades | hypermetric | hypermetric, slow (vertical) | hypermetric | hypermetric, mild | no | hypermetric, mild | no | slow saccades |
| Cerebellar dysarthria | moderate | moderate | mild | moderate | mild | moderate | moderate | moderate |
| Gait ataxia | moderate | moderate | mild | moderate | moderate | moderate | severe | moderate |
| Appendicular ataxia | moderate | moderate | mild | mild | mild | mild | moderate | moderate |
| Tendon reflexes: UL | increased | increased | increased | increased | increased | increased triceps | increased | increased |
| Tendon reflexes: LL (knee) | increased | increased | increased | increased | increased | increased | increased | increased |
| Tendon reflexes: LL (ankle) | increased | normal | increased | increased | normal | normal | increased | increased |
| Plantar responses | extensor | normal | normal | normal | normal | normal | normal | normal/Babinski |
| Other features | cold and blue toes | wasting and fasciculations proximal leg muscles, cold and blue fingers and toes | cold and blue fingers and toes | inspiratory stridor | pes cavus | fasciculations leg muscles. inspiratory stridor and vocal cord paresis | mild lower limb spasticity, slight rest tremor, pes cavus | episodic diplopia, pes cavus |
| EMG | motor neuron involvement | motor neuron involvement | not done | not done | not done | motor neuron involvement | not done | normal |
| Cerebellar atrophy seen on MRI or CT | severe | severe | severe | severe | severe | severe | not done | severe |
| Tortuosity of conjunctival vessels | absent | absent | absent | present | present | present | absent | absent |
Homozygosity mapping using a 10K SNP array identified five homozygous regions on chromosomes 3, 6, 10, 12, and 16. Only the region on chromosome 3 was confirmed by further fine mapping with short tandem repeat markers, which showed a locus of approximately 10.5 Mb on chromosome 3p21.32-p22.3 with a LOD score of 3.6 in this Dutch family. This locus could be further confirmed and fine mapped with the use of a 6.0 SNP array (Affymetrix) interrogating over 900,000 SNPs. The multipoint LOD score calculations and haplotype analysis were performed with GeneHunter (version 2.1, release 5.05) in the easyLINKAGE software package and Haplopainter software. Within the major locus more than 80 genes are located, of which 15 candidate genes were sequenced by traditional Sanger sequencing, selected on the basis of their known function and cerebellar expression. No mutations could be identified in these genes. Subsequently, we targeted the entire 12.5 Mb region on chromosome 3, as well as nine smaller homozygous regions on chromosomes 1, 2, 3, 4, and 14, for array-based sequence capture followed by next-generation sequencing (Table S1a). The array design included all known exons, untranslated regions (UTRs), microRNAs, and highly conserved regions (PhastCons Conserved Elements, 28-way MostCons Plac Mammal Multiz Alignment, LOD score ≥ 100) for all homozygous regions.3,4 An additional 30 base pair (bp) sequences flanking all exons were added to the regions that were captured on the array so as to enable the detection of splice-site mutations. Targets smaller than 250 bp, which is, based upon our experience, the minimum size for the DNA capture protocol used, were enlarged by extending both ends of the region. The targeted sequences comprise 1,905,376 bp in total (Table S1b), and include 1245 exons from 117 RefSeq genes and 187 UCSC genes, as well as seven microRNAs and noncoding RNAs. After stringent probe selection by NimbleGen (Roche NimbleGen, Madison, WI, USA) (uniqueness tested by Sequence Search and Alignment by Hashing Algorithm [SSAHA]), a total of 2,178,492 bases (Table S1c) were represented on an array, with 385,000 oligonucleotide probes targeting the regions of interest. Sequence capture was performed in accordance with the manufacturer's instructions (Roche NimbleGen), with the use of the Titanium optimized protocol. In brief, 5 μg of genomic DNA of the proband (Figure 1A) was used in the preparation of the DNA library for sequence-capture hybridization. A final amount of 3 μg prehybridization ligation-mediated-PCR-amplified DNA was hybridized to the customized array, eluted after 72 hr of hybridization, and amplified by posthybridization LM-PCR. The amplified captured sample was then used as input for emulsion PCR amplification and subsequent sequencing with the use of a Roche 454 GS FLX sequencer with Titanium series reagents.
