

Abstract
The tight junction, or zonula occludens, is a specialized cell-cell junction that regulates epithelial and endothelial permeability, and it is an essential component of the blood-brain barrier in the cerebrovascular endothelium. In addition to functioning as a diffusion barrier, tight junctions are also involved in signal transduction. In this study, we identified a homozygous mutation in the tight-junction protein gene JAM3 in a large consanguineous family from the United Arab Emirates. Some members of this family had a rare autosomal-recessive syndrome characterized by severe hemorrhagic destruction of the brain, subependymal calcification, and congenital cataracts. Their clinical presentation overlaps with some reported cases of pseudo-TORCH syndrome as well as with cases involving mutations in occludin, another component of the tight-junction complex. However, massive intracranial hemorrhage distinguishes these patients from others. Homozygosity mapping identified the disease locus in this family on chromosome 11q25 with a maximum multipoint LOD score of 6.15. Sequence analysis of genes in the candidate interval uncovered a mutation in the canonical splice-donor site of intron 5 of JAM3. RT-PCR analysis of a patient lymphoblast cell line confirmed abnormal splicing, leading to a frameshift mutation with early termination. JAM3 is known to be present in vascular endothelium, although its roles in cerebral vasculature have not been implicated. Our results suggest that JAM3 is essential for maintaining the integrity of the cerebrovascular endothelium as well as for normal lens development in humans.
Main Text
Tight junctions are composed of a complex of transmembrane proteins, which include claudins, occludins and JAMs (junctional adhesion molecules), and cytoplasmic plaque proteins, which include various scaffolding, adaptor and regulatory proteins.1 The molecular composition of the tight junction varies among different tissue types.2 In the cerebrovascular endothelium, tight junctions are an essential component of the blood-brain barrier.
There are several human genetic disorders resulting from mutations in genes that encode tight-junction proteins. These include familial hypercholanemia (MIM 607748),3 autosomal-recessive deafness 29 (DFNB29; MIM 605608),4 and 49 (DFNB49; MIM 610153),5 and Nance-Horan syndrome (MIM 302350).6 The wide range of clinical presentation of these disorders underscores the variable functions and molecular composition of tight junctions in different organs. However, no disorder of tight junctions associated with severe hemorrhage has been reported to date, despite the fact that tight junctions are essential to the integrity of vascular endothelial cells in various organs. Here we report a mutation in the tight-junction gene JAM3 in a large pedigree displaying a unique autosomal-recessive syndrome with severe hemorrhagic destruction of the brain as a cardinal feature.
In the United Arab Emirates, we ascertained a large consanguineous pedigree with its origin in Baluchistan (the region involving southeastern Iran, western Pakistan, and southwestern Afghanistan). In this pedigree, eight individuals in four branches showed a unique clinical syndrome characterized by hemorrhagic destruction of the brain parenchyma, subependymal calcification, and congenital cataracts (family OTH10700; Figure 1). All surviving patients developed generalized spasticity, exaggerated deep-tendon reflexes, and seizures. Hepatomegaly was documented in three patients, and of the four patients who had abdominal ultrasound, three were found to have renal anomalies. Thrombocytopenia was seen in one patient in the setting of bacterial meningitis, and no patient had persistently abnormal neutrophil counts (clinical findings are summarized in Table 1). Screening for congenital infections (serological examination for cytomegalovirus, toxoplasmosis, and rubella, plus urine examination for cytomegalovirus particles) was negative in individuals VIII-17, VIII-21, and VIII-22. Of note, there is a second autosomal-recessive disorder, short rib-polydactyly syndrome, type III (MIM 263510), that segregates independently in this family, and one individual (VIII-21) was affected by both conditions. Clinical description of part of this pedigree was previously published.7 All affected individuals who had brain imaging showed evidence of a destructive brain disorder (Figure 2). Individuals VIII-21, VIII-22, and VIII-10 had multifocal intraparenchymal hemorrhage with associated liquefaction, predominantly in the cerebral white matter and basal ganglia (Figures 2A–2D). A series of T1-weighted sagittal brain MR images of individual VIII-10 obtained at the age of 4 weeks are presented as a supplemental movie (Movie S1). Individual VIII-27 had massive cystic destruction of the cerebral white matter and basal ganglia, resulting in large ventricles (Figure 2E). Individual VIII-29 had a porencephalic cyst centered in the left frontal subcortical white matter, enlarged lateral ventricles, and reduced white matter volume, indicative of destructive changes (Figure 2F). Subependymal, basal-ganglia, and white-matter calcification was evident in all patients who underwent a brain CT scan (VIII-21, VIII-22, and VIII-29; Figures 2B, 2C, and 2F, respectively; in addition, a brain CT image of individual VIII-17 was published in Al-Gazali et al.7 [this individual is referred to as case 3 in the paper]). Abnormalities of the posterior fossa were seen in some cases: individual VIII-21 had a thin midbrain and pons; individual VIII-10 had mildly atrophic pons, cerebellar vermis, and hemispheres; individual VIII-27 had small pons, medulla, and cerebellar hemispheres, a small, upwardly rotated vermis, and a large fourth ventricle; and individual VIII-29 had a small right cerebellar hemisphere. It could not be determined whether these posterior fossa findings were the result of primary injury or were secondary to supratentorial lesions. No definitive evidence of a developmental cortical malformation was noted in any of the patients, although the severe destructive lesions made it impossible to rule out a coexisting developmental malformation. The distribution of both hemorrhage and cystic changes is most suggestive of a disorder involving small vessels. The exact time of onset of the intracranial lesions is difficult to determine. However, the imaging characteristics suggested lesions of varying ages. The signal characteristics of hemorrhagic lesions on MRI of individuals VIII-21 (MRI was performed postmortem because the patient died immediately after birth) and VIII-10 (initial MRI was performed at 4 days of age) were suggestive of acute or subacute lesions. Individual VIII-27 was noted to have enlarged ventricles on a third-trimester ultrasound and large areas of hemispheric porencephaly on postnatal MRI, suggesting very remote hemorrhage or infarction.
