

Abstract
Limb-girdle muscular dystrophy (LGMD) is a genetically heterogeneous group of inherited muscular disorders manifesting symmetric, proximal, and slowly progressive muscle weakness. Using Affymetrix 250K SNP Array genotyping and homozygosity mapping, we mapped an autosomal-recessive LGMD phenotype to the telomeric portion of chromosome 8q in a consanguineous Turkish family with three affected individuals. DNA sequence analysis of PLEC identified a homozygous c.1_9del mutation containing an initiation codon in exon 1f, which is an isoform-specific sequence of plectin isoform 1f. The same homozygous mutation was also detected in two additional families during the analysis of 72 independent LGMD2-affected families. Moreover, we showed that the expression of PLEC was reduced in the patient's muscle and that there was almost no expression for plectin 1f mRNA as a result of the mutation. In addition to dystrophic changes in muscle, ultrastructural alterations, such as membrane duplications, an enlarged space between the membrane and sarcomere, and misalignment of Z-disks, were observed by transmission electron microscopy. Unlike the control skeletal muscle, no sarcolemmal staining of plectin was detected in the patient's muscle. We conclude that as a result of plectin 1f deficiency, the linkage between the sarcolemma and sarcomere is broken, which could affect the structural organization of the myofiber. Our data show that one of the isoforms of plectin plays a key role in skeletal muscle function and that disruption of the plectin 1f can cause the LGMD2 phenotype without any dermatologic component as was previously reported with mutations in constant exons of PLEC.
Main Text
Limb-girdle muscular dystrophy (LGMD [MIM 253600 and MIM 159000]) is a clinically well characterized, inherited form of muscular dystrophies with predominantly proximal or generalized progressive muscle weakness of symmetric nature.1,2 Both genders are equally affected, and the age at onset is usually between 5 and 30 years. Early motor milestones are usually normal; however, in some affected individuals, the initial walking age may be delayed. LGMDs are known to be genetically heterogeneous, and both autosomal-dominant (LGMD1) and -recessive (LGMD2) modes of inheritance are described. Autosomal-recessive LGMDs are more prevalant (90%) than the autosomal-dominant forms (10%) of the disease.3 To date, mutations in at least 15 genes have been identified as being responsible for the LGMD2 phenotype.4 These genes and proteins have variable functions across the muscle fiber, ranging from structural to enzymatic. The genetic etiology of many LGMD cases is not yet known, and there are still quite a number of families that are not linked to any of the known loci.
We identified a consanguineous Turkish family with four affected individuals with progressive muscle weakness (Figure 1A). The index case individual (family 1, patient VI:1) was a 19-year-old male with delayed independent walking at 3 years of age. Thereafter, he developed additional signs of weakness, with occasional falls and difficulties in climbing stairs. Until 14 years of age, he remained relatively stable. He then became progressively weak. At his final follow-up (19 years of age) he demonstrated a prolonged Gowers sign, with 12 s and generalized muscle weakness, with proximal muscle strength of 3+/5 on the MRC (Medical Research Council) scale (Figure 1B). He could not jump and walked with increased lumbar lordosis. Muscle hypertrophy was not observed. There was no fluctuation of symptoms throughout the day. Serum creatine kinase (CK) at the age of 19 years was 5584 U/l (n < 191). Echocardiogram showed normal anatomy and heart function. Pulmonary function tests did not show any restriction. There were no ocular or bulbar signs. Intelligence was normal, and the patient recently graduated from high school. A routine electromyogram (EMG) examination was clearly myopathic. Muscle biopsy showed dystrophic features with variation in fiber size, internal nuclei, scattered basophilic and few necrotic fibers, and mild endomysial fibrosis (Figure 1C). There was a predominance of type 2 fibers, and merosin, dystrophin, sarcoglycan (alpha, beta, gamma, and delta), and dystroglycan (alpha and beta) expression was normal on immunocytochemistry (Figure S1, available online).
Figure 1.
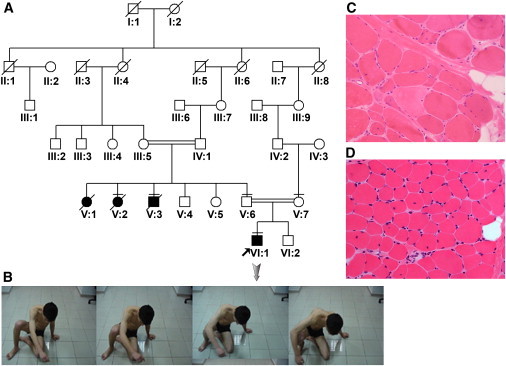
Clinical and Pathologic Findings
(A) Pedigree of family 1 with LGMD2. DNA was obtained from individuals III:5, IV:1, V:2, V.3, V:4, V:5, V:6, V:7, VI:1, VI:2. The individuals with a horizontal line (V:2, V:3, V:6, V:7, VI:1) in the pedigree are studied for homozygosity mapping.