The sample was sequenced by using one-quarter plate of a Roche sequencing run, yielding 83.7 Mb of sequence data. Approximately 95.0% of the sequence data mapped back to unique regions of the human genome (hg18, NCBI build 36.1), with the use of the Roche Newbler software (version 2.3). Of all mapped reads, 93.5% were located on or near the targeted regions (i.e., within 500 bp proximity) (Table S2). This was sufficient to reach an average of 32-fold coverage for all target regions. For the region of interest, less than 1% of all targeted sequences were not covered, and only 4.2% of the target sequence was covered fewer than ten times (11% was covered less than 15-fold).
The Roche software detected a total of 3917 high-confidence variants, identifying the variant in at least three reads. We used a custom-made data analysis pipeline as described by Hoischen et al.5 to annotate detected variants with various types of information, including known SNPs, amino acid substitutions, genomic location, and evolutionary conservation. A total of 3352 variants were found to correspond to known SNPs, and 169 variants overlapped with a known polymorphic region (dbSNP130) and were therefore considered not likely to be disease-causing variants. Of all remaining 396 variants, there were three potential splice-site variants, two synonymous coding variants, and five nonsynonymous coding variants; of these ten variants, only four were called as homozygous variants (i.e., > 80% variant reads) (Tables S2 and S3). Of the four remaining candidates, only two variants were conserved during evolution,6 one of which had previously been reported in our internal variant database in a patient with a different disorder. The remaining single candidate was a homozygous T>G change in ANO10 that was highly conserved (phyloP score: 5.26) and also scored high on the Grantham scale (145) (Table S3).
ANO10 consists of 13 exons, 12 of which are coding, spanning 2734 bp and 660 amino acids (NM_018075.3). All exons of ANO10 were covered by sequence reads from the targeted next-generation sequencing experiment (Figure 1B). The homozygous T>G change at position 1529 that was detected by next-generation sequencing occurred in exon 10 and is predicted to result in an amino acid substitution of leucine by arginine at codon 510 (p.Leu510Arg). The mutated nucleotide (at position g.43,571,913; hg18, NCBI build 36.1) was covered by 18 unique sequence reads. In 17 reads, the mutant allele was detected, indicating that the mutation is present in a homozygous state (Figure 1C). The c.1529T>G (p.Leu510Arg) mutation was confirmed by conventional Sanger sequencing and cosegregated with the disease in this family (Figures 1A and 1D). The mutation was not present in over 300 control alleles (Table 2). This residue is conserved across multiple vertebrate species (Figure 1E) and is located in the DUF590 domain, a domain of unknown function.
Table 2.
Overview of the Mutations Identified in ANO10
| Family | Proband | No. of Affected Siblings with the Mutation | Mutation (cDNA Level) | Mutation (Protein Level) | Frequency in Control Alleles |
|---|---|---|---|---|---|
| A | VII.1 | 3 | c.1529T>G/c.1529T>G | p.Leu510Arg (homo) | 0/300 |
| B | II.5 | 3 | c.1150_1151del/ c.1150_1151del | p.Leu384fs (homo) | 0/300 |
| C | II.3 | 2 | c.1476+1G>T/c.1604del | a/p.Leu535X (hetero) | 0/300 |
The effect of the splice-site mutation at the protein level has not been studied
Next, we analyzed this gene by conventional Sanger sequencing in a consanguineous family (family B) with three affected siblings of Romani ethnic origin from Serbia (primer sequences are listed in Table S4). In family B an independent linkage analysis was performed, and the candidate region overlapped with the whole region in the Dutch family initially studied (data not shown). A homozygous 2 bp deletion (TT) at position 1150 (c.1150_1151del) was identified, leading to a frameshift (p.Leu384fs) in exon 6 (Table 2). This frameshift mutation cosegregated with the disease in this family (Figure 2) and introduces a stop codon at position 474. The mutation was not present in the dbSNP130 database and was not detected in over 300 control alleles. The affected siblings in this second cerebellar ataxia family also manifested tortuosity of conjunctival vessels and intellectual disability in two siblings (Table 1). Furthermore, ANO10 mutation analysis was performed in a large cohort of 282 index patients with cerebellar ataxia with presumable autosomal-recessive inheritance. In most patients of this cohort, Friedreich ataxia (FA [MIM 22930]) had already been excluded by mutation analysis of FRDA. Two compound-heterozygous mutations, a splice-site mutation in exon 9 (c.1476+1G>T) and a nonsense mutation in exon 10 (c.1604del [p.Leu535X]), were identified in a single family (family C) with two affected siblings of French descent (Figure 2). The phenotype of these patients is almost identical to the phenotype of the Dutch family (Table 1). An overview of the mutations identified in ANO10 is presented in Table 2. So far, we have identified 29 different SNPs in this gene.