Figure 1.
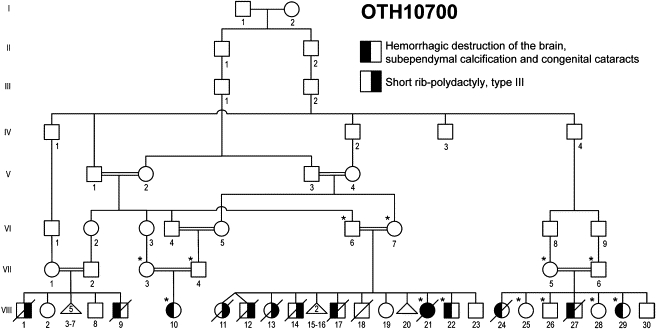
Pedigree Drawing of the Family OTH10700
Filled symbols denote affected individuals. The left half of the symbol is filled for the syndrome of hemorrhagic destruction of the brain, subependymal calcification, and congenital cataracts, and the right half of the symbol is filled for short rib-polydactyly syndrome, type III. Genotyped individuals are indicated with an asterisk.
Table 1.
Summary of Clinical Findings of the Family OTH10700
| Individual | Gender | Age | Birth Head Circumference | Current Head Circumference | Seizures | Ophthalmological Findings | Cardiac Findings | Hepatomegaly | Renal Findings | Thrombocytopenia | Other Organ Involvement |
|---|---|---|---|---|---|---|---|---|---|---|---|
| VIII-17 | Male | Died at 9 days | 35 cm (−0.4 SD) | Not applicable | No information | Bilateral congenital cataracts | Patent ductus arteriosus and ventricular septal defect | Yes | Normal by renal ultrasound | No | No information |
| VIII-21 | Female | Died a few minutes after birth | 34 cm (−0.5 SD) | Not applicable | No information | Bilateral congenital catarcts | No information | Yes | Bilateral cystic dysplasia | No information | Short rib polydactyly, type III |
| VIII-22 | Male | 3 years 4 months | 33 cm (−1.2 SD) | 45.5 cm (−2.7 SD) | Yes | Bilateral congenital cataracts and microphthalmia | No information | No information | No information | No | Small phallus, undescended testes |
| VIII-10 | Female | 1 year 2 months | No information | 38.8 cm (−5.4 SD) | Yes | Bilateral congenital cataracts | Systolic heart murmur, normal cardiac ultrasound | Yes | No information | Yes (during bacterial meningitis) | No information |
| VIII-27 | Male | Died at 2 months | No information | Not applicable | No information | Bilateral congenital cataracts, slightly pale optic discs, normal retina | Normal cardiac ultrasound | No information | Small ectopic right kidney in the iliac fossa | No information | No information |
| VIII-29 | Female | 6 years 1 month | No information | 41 cm (−8.1 SD) | Yes | Bilateral congenital cataracts | No heart murmur | No information | Ectopic, malrotated left kidney in the iliac fossa | No | No information |
Individual ID numbers correspond to those in Figure 1.
Figure 2.
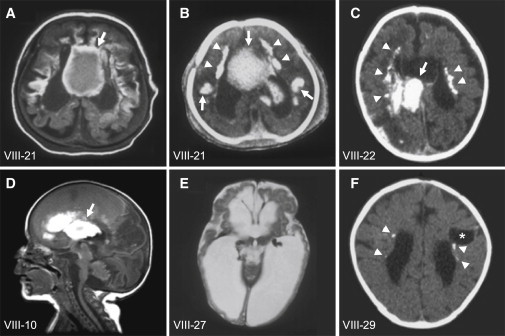
Brain Imaging of the Family OTH10700
A T1-weighted axial brain MRI (A) and brain CT (B) of individual VIII-21, a brain CT of individual VIII-22 (C), and a T1-weighted sagittal brain MRI of individual VIII-10 (D) show multifocal intraparenchymal hemorrhage, predominantly in the cerebral white matter and basal ganglia (arrows). A T2-weighted axial brain MRI of individual VIII-27 (E) shows massive cystic destruction of the cerebral white matter and basal ganglia and resulting large lateral ventricles. A brain CT of individual VIII-29 (F) shows a porencephalic cyst centered in the left frontal subcortical white matter (asterisk), enlarged lateral ventricles, and reduced white-matter volume, indicative of destructive changes. Subependymal calcification is evident on CT (B, C and F, arrowheads). Figure 2A is from the same MRI series previously published in Al-Gazali et al.7 (this individual is referred to as case 4 in the paper).