(B) Clinical presentation of patient VI:1 (proband), who has difficulty in raising from the floor.
(C) Muscle biopsy of patient VI:1 from family 1, showing prominent variation in fiber size, few fibers with internal nuclei, necrotic fibers, and mild increase in endomysial and perimysial connective tissue (hematoxylin and eosin stain [H&E], original magnification 20×).
(D) Muscle biopsy of patient 20 from family 2, at the age of 4 yrs. Variation in fiber size, fibers with internal nuclei, and focal increase in endomysial connective tissue are seen (H&E, original magnification 20×).
There were three additional family members with similar muscle complaints. We examined one uncle, aged 36 years (family 1, patient V:3), at home (examined by H.T.) one year ago. This individual was basically bedridden, with near-total atrophy of muscles and evident multiple contractures, and he had stopped walking at 24 years of age. He was originally diagnosed with muscular dystrophy of unknown type at another center, and he declined further clinical evaluation and expired recently. From a clinical point of view, two of the patients (family 1, patients V:3 and VI:1) described herein had early-onset limb-girdle muscle symptoms followed by years of plateau. Our index case individual (patient VI:1) was experiencing progressive muscle weakness, which started to escalate after 14 years of age. In this specific family, there was one individual (patient V:1; Figure 1A) with similar muscle symptoms who died before the diagnosis of the index case. For genetic work up, DNA samples were obtained after informed consent was secured from the patients and relevant family members. The study protocol was approved by the Hacettepe University Faculty of Medicine Ethics Commitee (HEK 07/186).
We initially excluded all known LGMD2 loci by linkage analysis. Genome-wide linkage and homozygosity mapping was carried out with the 50K XbaI SNP array (Affymetrix). We could not identify a unique homozygous region in all of the affected individuals despite several approaches, including parametric linkage analysis (Figure S2). As shown in the family pedigree (Figure 1A), there are multiple and distant consanguineous marriages. Thus, multiple meiosis may result in a very small homozygous region that may have been missed as a result of the low resolution of the 50K SNP array. Therefore, we proceeded with GeneChip Mapping 250K NspI SNP arrays from Affymetrix, which have a higher resolution. Both parents (individuals V:6 and V:7; Figure 1A) and three affected members of the pedigree (patients V:2, V:3, and VI:1; Figure 1A) were included in the analysis. Subsequently, we identified a single homozygous stretch spanning 3.7 Mb between SNP marker rs7819473 and the telomere on chromosome 8q24 (Figure 2A).5,6 According to the NCBI Map Viewer (version 37.1) and the Ensembl database, the common interval contained approximately 130 known genes. Among these, plectin (PLEC [MIM 601282]) was a highly relevant candidate gene, given that it has already been associated with peculiar forms of muscular dystrophy.7–9 However, mutations in PLEC cause various forms of epidermolysis bullosa (EB [MIM 131900]), including EB simplex with muscular dystrophy (EBS-MD [MIM 226670]),7 EB simplex with pyloric atresia (EBS-PA [MIM 612138]),10,11 and autosomal-dominant inherited EB simplex Ogna (EBS-OG [MIM 131950]).12 Mutations associated with these disorders are generally located close to the C-terminal part of the protein containing the globular domain, which is a potential binding site for intermediate filaments such as keratin, desmin, or vimentin.
Figure 2.