Figure 2.
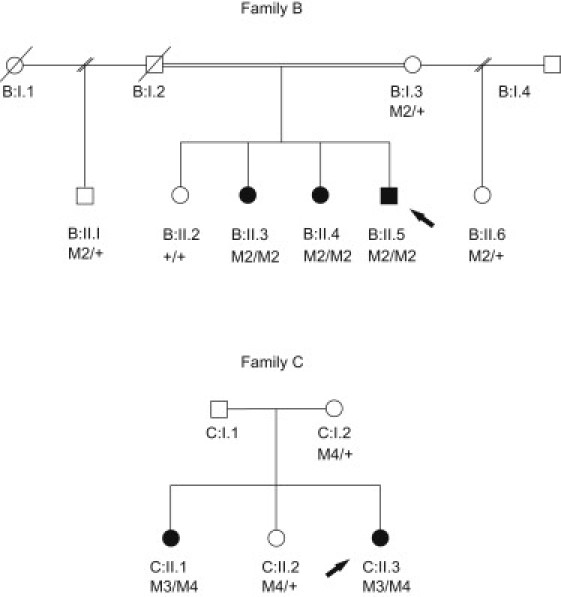
Pedigree Structure of Two Additional Families, B and C
Pedigrees for family B, from Serbia, and family C, from France, together with the segregation of the mutations identified in these families. M2/M2 indicates homozygous carriers of the p.Leu384fs mutation, and M2/+ indicates heterozygous carriers, whereas +/+ indicates individuals with two wild-type alleles. M3/M4 indicates compound-heterozygous carriers for the c.1476+1G>T and the p.Leu535X mutations, and M4/+ indicates heterozygous carriers for the p.Leu535X mutation.
For family B, the degree of consanguinity is not known. Unfortunately, DNA was not available for individual C:I.1.
In adult tissues, ANO10 has the highest expression in the brain, as shown by quantitative PCR (qPCR) (Figure 3A). Medium expression levels were found in the retina and heart, and low expression levels were found in other tissues tested. Comparing the ANO10 expression levels between several adult brain areas showed the highest expression levels in the frontal and occipital cortices and in the cerebellum (Figure 3B). In addition, we found that the expression of ANO10 in the fetal brain is lower than in the adult whole brain, indicating a specific function for ANO10 in the adult mature brain and especially in the cerebral cortex and the cerebellum, rather than in brain development, although a role of this gene in brain development cannot be excluded. The expression profile is consistent with the relatively late onset of ataxia. However, ANO10, as well as ANO2 (MIM 610109) and ANO8 (MIM 610216), were detected with similar distributions in the mantle layer of the neural tube and in the dorsal root ganglia at embryonic day 14.5 in murine embryos.7 Furthermore, expression studies during cephalic development in the mouse seems to show embryonic expression of ANO10 during brain development.8
Figure 3.