We collected peripheral blood samples from the affected children and parents after obtaining written informed consent according to the protocols approved by the participating institutions. All the research procedures were in accordance with the ethical standards of the responsible national and institutional committees on human-subject research. A total of 13 individuals (indicated with an asterisk in Figure 1) were genotyped via Illumina HumanHap550-Duo SNP microarray. Only one block of homozygosity on chromosome 11q25 emerged as being shared by all affected individuals tested (VIII-21, VIII-22, VIII-10, and VIII-29). The list of homozygous blocks is presented in Table S1. The candidate interval was defined by the recombinant SNP markers rs1627835 and rs1893054 and was 8.5 cM (3.2 Mb) in size. One of the unaffected siblings, VIII-26, shared homozygosity in the proximal portion of this interval, and this region was eliminated from the candidate interval. Thus, the minimal critical interval was defined by SNP markers rs12271299 and rs1893054 and measured 1.9 cM (1.1 Mb) (Figure 3A). Because of computational limitations, LOD scores across this region were calculated with a set of 175 SNP markers (every eighth marker out of the 1400 markers encompassed the minimal critical interval) with SimWalk2.8 The calculation was made under the assumption of a recessive mode of inheritance with 100% penetrance and a disease allele prevalence of 0.0001, and a maximum multipoint LOD score of 6.15 was obtained (Figure 3B). A second calculation involving a different set of 175 SNP markers in the same interval gave comparable results, with a slightly narrower peak and a maximum multipoint LOD score of 5.88 (data not shown). Sequence analysis of the coding exons of the nine protein-coding RefSeq genes in the linked interval (Figure 3C; see also Table S2) revealed a candidate mutation in JAM3 (Junctional Adhesion Molecule 3; MIM 606871; NM_032801.3) only. The homozygous nucleotide change occurred at the first base position of intron 5 (c.747+1G>T; Figure 4A). All affected children were homozygous for this change and the parents, who are obligate carriers, were all heterozygous. Two different web-based programs, Neural Network9 and NetGene2,10 predict that the c.747+1G>T nucleotide change abolishes the wild-type splice donor site and reveal a cryptic splice donor site 19 base pairs downstream into intron 5 (Figure 4A). Sequences of the primers for coding exons of JAM3 were designed with Primer311 and are presented in Table S3. We sequenced the intron 5 splice donor site of JAM3 in a total of 370 control individuals (740 chromosomes), including 270 controls of European descent (Coriell Institute for Medical Research) and 100 United Arab Emirates nationals, and did not find the same nucleotide change, either in a homozygous or a heterozygous state.
Figure 3.
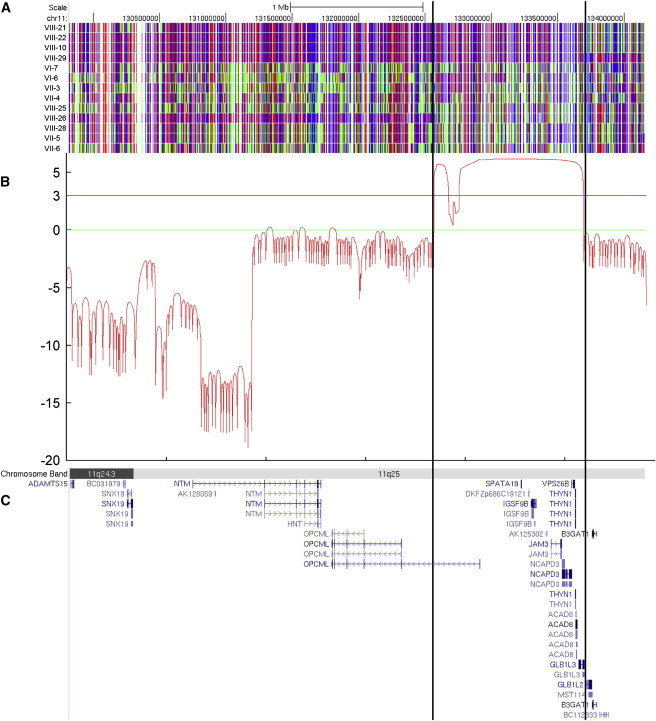
Homozygosity Mapping and Linkage Analysis of the Family OTH10700
(A) SNP genotyping results on chromosome 11q25. A block of homozygosity shared only by the affected individuals tested (VIII-21, VIII-22, VIII-10, and VIII-29) is evident and delineated by the vertical lines. Red and blue indicate homozygous SNP markers, and green indicates heterozygous SNP markers.