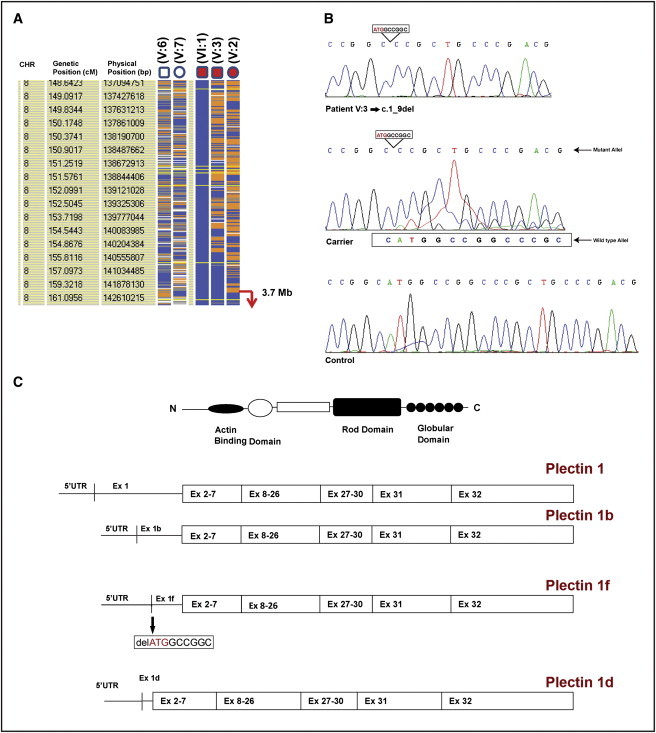
Disease Localization and Molecular Analysis
(A) Homozygosity data of the critical region on chromosome 8qtel. Genotype files (CHP) were generated with Affymetrix GTYPE software and were transferred to the VIGENOS (Visual Genome Studio) program (Hemosoft, Ankara), which facilitates visualization of a large amount of genome data in comprehensible visual screens.5,6 CHR indicates chromosome number. Genetic (cM) and physical (bp) position information were obtained from Affymetrix GTYPE. Parents (indicated as V:6 and V:7) and three affected individuals (indicated as V:2, V:3, and VI:1) from family 1 were included in the analysis. Affected individual VI:1 was chosen as an index case for the construction of genome-wide haplotypes. Homozygous genotypes identical to the genotype data obtained from the index case are shown in blue. Contrasting homozygous genotypes are shown in white, whereas heterozygous SNPs appear orange. Noninformativeness due to heterozygous genotypes in parent-child trios is indicated in yellow. Gray indicates no call. The overlapping homozygous stretches of three affected case individuals are approximately 3.7 Mb and are marked with an arrow in the telomeric part of the chromosome.
(B) Mutation detection by DNA sequence analysis of patient V:3 (homozygous c.1_9del mutation), his mother (heterozygous carrier), and a control subject. All amplification and sequencing primers used in this study are available in Table S1.
(C) Schematic representation of functional domains of plectin protein and four plectin isoforms (plectins 1, 1b, 1f, and 1d), which have unique N-terminal sequences. Exon 1f of PLEC (plectin isoform 1f) is mutated, with a 9 bp deletion including the initiation codon (c.1_9del).
PLEC is expressed in a wide variety of tissues, including skin and muscle.13 Because of its widespread distribution and multiple binding activities, plectin is one of the most important cytolinkers.14 Moreover, PLEC is a unique gene because of its unusual 5′ transcript diversity.13 It has a complex organization of 32 exons located on chromosome 8q24.15–18 Although exons 2–32 are constant exons, PLEC has eight alternative first exons that are spliced into a common exon 2.16 Additionally, variable first exons of PLEC are of different lengths and have no sequence similarity that makes the gene unique in the genome. It was previously reported that there was no conserved motif upstream of the variable exons and that the positions of the first exons correspond to the location of CpG islands, which implies that PLEC has variable multiple promoter regions.13,16 An alternative promoter region and alternative cis-splicing of PLEC can explain the tissue- or cell-type-specific expression, although there is no known mechanism for understanding the cell-tissue specifity of the gene.
As a result of distinct first-exon sequences, a total of eight plectin isoforms (plectins 1, 1a, 1b, 1c, 1d, 1e, 1f, and 1g) have been identified with different N termini and specific functions. Among them, plectin 1a associates with keratin intermediate filaments in basal keratinocytes, which supports the association of plectin deficiencies with skin disorders.19 Plectins 1, 1b, 1d, and 1f are also substantially expressed in muscle tissue.20 As already known, PLEC mutations have been associated with EB simplex with muscular dystrophy, which is an autosomal-recessive disorder characterized by skin blistering and generally late-onset progressive muscle dystrophy.7 Plectin deficiency also leads to myasthenic syndrome along with late-onset myopathy and EBS or congenital muscular dystrophy with late-onset myasthenic symptoms and EBS, conveying its substantial role on the neuromuscular junction.8,9 However, no mutation in PLEC has been identified as being associated with muscular dystrophy without a skin disorder. In addition to this, no mutation has been reported in alternative first exons of PLEC corresponding to plectin isoforms.
DNA sequencing of exons and flanking introns of the affected individuals (family 1, patients V:2, V:3, and VI:1) did not detect any mutation in exons 2–32 of plectin-containing functional domains. However, DNA sequencing of alternative first exons (exons 1, 1b, 1d, and 1f), as well as part of their 5′ UTR and flanking intronic sequences, revealed homozygous 9 bp deletion (c.1_9del) containing the initiation codon in exon 1f of the gene (plectin isoform 1f [NM_201378.2]) (Figures 2B and 2C). The parents from two branches of the family were heterozygous, and healthy siblings were either heterozygous carriers or completely normal for this deletion (Figure 1A; individual V:4, normal; individuals V:5 and VI:2, heterozygous). The mutation was not detected in 342 alleles of healthy Turkish controls by DNA sequence analysis.
