

Abstract
Germline mutations in FASL and FAS impair Fas-dependent apoptosis and cause recessively or dominantly inherited autoimmune lymphoproliferative syndrome (ALPS). Patients with ALPS typically present with no other clinical phenotype. We investigated a large, consanguineous, multiplex kindred in which biological features of ALPS were found in the context of severe bacterial and viral disease, recurrent hepatopathy and encephalopathy, and cardiac malformations. By a combination of genome-wide linkage and whole-exome sequencing, we identified a homozygous missense mutation in FADD, encoding the Fas-associated death domain protein (FADD), in the patients. This FADD mutation decreases steady-state protein levels and impairs Fas-dependent apoptosis in vitro, accounting for biological ALPS phenotypes in vivo. It also impairs Fas-independent signaling pathways. The observed bacterial infections result partly from functional hyposplenism, and viral infections result from impaired interferon immunity. We describe here a complex clinical disorder, its genetic basis, and some of the key mechanisms underlying its pathogenesis. Our findings highlight the key role of FADD in Fas-dependent and Fas–independent signaling pathways in humans.
Main Text
Germline mutations in FASL1 (MIM 134638) and FAS2,3 (MIM 134637) are genetic etiologies of autoimmune lymphoproliferative syndrome (ALPS [MIM 601859]) in humans. ALPS typically begins in early childhood (median age at onset: 2 yrs), with lymphadenopathy and/or splenomegaly. About half the patients have autoimmune disease, autoimmune cytopenias being particularly common.4,5 The patients do not suffer from unusually severe infection, unless they are on immunosuppressive treatment,4 and display no overt developmental phenotype. We investigated the molecular basis of a condition affecting at least four members of an extended consanguineous kindred of Pakistani origin (Figure 1A) with biological features of ALPS (high-circulating CD4−CD8−TCRαβ+ T-cell [DNT] counts, and elevated IL-10 and FasL serum levels), no clinical features of ALPS, a complex infectious phenotype with both viral and bacterial infections, and congenital cardiovascular malformations (Table 1 and Table S1 available online). This study was approved by the local institutional review board. Written informed consent for participation in the study was obtained from all patients and family members studied.
Figure 1.

Characterization of an Inherited FADD Mutation
(A) Pedigree of the Pakistani family. Black symbols indicate patients. Gray symbols indicate siblings in the parents' generation who died early in childhood with clinical symptoms related to the disorder observed in P1–P4. Clinical information was incomplete for these members of the parents' generation. Haplotypes of FADD are indicated: M stands for c.315T>G, and WT for the wild-type allele.
(B) Schematic diagram of FADD protein showing the death effector domain (DED) and the death domain (DD). The location corresponding to the mutation is indicated by an arrow, and the predicted amino acid substitution is shown.
(C) Evolutionary conservation of the FADD region containing amino acid residue C105 (indicated by the arrow).
(D) FADD immunoblot in primary fibroblasts and EBV-B cells from patients and controls. Experiments were carried out with two different antibodies: a mouse monoclonal antibody against the C terminus of FADD (#610399, BD Biosciences) and a rabbit polyclonal antibody against the residues surrounding Ser194 in human FADD (#2782, Cell Signaling). Similar results were obtained with both antibodies. GAPDH was used as a loading control. A representative blot with the monoclonal antibody is shown (n = 6).
(E and F) FADD protein levels in primary fibroblasts (E) and EBV-B cells (F) determined on the basis of the intensity of the signal on immunoblots and normalized with respect to GAPDH levels. A mean of six experiments is shown. Error bars indicate the SEM.
(G and H) Effect of the C105W mutation on the stability of FADD DD folding and the Fas-FADD complex. (G) Differential scanning calorimetry (DSC) analysis of WT and C105W His6-FADD DD proteins. (H) Fas-FADD complex stability assay. The retention of FADD DD (WT or C105W) with immobilized His6-Fas DD was assessed with various concentrations of NaCl, as previously described19 (E: imidazole elution of remaining complex after the final NaCl concentration).
Table 1.