(B) LOD score graph of the candidate region. A maximum multipoint LOD score of 6.15 was obtained within the candidate interval.
(C) Genes within the candidate region, shown as the RefSeq track of the UCSC Genome Browser (NCBI build 36/hg18).
Figure 4.
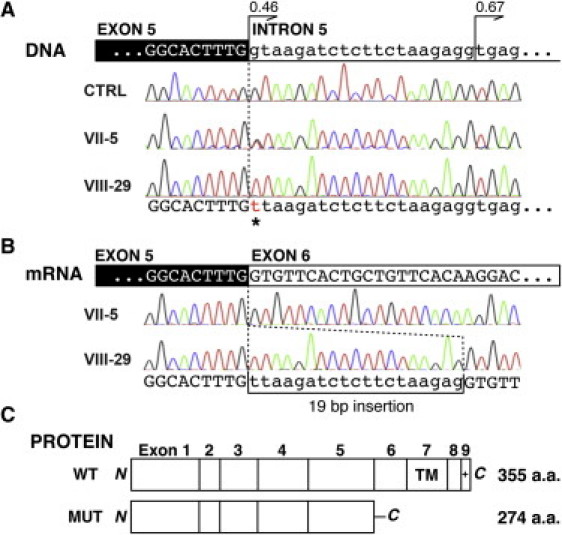
Mutation in JAM3
(A) Genomic DNA analysis of JAM3. Affected individual VIII-29 shows a G-to-T change in the canonical splice donor site of JAM3 intron 5 (c.747+1G>T; asterisk). Her mother (VII-5) is heterozygous for the mutation. A cryptic splice donor site 19 bp downstream is at least equally as potent as the wild-type donor site (confidence score 0.46 [wild-type] versus 0.67 [cryptic] by NetGene2). CTRL = normal control.
(B) mRNA analysis of JAM3. On the mRNA level, the mutation results in the usage of the downstream cryptic splice donor site and causes a 19 bp insertion in the affected individual (VIII-29). Only normal splice products are seen in her mother (VII-5).
(C) Predicted protein product of the mutant allele. The 19 bp insertion is expected to result in a truncating protein product with 25 aberrant amino acid residues in the C terminus (WT = wild-type protein, MUT = mutant protein). The truncation occurs proximal to the transmembrane domain (TM) and PDZ-binding motif (+).
RT-PCR experiments with transformed lymphoblast cell lines from one of the patients and her parent confirmed that this mutation disrupted normal splicing of JAM3. RNA was extracted from the cell lines, and cDNA was generated by reverse-transcriptase reaction, amplified by PCR, and sequenced (primer sequences are presented in Table S4). As predicted by the splice prediction programs, the patient's sample showed only the product in which the first 19 bases of intron 5 were inserted between exons 5 and 6 (Figure 4B). This insertion results in a frameshift, and 25 aberrant amino acids are incorporated before early termination (p.Val250LeufsX26; Figure 4C). The resulting protein is predicted to lack the transmembrane domain and PDZ-binding motif and therefore is expected to be nonfunctional. The parent's sample showed only wild-type sequence, and the abnormal splice product seen in the patient was not detected (Figure 4B). This might suggest that the abnormal splice product is less stable, and the normal splice product is preferentially amplified when both products are present.
The family we studied shows some similarities to cases of pseudo-TORCH syndrome (MIM251290) reported in the literature but has distinct clinical features as well. Pseudo-TORCH syndrome is characterized by congenital microcephaly, intracranial calcification, seizures, severe developmental delay, and sometimes thrombocytopenia and hepatomegaly.12 Another form of pseudo-TORCH syndrome has emerged and is characterized by band-like intracranial calcification, simplified cerebral gyral pattern, polymicrogyria, and progressive microcephaly but lacks hepatomegaly or thrombocytopenia.13–15 Recently, the latter group of patients was found to have mutations in another tight-junction gene, OCLN (occludin; MIM 602876), underscoring the importance of tight-junction proteins in the developing brain.16 However, the patients we studied did not show evidence of polymicrogyria or other developmental malformations of the cerebral cortex, and the patients with OCLN mutations did not have intracranial hemorrhage or congenital cataracts. No other reported patients show exactly the same clinical features as the family we report here, and we have not encountered a similar condition in our own experience with more than 1000 cases of congenital brain disorders. Perhaps the most similar patients in the literature are two brothers reported by Knoblauch et al.; these brothers had intracranial hemorrhage and calcification but no cataracts.17 Genomic DNA of these siblings was sequenced for the coding exons of JAM3, but no mutation was identified (data not shown).