We extended the analysis to 72 additional LGMD2 patients whose statuses were previously identified as unlinked to all known LGMD2 loci. The same homozygous 9 bp deletion in exon 1f was identified in two cases from two different families. One of the patients (family 2, patient 20) was a 4-yr-old boy of consanguineous parents with the onset of muscle weakness at 2 yrs. He had a lordotic gait with a Gowers sign around 4 s. The mental and fine-motor developmental milestones were normal. The CK level was 4159 U/l. His 25-yr-old paternal aunt had muscular dystrophy, proven by a muscle biopsy performed at 23 yrs of age. She was not analyzed by DNA sequencing for PLEC. The other patient (family 3, patient 47) was a 5-yr-old male child of second cousins with weakness beginning in early childhood. He had walked at 2.5 yrs of age. He always had difficulty climbing stairs and was behind his peers in running. He initially demonstrated a Gowers sign around 6 s. Years later, at the age of 13, the duration of the Gowers sign had not changed. Muscles were not hypertrophic. His serum CK was 3704 U/l. Both patients showed dystrophic changes in the muscle biopsy (Figure 1D; patient 20) and normal expression of merosin, dystrophin, and sarcoglycans (alpha, beta, gamma, and delta).
Although we did not identify a relationship between these three families, all of the families originated from neighboring cities in the Black Sea region of Turkey. Haplotype construction using polymorphic DNA markers linked to the PLEC locus further confirmed a common haplotype in these families and supported a founder effect (Figure S3). This mutation was not detected in 342 alleles of healthy Turkish controls, of which 120 come from the Black Sea region.
A clinical comparison of patients with exon 1f mutations from three LGMD2 families revealed that they had early-onset LGMD symptoms followed by years of plateau. The family history from family 1 may denote a type of LGMD with loss of ambulation in the late 20s. This indicates that this is a progressive disorder, as our index case may well be expected to deteriorate in the third to fourth decades of his life. All case individuals were examined by a dermatologist, and neither epidermolysis bullosa nor another dermatologic disorder was reported. The phenotype did not show muscle enlargement and was rather nonspecific as far as the selective muscle involvement in the clinical setting. None of the patients had myasthenic-syndrome-like symptoms such as ocular, facial, or bulbar involvement, although an involvement of the neuromuscular junction cannot be ruled out completely, because single-fiber EMG was not available.
For assessment of the expression of plectin 1f mRNA, a muscle biopsy was obtained from the right quadriceps of patient VI:1, and the expression of PLEC was initially analyzed by semiquantitative RT-PCR (Figure 3A). The relative densitometric intensity of the bands was measured by Scion image analysis software (Scion; Frederick, MD, USA). We detected a 3.2-fold reduction for the expression of PLEC when patient VI:1's muscle tissue was compared with control skeletal muscle tissue. The GAPDH (MIM 138400) level of expression was used for normalization. We suggest that the decreased expression of PLEC was due to the plectin 1f deficiency in the patient. Moreover, the level of expression of plectin 1f mRNA was determined by quantitative real-time PCR (qRT-PCR) with the use of the SYBR Green JumpStart Taq ReadyMix (S5193) on a Corbett Rotor-Gene 6000 instrument (Corbett Life Science). In this study, the relative expression of plectin 1f mRNA in control skeletal muscle and in patient VI:1 was 1 and 0.01, respectively (Figure 3B).21 We found that the defective plectin isoform 1f exhibited dramatically lower expression (100-fold) compared to normal skeletal muscle tissue, which corresponds to a few copies of the plectin 1f mRNA. These data might be evaluated as an insignificant expression of plectin 1f mRNA in the muscle tissue of the patient. In our study, translation will also not occur from the first ATG as a result of the 9 bp deleted sequence (ATGGCCGGC) containing a start codon.
Figure 3.
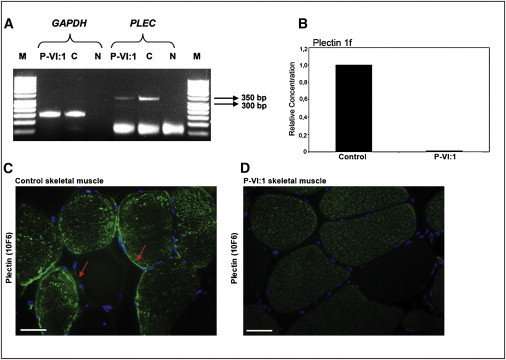
Expression of PLEC and IF Analysis of Plectin in Patient Muscle
(A) Semiquantitative analysis of PLEC in skeletal muscle tissue of patient VI:1 and control muscle. Total RNA was isolated from the muscle tissue of patient VI:1 and the control via the TRIZOL method. cDNA synthesis was performed with a QuantiTect Reverse Transcription Kit (QIAGEN). It was then amplified by PCR with primers specific for exons 20 and 23 (20F, 23R) (Table S2). The PCR products were assessed on a 3% agarose gel. The amplification product is 338 bp. GAPDH is an internal control. The amplification product of GAPDH is 150 bp. Significant reduction of PLEC expression in the patient's skeletal muscle is represented. P-VI:1, patient VI:1 (proband) from family 1; C, control; N, negative control; M, 50 bp DNA ladder (New England Biolabs).