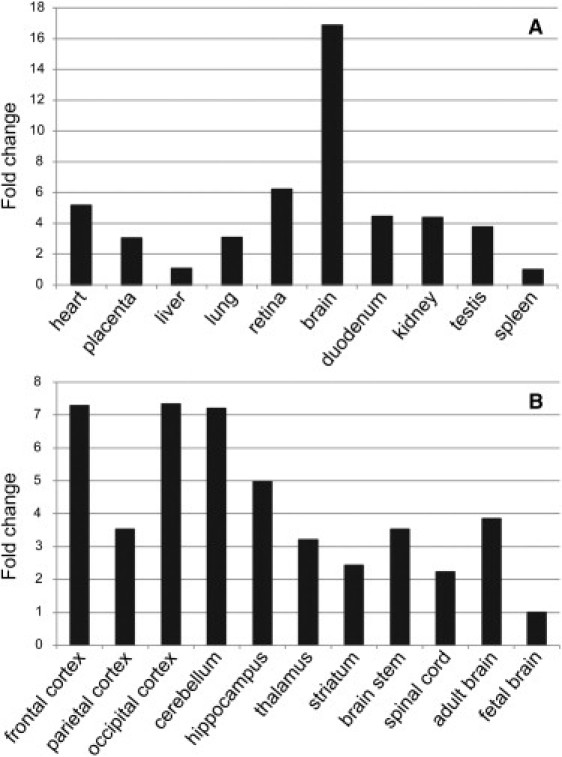
Expression of ANO10 in Different Human Tissues, Seen by qPCR
(A) Expression of ANO10 in different adult tissues, with the highest expression seen in the brain.
(B) Expression of ANO10 in different brain tissues, with the highest expression seen in the adult cerebellum and frontal and occipital cortices.
ANO10, also known as TMEM16K (transmembrane 16K), is a member of the human anoctamin (ANO) family, which comprises at least nine other proteins, all exhibiting eight transmembrane domains and a DUF590 domain of unknown function.9,10 Only very recently, it was proposed that some or all of the anoctamin genes code for cell- and tissue-specific calcium-activated chloride channels.11–13 In murine tissues, ANO10 is predominantly expressed in epithelial cells next to ANO1 (MIM 610108), ANO6 (MIM 608663), ANO7 (MIM 605096), ANO8, and ANO9, whereas ANO2, ANO3 (MIM 610110), ANO4 (MIM 610111), and ANO5 (MIM 608662) expression seem common in neuronal and muscle tissues. On the basis of results of the functional studies done by Schreiber et al., it appears likely that different anoctamins interact with each other.11
It is known that calcium-activated chloride channels have important physiological functions, including, among others, regulation of neuronal excitability.14 Whether ANO10 really codes for a calcium-regulated chloride channel remains to be confirmed. In the phylogenic tree of anoctamin genes, ANO1 is the member most distant from ANO10. A total of ∼25 amino acids are missing from the region coding for the reentrant loop between TM5 and TM6 in ANO10, compared to ANO1,10 possibly blocking the proper formation of a channel. It is also possible that the ANO10 product can function only in combination with other anoctamins to form heteromeric channels. Notwithstanding this, it is tempting to speculate that ANO10 indeed encodes a (subunit of a) calcium-regulated chloride channel. Deranged calcium signaling in Purkinje cells is one of the major mechanisms causing cerebellar ataxia.15,16 Several products of known ataxia-associated genes, such as calcium pumps and voltage-gated sodium or potassium channels, may increase or dampen calcium-signaling pathways in Purkinje cells.17–20 It could well be that the ANO10 product, which is postulated to be a calcium-dependent chloride channel, is also a player influencing calcium signaling in Purkinje cells, and a dysfunctional or absent ANO10 product may cause cerebellar ataxia via this mechanism. The identification of a putative chloride channel involved in a relatively pure, nonepisodic cerebellar ataxia may thus shed new light on the pathological mechanisms leading to cerebellar degeneration. Different molecular pathways, such as mitochondrial- and DNA-repair dysfunction, are already known to be involved. As mentioned, in autosomal-dominant cerebellar ataxias, involvement of calcium, sodium, and potassium channels has been demonstrated.19,21,22 However, to our knowledge, the involvement of a (calcium-activated) chloride channel in a cerebellar neurodegenerative disease has not yet been reported. Chloride channels are known to be involved in other diseases, such as myotonia congenita (MIM 160800 and MIM 255700), Dent disease (MIM 300009), cystic fibrosis (MIM 219700), and Bartter syndrome (MIM 602522).23–26 At present, however, the basic function of ANO10 is poorly understood, and so far most functional studies of anoctamins have been performed with ANO1.