JAMs are immunoglobulin superfamily proteins that localize to tight junctions of epithelial and endothelial cells.18 They are implicated in a wide range of physiological and pathological functions through cell adhesion involving homophilic and heterophilic interactions. These activities include barrier function, leukocyte migration, platelet activation, and angiogenesis (reviewed by Mandell and Parkos19). Full-length human JAM3 is an approximately 43 kDa protein with two extracellular immunoglobulin-like domains, a transmembrane domain, and a C-terminal PDZ-binding motif.20 Human JAM3 is expressed at a high level in the brain, kidney, and placenta.20 It is interesting to note that three out of four affected individuals who had abdominal imaging (VIII-21, VIII-27, and VIII-29) were found to have abnormalities in one or both kidneys (Table 1).
Engineered mice deficient for the mouse ortholog of JAM3 (Jam3; also known as JAM-C) have been reported21 and show growth retardation and increased susceptibility to multilobar pneumonia and related death.22 They have nuclear cataracts23 and defects in multiple organ system functions, including spermatid differentiation,21 peripheral nerve conduction,24 and granulocyte homeostasis.22 Abnormalities in the central nervous system have not been reported in these mice, however. Jam3 (referred to as JAM-2 in Aurrand-Lions et al.25) is reported to be absent in vasculature of the adult mouse brain,25 and in situ hybridization of developing and adult mouse brains shows only a low level of Jam3 expression (Allen Mouse Brain Atlas and Allen Developing Mouse Brain Atlas). This is in sharp contrast to the high level of expression of JAM3 in the human adult brain,20 suggesting that the tissue distribution of orthologous tight-junction proteins varies significantly between species. In the peripheral nervous system, Jam3 is localized to Schwann cells at junctions between adjoining myelin end loops, and JAM3 has a similar localization pattern in human sural nerves.24 The presence of exaggerated deep-tendon reflexes in the patients we studied suggests that severe defects in nerve conduction are rather unlikely, but mild conduction defects cannot be ruled out without electrophysiological studies.
Cataracts are seen in Jam3-deficient mice as well as the patients presented here. Of note, NHS, the gene mutated in Nance-Horan syndrome, which is characterized by congenital cataracts, dental anomalies, and intellectual disability, has been shown to localize to tight junctions and be expressed in the developing lens epithelium.26 Thus, tight-junction proteins seem to play an important role in normal lens development. In addition, Jam3 and JAM3 are found in the mouse23 and human retina,27 respectively. However, Jam3-deficient mice have normal retinal morphology,23 and we did not find evidence of retinal pathology in the one patient who had undergone a documented fundoscopic examination (VIII-27). The role of JAM3 in retinal development is yet to be elucidated, but it has been shown to regulate transmigration of granulocytes in human retinal pigment epithelium, and it might play a role in retinal inflammatory processes.27
Three of the patients we studied had various renal abnormalities, and one had congenital heart disease (Table 1). Jam3-deficient mice have no apparent abnormalities in the heart or kidneys, however.28 A patient with hypoplastic left-heart and congenital thrombocytopenia was reported to have a paracentric inversion in 11q with a breakpoint in or near JAM3.28 Studies of additional patients with mutations in JAM3 would help determine its potential role in human cardiac and renal development. Although JAM3 is present on the surface of platelets,29 we believe the most likely cause of the intracerebral hemorrhage in these patients was malfunctioning tight junctions of cerebrovascular endothelial cells. The absence of a systemic bleeding diathesis in any of the patients argues against abnormalities of platelets or coagulation. In addition, three of the patients were confirmed to have a normal platelet count. Thrombocytopenia was noted in another patient, but in the setting of severe meningitis. However, there is also evidence that disruption of JAM3 function leads to a decrease, rather than an increase, in permeability of human dermal microvascular endothelial cells30 and human umbilical-vein endothelial cells.31 Therefore, the effect of the JAM3 mutation we identified might not simply be increased vascular permeability throughout the body, and it might argue for a unique function of JAM3 in the cerebrovascular endothelium. Intraparenchymal hemorrhage without definable cause is sometimes seen in term neonates,32 and it would also be of interest to investigate the role of JAM3 in such cases without other organ involvement.
Interestingly, mutations in COL4A1 (collagen, type IV, alpha 1; MIM 120130), which encodes a vascular basement membrane protein, lead to clinical presentations that overlap with those of the patients we studied and include antenatal intracranial hemorrhage,33 porencephaly (MIM 175780),34 brain small-vessel disease with hemorrhage or vascular leukoencephalopathy (MIM 607595),35,36 and hereditary angiopathy with nephropathy, aneurysms, and muscle cramps (MIM 611773).37 Ocular abnormalities including congenital cataracts have also been reported.36 Even though COL4A1 is expressed in blood vessels of numerous organs,38 the brain is the most susceptible organ for hemorrhage in the presence of COL4A1 mutations. This might further support a small-vessel origin of intracranial pathology of the patients presented here.