(B) Analysis of plectin 1f mRNA expression by qRT-PCR. The exon 1f part of PLEC- and GAPDH-specific primers (Table S3) were used for the qRT-PCR reaction, and the experiments were performed in triplicate. PCR efficiency was 100%. Ct values of both the calibrator (control) and the muscle sample of the patient are normalized to GAPDH as an internal control. The patient and control samples were analyzed in the same run for the exclusion of between-run variations. A no-template control of nuclease-free water was included in each run. The experiments were repeated twice for the same samples. The results were analyzed with Corbett Rotor-Gene 6000 series software, version 1.7. Relative quantitation of gene expression was performed via the delta-delta CT method with the use of two standard curves.21 The expression level of plectin 1f mRNA was normalized with the reference gene GAPDH, and a control sample was used as a calibrator. Data represent a 100-fold decreased expression of plectin 1f mRNA in patient VI:1's (P-VI:1) skeletal muscle with respect to the control sample.
(C and D) Immunofluorescence analysis of plectin in control muscle and patient VI:1 (P-VI:1), respectively. Cross sections of the muscle tissues were stained with plectin 10F6 (mAb) antibody. Strong sarcoplasmic staining, but no sarcolemmal staining, was detected in the patient muscle when compared to the control sample. Arrows show the sarcolemmal staining of type 2 fibers (fibers with sarcoplasmic staining) and type 1 fibers in control skeletal muscle. Scale bars represent 50 μm.
In addition to these findings, we decided to determine the immunolocalization of plectin in skeletal muscle of patient VI:1 and the control sample. We performed immunofluorescence staining of the muscle tissue (Figures 3C and 3D). Because there were no commercially available isoform-specific plectin antibodies, we used 10F6 mAb, which is against the rod domains of all plectin isoforms. The plectin 10F6 antibody was a gift from Dr. Gerhard Wiche (University of Vienna).
It was previously proposed that plectin 1f is a sarcolemma-associated protein, and it could be localized beneath the sarcolemma. With pan-plectin antiserum, strong subsarcolemmal and moderate sarcoplasmic staining was revealed in the small-diameter fibers, whereas faint sarcoplasmic and sarcolemmal staining was observed in the larger-diameter fibers.20 It was previously reported that the expression of plectin 1f depends on the fiber type of the muscle, and it was especially localized in type 2A fibers (one of the fast twitch fibers), with moderate sarcoplasmic staining and rare and weak staining of the membrane, with the use of plectin-1f-specific antibody.20 In our studies, we showed strong sarcoplasmic staining but irregular and weak sarcolemmal staining of type 2 fibers, but only rare and faint staining for type 1 fibers in cross-sections of control skeletal muscle, with the use of 10F6 mAb (Figure 3C). Contrary to the control muscle, even though there was cytoplasmic staining of type 2 fibers, no sarcolemmal staining of plectin was detected in the patient muscle (Figure 3D). For this reason, we suggest that the loss of sarcolemmal staining of plectin in patient's muscle might result from the absence of plectin 1f.
To demonstrate the specific ultrastructural features of the patient muscle, we performed transmission electron microscopy (TEM-Jeol, JEM1400). There was a slight variation in muscle fiber size, and hypertrophic and atrophic fibers were observed. An increase in endomysial and perimysial connective tissue was noted. Internal nuclei were seen in some fibers. The dilated tubular system from skeletal muscle of index case was present in some fiber cross-sections, the sarcolemma displayed duplications (separation of membranes) (Figures 4A and 4B), and the presence of an empty space between the sarcolemma and the contractile elements (sarcomere) was observed (Figure 4B). The empty spaces generally consisted of swollen sarcoplasmic reticuli cisternae and glycogen (Figure 4C). The longitudinal sections revealed focal loss of myofibrillar organization in some areas (Figures 4D and 4E). The width of the sarcomere unit was thinner than normal, as well. Degenerating mitochondria were noted in atrophic fibers, although the mitochondria number and distribution revealed an even pattern. Given that plectin 1f is a sarcolemma-associated protein, a variably enlarged space between the sarcolemma and the sarcomere might be the result of plectin 1f deficiency due to the broken anchorage between these structures. It has been proposed that there is an interaction between plectin 1f and plectin 1d that is associated with Z-lines.22 In this situation, a deficiency of plectin 1f can cause the loss of linkage between plectin 1f and plectin 1d, which might explain the disorganization or misalignment of Z-lines. The presence of duplication, the empty space between the sarcolemma and the contractile elements, and the focal myofibrillar disorganization were the striking ultrastructural features in our case, which were very similar to the findings reported earlier by McMillan et al. in EBS-MD patients with a homozygous plectin-exon 32 rod domain mutation (p.Arg2465X) in PLEC.23
Figure 4.