Clinical Features of Four Patients in the Family
| Patient | P1 | P2 | P3 | P4 | |
|---|---|---|---|---|---|
| Gender | male | female | female | female | |
| Cardiovascular malformation | none | none | yes (pulmonary atresia) + ventricular septal defect | yes (L-sided SVC draining to left atrium) | |
| Functional hyposplenism | spleen size | NA | normal | normal | normal |
| Howell-Jolly bodies | NA | NA | yes | yes | |
| invasive pneumococcal infection | yes (meningitis, 4 mo) | yes (septicemia, 14 mo) | noa | noa | |
| Features of febrile episodes | documented viral trigger | VZV | VZV, HHV6 | MMR vaccine, astrovirus, parainfluenza virus 2, EBV | |
| encephalopathy | yes | yes | yes | yes | |
| seizures | yes | yes | yes | yes | |
| liver dysfunction (maximal ALT, IU/L) | yes (NA) | yes (NA) | yes (1042) | yes (383) | |
| ALPS phenotype | autoimmune disease | no | no | no | no |
| presence of autoantibodies | NA | NA | nob | yesb (antierythrocyte antibodies intermittently present) |
|
| lymphadenopathy or splenomegaly | no | no | no | no | |
| percentage of CD4−CD8−TCRαβ+ T cells | NA | NA | NA | 7.8 | |
| lymphocyte apoptosis | NA | NA | NA | impaired | |
| serum FasL | NA | NA | NA | high | |
| serum IL-10 | NA | NA | NA | high | |
| CNS outcome | neurodevelopmental progress | NA | within normal limits | within normal limits | within normal limits |
| cerebral atrophy on CT/MRI (age) | yes (4 mo) | NA | yes (3 mo) | mild (5 mo) + increased at 2.5 yr + spotty calcification of subcortical white matter | |
| Liver histology | NA | mild chronic portal inflammation with bridging fibrosis | NA | mild chronic portal inflammation with mild fibrosis | |
| Follow-up data | died at 4 mo | died at 14 mo | died at 4 yr and 4 mo. | alive and currently 2 yr and 9 mo. |
Abbreviations are as follows: ALT, alanine transaminase; FasL, Fas ligand; HHV6, Human Herpesvirus Six; IL-10, interleukin 10; NA, not assessed.
On antibacterial prophylaxis from early infancy.
Autoantibody screen including antinuclear, anti-smooth muscle, antimitochondrial, and anti-gastric parietal cell antibodies, direct antiglobulin test.
The affected children suffered from recurrent, stereotypical episodes of fever, encephalopathy, and mild liver dysfunction (modestly elevated transaminases without cholestasis, metabolic derangement, or synthetic defects), sometimes accompanied by generalized seizures that were difficult to control. Episodes lasted several days, sometimes requiring intensive care, and cranial imaging in three patients (P1, P3, and P4 [IV.2, IV.4, and IV.5 in Figure 1]) suggested atrophy, despite subsequent recovery in two of these patients (P1 and P3). For some of the episodes, it was possible to identify a viral trigger: varicella zoster virus (VZV), measles mumps rubella (MMR) attenuated vaccine, parainfluenza virus, and Epstein-Barr virus (EBV). The index case, P3 (IV.4 in Figure 1), died at the age of 4 yrs, during such an episode. In addition, fatal invasive pneumococcal disease occurred in two children (P1 and P2 [IV.2 and IV.3 in Figure 1]), and Howell-Jolly bodies were detected in the two remaining patients (P3 and P4 [IV.4 and IV.5 in Figure 1]), despite the presence of a spleen (indicating functional hyposplenism).6–8 This clinical syndrome has never before been described (no MIM number). In the previous generation, another five family members had died in childhood, two with “epilepsy” and two from infection (pneumonia and measles, respectively) (data not shown), suggesting that there may have been up to nine patients with this disorder in these two generations of this kindred.
We hypothesized that this syndrome would segregate as an autosomal-recessive trait. We investigated its genetic etiology, by combining genome-wide linkage analysis by homozygosity mapping with whole-exome sequencing.9–11 For the linkage analysis, we genotyped three affected patients (P2, P3, and P4), their parents (III.1, III.2, III.3, and III.4), and one healthy sibling (IV.1) with the Affymetrix 500K SNP Array or the Affymetrix 6.0 Array. No material was available for P1. Homozygous regions were detected with the HomozygosityMapper genome browser. Two regions (> 1 Mb) were homozygous in the three affected patients and heterozygous in other members of the family: an 8 Mb region on chromosome 11 (between rs4930243 and rs7926553; bp position: g.68,499,874– g.76,470,079; hg18, NCBI 36.1) and a 9 Mb region on chromosome 18 (between rs9959449 and rs12327522; bp position: g.60,703,632–g.70,387,399; hg18, NCBI 36.1). The regions were further confirmed by typing microsatellite markers (Figure S1).