Acknowledgments
We would like to thank the family for their participation in this research. SNP genotyping was performed at The W.M. Keck Foundation Biotechnology Resource Laboratory at Yale University through the National Institutes of Health Neuroscience Microarray Consortium. We thank the Partners HealthCare Center for Personalized Genetic Medicine for establishing the lymphoblast cell lines. We thank Brenda J. Barry for coordinating patient studies and Philippe Schröter and Juliane Artelt for help with cell culture. This research was supported by grants from the National Institute of Neurological Disorders and Stroke (5R01NS035129-13) and Fogarty International Center (5R21TW008223-02) to C.A.W. and by the Nancy Lurie Marks Family Foundation, the Dubai Harvard Foundation for Medical Research, the Simons Foundation, and the Manton Center for Orphan Disease Research. G.H.M. was supported by the Young Investigator Award of the National Alliance for Research on Schizophrenia and Depression (NARSAD) as a NARSAD Lieber Investigator. C.A.W. is an Investigator of the Howard Hughes Medical Institute.
Contributor Information
Lihadh Al-Gazali, Email: l.algazali@uaeu.ac.ae.
Christopher A. Walsh, Email: christopher.walsh@childrens.harvard.edu.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Allen Mouse Brain Atlas, http://mouse.brain-map.org/
Allen Developing Mouse Brain Atlas, http://developingmouse.brain-map.org/
Berkeley Drosophila Genome Project: Splice Site Prediction by Neural Network, http://www.fruitfly.org/seq_tools/splice.html
NCBI Reference Sequence (RefSeq), http://www.ncbi.nlm.nih.gov/RefSeq/
NetGene2, http://www.cbs.dtu.dk/services/NetGene2/
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/omim/
Primer3, http://frodo.wi.mit.edu/primer3/
UCSC Genome Browser, http://genome.ucsc.edu/
References
- 1.Ebnet K. Organization of multiprotein complexes at cell-cell junctions. Histochem. Cell Biol. 2008;130:1–20. doi: 10.1007/s00418-008-0418-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vestweber D. Molecular mechanisms that control endothelial cell contacts. J. Pathol. 2000;190:281–291. doi: 10.1002/(SICI)1096-9896(200002)190:3<281::AID-PATH527>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 3.Carlton V.E., Harris B.Z., Puffenberger E.G., Batta A.K., Knisely A.S., Robinson D.L., Strauss K.A., Shneider B.L., Lim W.A., Salen G. Complex inheritance of familial hypercholanemia with associated mutations in TJP2 and BAAT. Nat. Genet. 2003;34:91–96. doi: 10.1038/ng1147. [DOI] [PubMed] [Google Scholar]
- 4.Wilcox E.R., Burton Q.L., Naz S., Riazuddin S., Smith T.N., Ploplis B., Belyantseva I., Ben-Yosef T., Liburd N.A., Morell R.J. Mutations in the gene encoding tight junction claudin-14 cause autosomal recessive deafness DFNB29. Cell. 2001;104:165–172. doi: 10.1016/s0092-8674(01)00200-8. [DOI] [PubMed] [Google Scholar]
- 5.Riazuddin S., Ahmed Z.M., Fanning A.S., Lagziel A., Kitajiri S., Ramzan K., Khan S.N., Chattaraj P., Friedman P.L., Anderson J.M. Tricellulin is a tight-junction protein necessary for hearing. Am. J. Hum. Genet. 2006;79:1040–1051. doi: 10.1086/510022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Burdon K.P., McKay J.D., Sale M.M., Russell-Eggitt I.M., Mackey D.A., Wirth M.G., Elder J.E., Nicoll A., Clarke M.P., FitzGerald L.M. Mutations in a novel gene, NHS, cause the pleiotropic effects of Nance-Horan syndrome, including severe congenital cataract, dental anomalies, and mental retardation. Am. J. Hum. Genet. 2003;73:1120–1130. doi: 10.1086/379381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Al-Gazali L.I., Sztriha L., Dawodu A., Varady E., Bakir M., Khdir A., Johansen J. Complex consanguinity associated with short rib-polydactyly syndrome III and congenital infection-like syndrome: a diagnostic problem in dysmorphic syndromes. J. Med. Genet. 1999;36:461–466. [PMC free article] [PubMed] [Google Scholar]
- 8.Sobel E., Sengul H., Weeks D.E. Multipoint estimation of identity-by-descent probabilities at arbitrary positions among marker loci on general pedigrees. Hum. Hered. 2001;52:121–131. doi: 10.1159/000053366. [DOI] [PubMed] [Google Scholar]