Dominant mutations in ANO5 (also known as GDD1) have been associated with a rare skeletal disorder called gnathodiaphyseal dysplasia (GDD [MIM 166260]).27 Recently, recessive mutations in ANO5 have been identified in patients with limb girdle muscular dystrophy type 2L (LGMD2L [MIM 611307]) and distal nondysferlin Miyoshi myopathy (MMD3 [MIM 613319]).28 So far, no other members of the anoctamin gene family have been associated with genetic diseases in human.
In conclusion, we have identified mutations in ANO10 associated with autosomal-recessive cerebellar ataxia. This report illustrates that the combination of homozygosity mapping together with targeted array-based sequencing is a powerful tool for identifying causative genes for autosomal-recessive diseases. Mutations in ANO10 have so far been identified in three different families, with a total of eight patients, originating from the Netherlands, Serbia, and France. The phenotype of the patients is fairly similar (Table 1). Additional functional studies are to be awaited in order to understand how dysfunction of the ANO10-encoded putative chloride channel results in cerebellar ataxia.
Acknowledgments
We are grateful to the patients for their participation, to the referring clinicians, and especially to C. Haaxma, E. Kamping, E. Mundwiller, and the DNA and Cell Bank of the CR-ICM for their clinical and technical support. This work was supported by a grant from the Netherlands Organization of Health Research and Development (ZonMW RM000085 to N.K.). The next-generation sequencing platforms have been funded in part by the Netherlands Organization for Health Research and Development (ZonMW grants 917-66-36 and 911-08-025 to J.V.). Targeted sequence-capture experiments were performed in part by funding to H.S. from the European Community's Seventh Framework Programme FP7/2007-2013 under grant agreement number 223143 (project acronym: TECHGENE). Furthermore, we would like to thank the Verum Foundation (support to A.B.), the French National Agency for Research (support to A.D., M.K., and G.S.), and Agence Nationale pour la Recherche-Maladies Neurologiques et Psychiatriques (ANR-09-MNPS-001-01; support to M.K., A.D., and A.B.).
Contributor Information
Sascha Vermeer, Email: s.vermeer@antrg.umcn.nl.
Nine Knoers, Email: n.knoers@antrg.umcn.nl.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
dbSNP, build 130, http://www.ncbi.nlm.nih.gov/projects/SNP/snp_summary.cgi?build_id=130
easyLINKAGE, http://compbio.charite.de/genetik/hoffmann/easyLINKAGE/
HaploPainter, http://haplopainter.sourceforge.net/
Integrative Genomics Viewer, http://www.broadinstitute.org/igv
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
University of California-Santa Cruz (UCSC) Genome Bioinformatics, http://www.genome.ucsc.edu
References
- 1.Anheim M., Fleury M., Monga B., Laugel V., Chaigne D., Rodier G., Ginglinger E., Boulay C., Courtois S., Drouot N. Epidemiological, clinical, paraclinical and molecular study of a cohort of 102 patients affected with autosomal recessive progressive cerebellar ataxia from Alsace, Eastern France: implications for clinical management. Neurogenetics. 2010;11:1–12. doi: 10.1007/s10048-009-0196-y. [DOI] [PubMed] [Google Scholar]
- 2.Harding A.E. Classification of the hereditary ataxias and paraplegias. Lancet. 1983;1:1151–1155. doi: 10.1016/s0140-6736(83)92879-9. [DOI] [PubMed] [Google Scholar]