In parallel to the linkage analysis, we sequenced the exome of P3 with the SureSelect Human All Exon Kit (Agilent Technologies). The sequences obtained were aligned to the reference genome (hg18 build) with the Burrows-Wheeler Aligner. Three open-source packages were used for downstream processing and variant calling: the Genome Analysis Toolkit (GATK),12 SAMtools,13 and Picard Tools. Substitution calls were made with the GATK UnifiedGenotyper. All calls with a read coverage < 4× and a phred-scaled SNP quality of < 30 were filtered out. All the variants were annotated with SeattleSeq SNP Annotation. In total, we identified 23,146 variations. Of these, 67 variants were found in the chromosome 11 candidate region, and 14 variants were found in the chromosome 18 candidate region (Table S2). Comparisons with the NCBI dbSNP build 129, the 1000 Genomes Project database, and our in-house database (composed of 70 exomes) identified only one nonsynonymous variant that had not been reported before and mapped to the chromosome 11 region. No previously unreported nonsynonymous variant mapping to the linked region on chromosome 18 was identified. The variant identified was a missense homozygous c.315T>G in exon 2 of FADD (NM_003824.3, MIM 602457) that changes cysteine at amino acid position 105 to tryptophan, p.C105W (referred to as C105W hereafter) (Figure 1B and Figure S2). Cysteine at amino acid position 105 is highly conserved throughout evolution (Figure 1C). We validated this variant by Sanger sequencing on genomic DNA from peripheral blood and on cDNA from EBV-transformed B cells (EBV-B). This variant segregated with the disease status in all family members examined (Figure 1A) and was not found in 282 Pakistani controls, suggesting that it is not an irrelevant polymorphism.
FADD was first described as an adaptor protein interacting with the apoptosis-inducing surface receptor Fas.14 It has since been shown to interact with various partners and to participate in many other cellular processes, such as autophagy, inflammation, innate immunity, cell proliferation, and tumor development.15–18 Thus, we hypothesized that the FADD mutation was responsible for the complex clinical phenotype of the affected individuals. To test this hypothesis, we assessed the expression of FADD mRNA levels in EBV-B cells by quantitative RT-PCR. We found that FADD mRNA levels in the EBV-B cells from P3 (C105W/C105W) were similar to those in the EBV-B cells from IV.1 (WT/WT) (data not shown). However, FADD protein levels were clearly lower in primary fibroblasts from P4 (∼16%) than in primary fibroblasts from a healthy control (Figures 1D and 1E). Similarly, FADD protein levels were lower in the EBV-B cells from P3 (∼21%) and III.1 (WT/C105W) (∼62%) than in the EBV-B cells from IV.1 (Figures 1D and 1F). Residue C105 is located in alpha-helix 1 of the FADD death domain (DD), at the interface of the Fas-FADD complex.19 We tested the effect of the C105W mutation on FADD folding stability and Fas-FADD interaction with recombinant Fas DD and FADD DD proteins. The C105W mutant protein was folded, as shown by differential scanning calorimetry (DSC), but the folding stability of the mutant protein was lower than that of WT FADD, by ∼10°C (Figure 1G). We then assessed the effect of the mutation on Fas-FADD complex stability by using a gel copurification assay in the presence of increasing concentrations of NaCl (Figure 1H). Binding levels for C105W FADD and Fas were clearly lower than those for WT FADD, suggesting that the primary Fas-FADD complex was less stable. Thus, the C105W mutation in FADD strongly decreases steady-state protein levels and impairs the interaction of the residual FADD protein with Fas.
FADD forms a critical bridge between the death receptor Fas and caspase-8 (MIM 601763), in a multimolecular array known as the death-inducing signaling complex (DISC).20,21 We therefore studied Fas-induced apoptosis in the patients' cells. Phytohemagglutinin-stimulated peripheral-blood lymphocytes (PHA blasts) from P4 displayed impaired apoptosis (viability >50% higher than for controls) when challenged with cross-linked Fas ligand (FasL), similar to Fas-deficient cells (Figure 2A). In addition, EBV-B cells from P3 displayed impaired apoptosis when Fas was crosslinked with agonistic Fas-specific antibody (Apo1.3) (Figure 2B). In keeping with this finding, investigations of the surviving patient P4 revealed several features classically associated with ALPS due to Fas2,3 or FasL1 deficiency and more rarely associated with deficiencies of caspase-8 or caspase-10 (MIM 601762).4,22,23 P4 displayed an expansion of the population of mature CD4−CD8−TCRαβ+ T cells (DNT) in peripheral blood, as well as elevated serum levels of IL-10 and soluble FasL (Table 1 and Figures 2C and 2D).24,25 Our data confirm that the FADD C105W variant impairs apoptotic function both in vitro and in vivo, resulting in biological features of ALPS despite the lack of clinically evident autoimmunity or lymphoproliferation (although P4 did have transiently detectable antierythrocyte autoantibodies and P2, P3, and P4 displayed peripheral lymphocytosis [Table S1]). T cell proliferation in response to various stimuli was normal (Figure 2E), in contrast with findings for caspase-8 deficient individuals22 and mice with FADD deficiency specific to T or B cells.26 A recent study in mice suggested that FADD deficiency may decrease T cell proliferation as a result of an excess of necroptosis.16 To examine whether C105W affected necroptosis, we added an inhibitor of necroptosis, necrostatin 1, to CD3- and CD28-stimulated proliferation assays. Necrostatin-1 did not affect proliferative responses in this system (Figure S3).