- 9.Reese M.G., Eeckman F.H., Kulp D., Haussler D. Improved splice site detection in Genie. J. Comput. Biol. 1997;4:311–323. doi: 10.1089/cmb.1997.4.311. [DOI] [PubMed] [Google Scholar]
- 10.Brunak S., Engelbrecht J., Knudsen S. Prediction of human mRNA donor and acceptor sites from the DNA sequence. J. Mol. Biol. 1991;220:49–65. doi: 10.1016/0022-2836(91)90380-o. [DOI] [PubMed] [Google Scholar]
- 11.Rozen S., Skaletsky H.J. Primer3 on the WWW for general users and for biologist programmers. In: Krawetz S., Misener S., editors. Bioinformatics Methods and Protocols: Methods in Molecular Biology. Humana Press; Totowa, New Jersey: 2000. pp. 365–386. [DOI] [PubMed] [Google Scholar]
- 12.Reardon W., Hockey A., Silberstein P., Kendall B., Farag T.I., Swash M., Stevenson R., Baraitser M. Autosomal recessive congenital intrauterine infection-like syndrome of microcephaly, intracranial calcification, and CNS disease. Am. J. Med. Genet. 1994;52:58–65. doi: 10.1002/ajmg.1320520112. [DOI] [PubMed] [Google Scholar]
- 13.Abdel-Salam G.M., Zaki M.S. Band-like intracranial calcification (BIC), microcephaly and malformation of brain development: a distinctive form of congenital infection like syndromes. Am. J. Med. Genet. A. 2009;149A:1565–1568. doi: 10.1002/ajmg.a.32894. [DOI] [PubMed] [Google Scholar]
- 14.Abdel-Salam G.M., Zaki M.S., Saleem S.N., Gaber K.R. Microcephaly, malformation of brain development and intracranial calcification in sibs: pseudo-TORCH or a new syndrome. Am. J. Med. Genet. A. 2008;146A:2929–2936. doi: 10.1002/ajmg.a.32549. [DOI] [PubMed] [Google Scholar]
- 15.Briggs T.A., Wolf N.I., D'Arrigo S., Ebinger F., Harting I., Dobyns W.B., Livingston J.H., Rice G.I., Crooks D., Rowland-Hill C.A. Band-like intracranial calcification with simplified gyration and polymicrogyria: A distinct “pseudo-TORCH” phenotype. Am. J. Med. Genet. A. 2008;146A:3173–3180. doi: 10.1002/ajmg.a.32614. [DOI] [PubMed] [Google Scholar]
- 16.O'Driscoll M.C., Daly S.B., Urquhart J.E., Black G.C., Pilz D.T., Brockmann K., McEntagart M., Abdel-Salam G., Zaki M., Wolf N.I. Recessive mutations in the gene encoding the tight junction protein occludin cause band-like calcification with simplified gyration and polymicrogyria. Am. J. Hum. Genet. 2010;87:354–364. doi: 10.1016/j.ajhg.2010.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Knoblauch H., Tennstedt C., Brueck W., Hammer H., Vulliamy T., Dokal I., Lehmann R., Hanefeld F., Tinschert S. Two brothers with findings resembling congenital intrauterine infection-like syndrome (pseudo-TORCH syndrome) Am. J. Med. Genet. A. 2003;120A:261–265. doi: 10.1002/ajmg.a.20138. [DOI] [PubMed] [Google Scholar]
- 18.Bazzoni G. The JAM family of junctional adhesion molecules. Curr. Opin. Cell Biol. 2003;15:525–530. doi: 10.1016/s0955-0674(03)00104-2. [DOI] [PubMed] [Google Scholar]
- 19.Mandell K.J., Parkos C.A. The JAM family of proteins. Adv. Drug Deliv. Rev. 2005;57:857–867. doi: 10.1016/j.addr.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 20.Arrate M.P., Rodriguez J.M., Tran T.M., Brock T.A., Cunningham S.A. Cloning of human junctional adhesion molecule 3 (JAM3) and its identification as the JAM2 counter-receptor. J. Biol. Chem. 2001;276:45826–45832. doi: 10.1074/jbc.M105972200. [DOI] [PubMed] [Google Scholar]
- 21.Gliki G., Ebnet K., Aurrand-Lions M., Imhof B.A., Adams R.H. Spermatid differentiation requires the assembly of a cell polarity complex downstream of junctional adhesion molecule-C. Nature. 2004;431:320–324. doi: 10.1038/nature02877. [DOI] [PubMed] [Google Scholar]
- 22.Imhof B.A., Zimmerli C., Gliki G., Ducrest-Gay D., Juillard P., Hammel P., Adams R., Aurrand-Lions M. Pulmonary dysfunction and impaired granulocyte homeostasis result in poor survival of Jam-C-deficient mice. J. Pathol. 2007;212:198–208. doi: 10.1002/path.2163. [DOI] [PubMed] [Google Scholar]