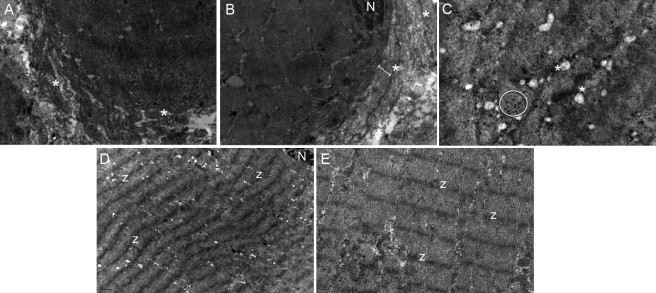
Transmission Electron Microscopy
Electron micrographs of the transverse (A–C) and longitudinal (D and E) sections exhibiting the dilated tubular system from skeletal muscle of patient VI:1.
(A and B) Duplication of the plasma membrane (separation of membranes) is observed (indicated by an asterisk), and the double arrow in (B) shows the empty space between the sarcolemma and sarcomere.
(C) Black glycogen inclusions (indicated within the circle) and dilated sarcoplasmic reticuli (asterisk) are present in the space between the sarcolemma and sarcomere.
(D) Mild disalignment of sarcomeres.
(E) Focal variability of Z lines. (N, nucleus; Z, Z lines).
Scale bars represent the following: (A, B, E) 0.5 μm; (C) 0.2 μm; (D) 1 μm.
Although PLEC-associated phenotypes clearly convey the substantial role of plectin in the heart, skin, and neuromuscular junction, potential roles of plectin 1f in these tissues still remain inconclusive because of a lack of functional data.7–9,24 The absence of any other tissue involvement except skeletal muscle in our patients suggests that plectin 1f has a major role in muscle.
Tissue-specific expression and isoform-dependent deficiency of plectin explain the muscular dystrophy phenotype without skin blistering in our patients. Plectin isoforms expressed in skeletal muscle at considerable levels have different and significant roles in muscle integrity. Plectin 1d is associated with Z-disks, whereas plectin 1b colocalizes with mitochondria, which plays a role for organelle shaping and forms a network between mitochondria and the cytoskeleton.20,25 Plectin 1 and 1f are sarcolemma-associated isoforms and are localized at costameres.20 It has been demonstrated that although exon 1 of the plectin 1 isoform binds to alpha-dystrobrevin, no interaction has been found between plectin 1f and alpha-dystrobrevin.26 According to their myoblast-differentiation experiments, Recniczek et al. found that the temporal expression profile of plectin 1f is highly similar to dystrophin and that there is a direct interaction between plectin 1f and dystrophin and beta-dystroglycan, which is a major costamere in the muscle.20 They suggested that there is an association between plectin 1f and the dystrophin-glycoprotein complex (DGC) via beta-dystroglycan. Plectin 1f binds to dystrophin via its functional actin binding domain and also binds to beta-dystroglycan by its plakin domain. The C-terminal part of the protein is an IF-binding domain that is available for binding to the desmin, a major intermediate filament in the muscle tissue. In addition to these proposed models, nothing is known about proteins specifically binding to the exon 1f part of the protein. We believe that the unknown LGMD etiology of a number of families may be associated with the disruption of these isoforms, in addition to plectin 1f.
In conclusion, we describe a pure LGMD2 that is caused by a homozygous deletion in exon 1f of PLEC. The overall clinical phenotype of our patients shows proximal weighted, generalized muscle weakness without any myasthenic features or skin involvement. On the basis of our index cases, this LGMD2 phenotype manifests in early childhood and shows very slow progress until the late teens. However, it may show rapid progression later on; loss of ambulation in the late 20s has been observed in one family. Additional cases are needed for confirmation of our observation. Plectin 1f is one of the particular scaffolds for costameric proteins, among other isoforms (plectins 1, 1b, and 1d). Our study emphasizes the importance of plectin isoforms and their different tasks in the formation of precise myofiber structure. This study will also shed light on the contribution of tissue- and/or cell-type-specific expression of gene isoforms for understanding of the pathologic mechanism of human diseases.