- 3.Siepel A., Bejerano G., Pedersen J.S., Hinrichs A.S., Hou M., Rosenbloom K., Clawson H., Spieth J., Hillier L.W., Richards S. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005;15:1034–1050. doi: 10.1101/gr.3715005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blanchette M., Kent W.J., Riemer C., Elnitski L., Smit A.F., Roskin K.M., Baertsch R., Rosenbloom K., Clawson H., Green E.D. Aligning multiple genomic sequences with the threaded blockset aligner. Genome Res. 2004;14:708–715. doi: 10.1101/gr.1933104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hoischen A., van Bon B.W., Gilissen C., Arts P., van Lier B., Steehouwer M., de Vries P., de Reuver R., Wieskamp N., Mortier G. De novo mutations of SETBP1 cause Schinzel-Giedion syndrome. Nat. Genet. 2010;42:483–485. doi: 10.1038/ng.581. [DOI] [PubMed] [Google Scholar]
- 6.Pollard K.S., Hubisz M.J., Rosenbloom K.R., Siepel A. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res. 2010;20:110–121. doi: 10.1101/gr.097857.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rock J.R., Futtner C.R., Harfe B.D. The transmembrane protein TMEM16A is required for normal development of the murine trachea. Dev. Biol. 2008;321:141–149. doi: 10.1016/j.ydbio.2008.06.009. [DOI] [PubMed] [Google Scholar]
- 8.Gritli-Linde A., Vaziri Sani F., Rock J.R., Hallberg K., Iribarne D., Harfe B.D., Linde A. Expression patterns of the Tmem16 gene family during cephalic development in the mouse. Gene Expr. Patterns. 2009;9:178–191. doi: 10.1016/j.gep.2008.11.002. [DOI] [PubMed] [Google Scholar]
- 9.Galindo B.E., Vacquier V.D. Phylogeny of the TMEM16 protein family: some members are overexpressed in cancer. Int. J. Mol. Med. 2005;16:919–924. [PubMed] [Google Scholar]
- 10.Hartzell H.C., Yu K., Xiao Q., Chien L.T., Qu Z. Anoctamin/TMEM16 family members are Ca2+-activated Cl- channels. J. Physiol. 2009;587:2127–2139. doi: 10.1113/jphysiol.2008.163709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schreiber R., Uliyakina I., Kongsuphol P., Warth R., Mirza M., Martins J.R., Kunzelmann K. Expression and function of epithelial anoctamins. J. Biol. Chem. 2010;285:7838–7845. doi: 10.1074/jbc.M109.065367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang Y.D., Cho H., Koo J.Y., Tak M.H., Cho Y., Shim W.S., Park S.P., Lee J., Lee B., Kim B.M. TMEM16A confers receptor-activated calcium-dependent chloride conductance. Nature. 2008;455:1210–1215. doi: 10.1038/nature07313. [DOI] [PubMed] [Google Scholar]
- 13.Caputo A., Caci E., Ferrera L., Pedemonte N., Barsanti C., Sondo E., Pfeffer U., Ravazzolo R., Zegarra-Moran O., Galietta L.J. TMEM16A, a membrane protein associated with calcium-dependent chloride channel activity. Science. 2008;322:590–594. doi: 10.1126/science.1163518. [DOI] [PubMed] [Google Scholar]
- 14.Hartzell C., Putzier I., Arreola J. Calcium-activated chloride channels. Annu. Rev. Physiol. 2005;67:719–758. doi: 10.1146/annurev.physiol.67.032003.154341. [DOI] [PubMed] [Google Scholar]
- 15.Carlson K.M., Andresen J.M., Orr H.T. Emerging pathogenic pathways in the spinocerebellar ataxias. Curr. Opin. Genet. Dev. 2009;19:247–253. doi: 10.1016/j.gde.2009.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kasumu A., Bezprozvanny I. Deranged Calcium Signaling in Purkinje Cells and Pathogenesis in Spinocerebellar Ataxia 2 (SCA2) and Other Ataxias. The Cerebellum. 2010 doi: 10.1007/s12311-010-0182-9. Published online May 18, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Saegusa H., Wakamori M., Matsuda Y., Wang J., Mori Y., Zong S., Tanabe T. Properties of human Cav2.1 channel with a spinocerebellar ataxia type 6 mutation expressed in Purkinje cells. Mol. Cell. Neurosci. 2007;34:261–270. doi: 10.1016/j.mcn.2006.11.006. [DOI] [PubMed] [Google Scholar]