- 23.Daniele L.L., Adams R.H., Durante D.E., Pugh E.N., Jr., Philp N.J. Novel distribution of junctional adhesion molecule-C in the neural retina and retinal pigment epithelium. J. Comp. Neurol. 2007;505:166–176. doi: 10.1002/cne.21489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Scheiermann C., Meda P., Aurrand-Lions M., Madani R., Yiangou Y., Coffey P., Salt T.E., Ducrest-Gay D., Caille D., Howell O. Expression and function of junctional adhesion molecule-C in myelinated peripheral nerves. Science. 2007;318:1472–1475. doi: 10.1126/science.1149276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aurrand-Lions M., Duncan L., Ballestrem C., Imhof B.A. JAM-2, a novel immunoglobulin superfamily molecule, expressed by endothelial and lymphatic cells. J. Biol. Chem. 2001;276:2733–2741. doi: 10.1074/jbc.M005458200. [DOI] [PubMed] [Google Scholar]
- 26.Sharma S., Ang S.L., Shaw M., Mackey D.A., Gecz J., McAvoy J.W., Craig J.E. Nance-Horan syndrome protein, NHS, associates with epithelial cell junctions. Hum. Mol. Genet. 2006;15:1972–1983. doi: 10.1093/hmg/ddl120. [DOI] [PubMed] [Google Scholar]
- 27.Economopoulou M., Hammer J., Wang F., Fariss R., Maminishkis A., Miller S.S. Expression, localization, and function of junctional adhesion molecule-C (JAM-C) in human retinal pigment epithelium. Invest. Ophthalmol. Vis. Sci. 2009;50:1454–1463. doi: 10.1167/iovs.08-2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ye M., Hamzeh R., Geddis A., Varki N., Perryman M.B., Grossfeld P. Deletion of JAM-C, a candidate gene for heart defects in Jacobsen syndrome, results in a normal cardiac phenotype in mice. Am. J. Med. Genet. A. 2009;149A:1438–1443. doi: 10.1002/ajmg.a.32913. [DOI] [PubMed] [Google Scholar]
- 29.Erpenbeck L., Rubant S., Hardt K., Santoso S., Boehncke W.H., Schon M.P., Ludwig R.J. Constitutive and functionally relevant expression of JAM-C on platelets. Thromb. Haemost. 2010;103:857–859. doi: 10.1160/TH09-08-0572. [DOI] [PubMed] [Google Scholar]
- 30.Orlova V.V., Economopoulou M., Lupu F., Santoso S., Chavakis T. Junctional adhesion molecule-C regulates vascular endothelial permeability by modulating VE-cadherin-mediated cell-cell contacts. J. Exp. Med. 2006;203:2703–2714. doi: 10.1084/jem.20051730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li X., Stankovic M., Lee B.P., Aurrand-Lions M., Hahn C.N., Lu Y., Imhof B.A., Vadas M.A., Gamble J.R. JAM-C induces endothelial cell permeability through its association and regulation of β3 integrins. Arterioscler. Thromb. Vasc. Biol. 2009;29:1200–1206. doi: 10.1161/ATVBAHA.109.189217. [DOI] [PubMed] [Google Scholar]
- 32.Volpe J.J. Saunders/Elsevier; Philadelphia, Pennsylvania: 2008. Neurology of the newborn. pp. 483–516. [Google Scholar]
- 33.de Vries L.S., Koopman C., Groenendaal F., Van Schooneveld M., Verheijen F.W., Verbeek E., Witkamp T.D., van der Worp H.B., Mancini G. COL4A1 mutation in two preterm siblings with antenatal onset of parenchymal hemorrhage. Ann. Neurol. 2009;65:12–18. doi: 10.1002/ana.21525. [DOI] [PubMed] [Google Scholar]
- 34.Gould D.B., Phalan F.C., Breedveld G.J., van Mil S.E., Smith R.S., Schimenti J.C., Aguglia U., van der Knaap M.S., Heutink P., John S.W. Mutations in Col4a1 cause perinatal cerebral hemorrhage and porencephaly. Science. 2005;308:1167–1171. doi: 10.1126/science.1109418. [DOI] [PubMed] [Google Scholar]
- 35.Gould D.B., Phalan F.C., van Mil S.E., Sundberg J.P., Vahedi K., Massin P., Bousser M.G., Heutink P., Miner J.H., Tournier-Lasserve E. Role of COL4A1 in small-vessel disease and hemorrhagic stroke. N. Engl. J. Med. 2006;354:1489–1496. doi: 10.1056/NEJMoa053727. [DOI] [PubMed] [Google Scholar]
- 36.Sibon I., Coupry I., Menegon P., Bouchet J.P., Gorry P., Burgelin I., Calvas P., Orignac I., Dousset V., Lacombe D. COL4A1 mutation in Axenfeld-Rieger anomaly with leukoencephalopathy and stroke. Ann. Neurol. 2007;62:177–184. doi: 10.1002/ana.21191. [DOI] [PubMed] [Google Scholar]
- 37.Plaisier E., Gribouval O., Alamowitch S., Mougenot B., Prost C., Verpont M.C., Marro B., Desmettre T., Cohen S.Y., Roullet E. COL4A1 mutations and hereditary angiopathy, nephropathy, aneurysms, and muscle cramps. N. Engl. J. Med. 2007;357:2687–2695. doi: 10.1056/NEJMoa071906. [DOI] [PubMed] [Google Scholar]
- 38.Sado Y., Kagawa M., Naito I., Ueki Y., Seki T., Momota R., Oohashi T., Ninomiya Y. Organization and expression of basement membrane collagen IV genes and their roles in human disorders. J. Biochem. 1998;123:767–776. doi: 10.1093/oxfordjournals.jbchem.a022003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.