Acknowledgments
We are very grateful to all of the patients and their families for their participation in the study. Special thanks to Kevin P. Campbell for critically reading the manuscript. We thank Gerhard Wiche for providing the plectin 10F6 mAb. We would like to acknowledge the Dubowitz Neuromuscular Centre, London, UK for the analysis of 50K SNP array data. This study was supported by the Scientific and Technological Research Council of Turkey (TÜBİTAK) (grant numbers 108S124 and SBAG-1774 to P.D).
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Ensembl, http://www.ensembl.org
NCBI Map Viewer, http://www.ncbi.nlm.nih.gov/mapview/
NCBI UniSTS, http://www.ncbi.nlm.nih.gov/unists/
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
References
- 1.Guglieri M., Straub V., Bushby K., Lochmüller H. Limb-girdle muscular dystrophies. Curr. Opin. Neurol. 2008;21:576–584. doi: 10.1097/WCO.0b013e32830efdc2. [DOI] [PubMed] [Google Scholar]
- 2.Guglieri M., Bushby K. How to go about diagnosing and managing the limb-girdle muscular dystrophies. Neurol. India. 2008;56:271–280. doi: 10.4103/0028-3886.43445. [DOI] [PubMed] [Google Scholar]
- 3.Bushby K.M.D., Beckmann J.S. The limb-girdle muscular dystrophies—proposal for a new nomenclature. Neuromuscul. Disord. 1995;5:337–343. doi: 10.1016/0960-8966(95)00005-8. [DOI] [PubMed] [Google Scholar]
- 4.Kaplan J.C. The 2009 version of the gene table of neuromuscular disorders. Neuromuscul. Disord. 2009;19:77–98. doi: 10.1016/j.nmd.2008.11.001. [DOI] [PubMed] [Google Scholar]
- 5.Kayserili H., Uz E., Niessen C., Vargel I., Alanay Y., Tuncbilek G., Yigit G., Uyguner O., Candan S., Okur H. ALX4 dysfunction disrupts craniofacial and epidermal development. Hum. Mol. Genet. 2009;18:4357–4366. doi: 10.1093/hmg/ddp391. [DOI] [PubMed] [Google Scholar]
- 6.Uz E., Alanay Y., Aktas D., Vargel I., Gucer S., Tuncbilek G., von Eggeling F., Yilmaz E., Deren O., Posorski N. Disruption of ALX1 causes extreme microphthalmia and severe facial clefting: expanding the spectrum of autosomal-recessive ALX-related frontonasal dysplasia. Am. J. Hum. Genet. 2010;86:789–796. doi: 10.1016/j.ajhg.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Uitto J., Pulkkinen L., Smith F.J.D., McLean W.H.I. Plectin and human genetic disorders of the skin and muscle. The paradigm of epidermolysis bullosa with muscular dystrophy. Exp. Dermatol. 1996;5:237–246. doi: 10.1111/j.1600-0625.1996.tb00124.x. [DOI] [PubMed] [Google Scholar]
- 8.Banwell B.L., Russel J., Fukudome T., Shen X.M., Stilling G., Engel A.G. Myopathy, myasthenic syndrome, and epidermolysis bullosa simplex due to plectin deficiency. J. Neuropathol. Exp. Neurol. 1999;58:832–846. doi: 10.1097/00005072-199908000-00006. [DOI] [PubMed] [Google Scholar]
- 9.Forrest K., Mellerio J.E., Robb S., Dopping-Hepenstal P.J.C., McGrath J.A., Liu L., Buk S.J.A., Al-Sarraj S., Wraige E., Jungbluth H. Congenital muscular dystrophy, myasthenic symptoms and epidermolysis bullosa simplex (EBS) associated with mutations in the PLEC1 gene encoding plectin. Neuromuscul. Disord. 2010;20:709–711. doi: 10.1016/j.nmd.2010.06.003. [DOI] [PubMed] [Google Scholar]
- 10.Pfendner E., Uitto J. Plectin gene mutations can cause epidermolysis bullosa with pyloric atresia. J. Invest. Dermatol. 2005;124:111–115. doi: 10.1111/j.0022-202X.2004.23564.x. [DOI] [PubMed] [Google Scholar]
- 11.Nakamura H., Sawamura D., Goto M., Nakamura H., McMillan J.R., Park S., Kono S., Hasegawa S., Paku S., Nakamura T. Epidermolysis bullosa simplex associated with pyloric atresia is a novel clinical subtype caused by mutations in the plectin gene (PLEC1) J. Mol. Diagn. 2005;7:28–35. doi: 10.1016/S1525-1578(10)60005-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Koss-Harnes D., Jahnsen F.L., Wiche G., Søyland E., Brandtzaeg P., Gedde-Dahl T., Jr. Plectin abnormality in epidermolysis bullosa simplex Ogna: non-responsiveness of basal keratinocytes to some anti-rat plectin antibodies. Exp. Dermatol. 1997;6:41–48. doi: 10.1111/j.1600-0625.1997.tb00144.x. [DOI] [PubMed] [Google Scholar]