- 18.van de Leemput J., Chandran J., Knight M.A., Holtzclaw L.A., Scholz S., Cookson M.R., Houlden H., Gwinn-Hardy K., Fung H.C., Lin X. Deletion at ITPR1 underlies ataxia in mice and spinocerebellar ataxia 15 in humans. PLoS Genet. 2007;3:e108. doi: 10.1371/journal.pgen.0030108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Waters M.F., Minassian N.A., Stevanin G., Figueroa K.P., Bannister J.P., Nolte D., Mock A.F., Evidente V.G., Fee D.B., Müller U. Mutations in voltage-gated potassium channel KCNC3 cause degenerative and developmental central nervous system phenotypes. Nat. Genet. 2006;38:447–451. doi: 10.1038/ng1758. [DOI] [PubMed] [Google Scholar]
- 20.Trudeau M.M., Dalton J.C., Day J.W., Ranum L.P., Meisler M.H. Heterozygosity for a protein truncation mutation of sodium channel SCN8A in a patient with cerebellar atrophy, ataxia, and mental retardation. J. Med. Genet. 2006;43:527–530. doi: 10.1136/jmg.2005.035667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhuchenko O., Bailey J., Bonnen P., Ashizawa T., Stockton D.W., Amos C., Dobyns W.B., Subramony S.H., Zoghbi H.Y., Lee C.C. Autosomal dominant cerebellar ataxia (SCA6) associated with small polyglutamine expansions in the alpha 1A-voltage-dependent calcium channel. Nat. Genet. 1997;15:62–69. doi: 10.1038/ng0197-62. [DOI] [PubMed] [Google Scholar]
- 22.Brusse E., de Koning I., Maat-Kievit A., Oostra B.A., Heutink P., van Swieten J.C. Spinocerebellar ataxia associated with a mutation in the fibroblast growth factor 14 gene (SCA27): A new phenotype. Mov. Disord. 2006;21:396–401. doi: 10.1002/mds.20708. [DOI] [PubMed] [Google Scholar]
- 23.Lloyd S.E., Pearce S.H., Fisher S.E., Steinmeyer K., Schwappach B., Scheinman S.J., Harding B., Bolino A., Devoto M., Goodyer P. A common molecular basis for three inherited kidney stone diseases. Nature. 1996;379:445–449. doi: 10.1038/379445a0. [DOI] [PubMed] [Google Scholar]
- 24.Koch M.C., Steinmeyer K., Lorenz C., Ricker K., Wolf F., Otto M., Zoll B., Lehmann-Horn F., Grzeschik K.H., Jentsch T.J. The skeletal muscle chloride channel in dominant and recessive human myotonia. Science. 1992;257:797–800. doi: 10.1126/science.1379744. [DOI] [PubMed] [Google Scholar]
- 25.Riordan J.R., Rommens J.M., Kerem B., Alon N., Rozmahel R., Grzelczak Z., Zielenski J., Lok S., Plavsic N., Chou J.L. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 1989;245:1066–1073. doi: 10.1126/science.2475911. [DOI] [PubMed] [Google Scholar]
- 26.Birkenhäger R., Otto E., Schürmann M.J., Vollmer M., Ruf E.M., Maier-Lutz I., Beekmann F., Fekete A., Omran H., Feldmann D. Mutation of BSND causes Bartter syndrome with sensorineural deafness and kidney failure. Nat. Genet. 2001;29:310–314. doi: 10.1038/ng752. [DOI] [PubMed] [Google Scholar]
- 27.Tsutsumi S., Kamata N., Vokes T.J., Maruoka Y., Nakakuki K., Enomoto S., Omura K., Amagasa T., Nagayama M., Saito-Ohara F. The novel gene encoding a putative transmembrane protein is mutated in gnathodiaphyseal dysplasia (GDD) Am. J. Hum. Genet. 2004;74:1255–1261. doi: 10.1086/421527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bolduc V., Marlow G., Boycott K.M., Saleki K., Inoue H., Kroon J., Itakura M., Robitaille Y., Parent L., Baas F. Recessive mutations in the putative calcium-activated chloride channel Anoctamin 5 cause proximal LGMD2L and distal MMD3 muscular dystrophies. Am. J. Hum. Genet. 2010;86:213–221. doi: 10.1016/j.ajhg.2009.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.