- 13.Elliott C.E., Becker B., Oehler S., Castañón M.J., Hauptmann R., Wiche G. Plectin transcript diversity: identification and tissue distribution of variants with distinct first coding exons and rodless isoforms. Genomics. 1997;42:115–125. doi: 10.1006/geno.1997.4724. [DOI] [PubMed] [Google Scholar]
- 14.Wiche G. Role of plectin in cytoskeleton organization and dynamics. J. Cell Sci. 1998;111:2477–2486. doi: 10.1242/jcs.111.17.2477. [DOI] [PubMed] [Google Scholar]
- 15.Fuchs P., Zörer M., Rezniczek G.A., Spazierer D., Oehler S., Castañón M.J., Hauptmann R., Wiche G. Unusual 5′ transcript complexity of plectin isoforms: novel tissue-specific exons modulate actin binding activity. Hum. Mol. Genet. 1999;8:2461–2472. doi: 10.1093/hmg/8.13.2461. [DOI] [PubMed] [Google Scholar]
- 16.Zhang T., Haws P., Wu Q. Multiple variable first exons: a mechanism for cell- and tissue-specific gene regulation. Genome Res. 2004;14:79–89. doi: 10.1101/gr.1225204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu C.G., Maercker C., Castañon M.J., Hauptmann R., Wiche G. Human plectin: organization of the gene, sequence analysis, and chromosome localization (8q24) Proc. Natl. Acad. Sci. USA. 1996;93:4278–4283. doi: 10.1073/pnas.93.9.4278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Andrä K., Lassmann H., Bittner R., Shorny S., Fässler R., Propst F., Wiche G. Targeted inactivation of plectin reveals essential function in maintaining the integrity of skin, muscle, and heart cytoarchitecture. Genes Dev. 1997;11:3143–3156. doi: 10.1101/gad.11.23.3143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Andrä K., Kornacker I., Jörgl A., Zörer M., Spazierer D., Fuchs P., Fischer I., Wiche G. Plectin-isoform-specific rescue of hemidesmosomal defects in plectin (-/-) keratinocytes. J. Invest. Dermatol. 2003;120:189–197. doi: 10.1046/j.1523-1747.2003.12027.x. [DOI] [PubMed] [Google Scholar]
- 20.Rezniczek G.A., Konieczny P., Nikolic B., Reipert S., Schneller D., Abrahamsberg C., Davies K.E., Winder S.J., Wiche G. Plectin 1f scaffolding at the sarcolemma of dystrophic (mdx) muscle fibers through multiple interactions with β-dystroglycan. J. Cell Biol. 2007;176:965–977. doi: 10.1083/jcb.200604179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Livak K.J., Schmittgen T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Δ ΔC(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 22.Konieczny P., Fuchs P., Reipert S., Kunz W.S., Zeöld A., Fischer I., Paulin D., Schröder R., Wiche G. Myofiber integrity depends on desmin network targeting to Z-disks and costameres via distinct plectin isoforms. J. Cell Biol. 2008;181:667–681. doi: 10.1083/jcb.200711058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McMillan J.R., Akiyama M., Rouan F., Mellerio J.E., Lane E.B., Leigh I.M., Owaribe K., Wiche G., Fujii N., Uitto J. Plectin defects in epidermolysis bullosa simplex with muscular dystrophy. Muscle Nerve. 2007;35:24–35. doi: 10.1002/mus.20655. [DOI] [PubMed] [Google Scholar]
- 24.Bolling M.C., Pas H.H., de Visser M., Aronica E., Pfendner E.G., van den Berg M.P., Diercks G.F., Suurmeijer A.J., Jonkman M.F. PLEC1 mutations underlie adult-onset dilated cardiomyopathy in epidermolysis bullosa simplex with muscular dystrophy. J. Invest. Dermatol. 2010;130:1178–1181. doi: 10.1038/jid.2009.390. [DOI] [PubMed] [Google Scholar]
- 25.Winter L., Abrahamsberg C., Wiche G. Plectin isoform 1b mediates mitochondrion-intermediate filament network linkage and controls organelle shape. J. Cell Biol. 2008;181:903–911. doi: 10.1083/jcb.200710151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hijikata T., Nakamura A., Isokawa K., Imamura M., Yuasa K., Ishikawa R., Kohama K., Takeda S., Yorifuji H. Plectin 1 links intermediate filaments to costameric sarcolemma through β-synemin, α-dystrobrevin and actin. J. Cell Sci. 2008;121:2062–2074. doi: 10.1242/jcs.021634. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.