Figure 2.
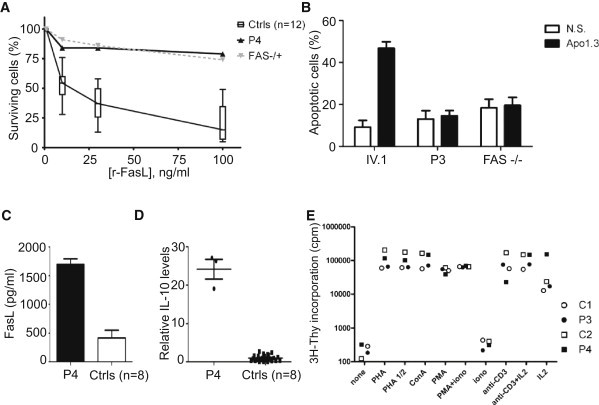
Characterization of the ALPS Phenotype
(A) PHA blasts from normal volunteers (Ctrls), patient P4, and a heterozygous Fas-deficient patient were stimulated with recombinant FasL (r-FasL), and cell viability was analyzed. The proportion of live cells in the presence of r-FasL is displayed as a percentage of live cells in the presence of enhancer alone, for each sample. Error bars indicate the SEM.
(B) EBV-B cells from P3, IV.1, and a Fas-deficient patient were treated with an antibody against Fas (Apo-1.3), in the presence of rabbit anti-mouse Ig (Ramig), for 6 hr and analyzed for early apoptosis. The percentage of cells that were apoptotic was calculated as the percentage of cells positive for AnnexinV and negative for propidium iodide staining. NS indicates that the cells were incubated with Ramig only. A mean of three independent experiments is shown. Error bars indicate the SEM.
(C) FasL levels in the serum of P4 and eight healthy controls (Ctrls). Error bars indicate the SEM.
(D) IL-10 levels in the serum of P4 and eight healthy controls. IL-10 values were normalized with respect to the mean of the control samples. Error bars indicate the SEM.
(E) Proliferation assay. Proliferative responses to various stimuli (PHA, 5 μg/ml; IL-2, 500 U/ml; concanavalin A, 5 μg/ml; anti-CD3 [OKT3], 0.1 μg/ml; PMA, 50 ng/ml; ionomycin, 0.1 μg/ml). P3 and P4 responded similarly in comparison to the two controls, C1 and C2.
The laboratory findings for the FADD-deficient patients were typical of ALPS, but clinical presentation was dominated by features absent from classical ALPS, leading to early death. These features included invasive pneumococcal disease (IPD), which killed both P1 and P2. Neither P3 nor P4 suffered from IPD, probably because they were on antibacterial prophylaxis from early infancy, but abnormally high counts of Howell-Jolly bodies were seen on their blood smears. The identification of Howell-Jolly bodies on peripheral-blood smears is the most reliable indicator of impaired splenic phagocyte function (including functional or anatomical asplenia).6–8 Moreover, asplenia (MIM 271400 and MIM 208530)—whether anatomical or functional, congenital or acquired—has frequently been associated with a strong predisposition to infections by encapsulated bacteria, such as IPD, in particular.6–8,27 For example, we showed that 15 of 20 patients with isolated congenital asplenia (ICA) suffered from invasive bacterial infections, caused mostly by Streptococcus pneumoniae (61%).27 Finally, B cell responses to vaccine antigens appeared to be intact in the patients (Table S1), ruling out other known causes of IPD.28 These observations suggest that functional hyposplenism underlies IPD in FADD-deficient patients.
In addition to IPD, FADD-deficient patients suffered from recurrent episodes of encephalopathy and liver dysfunction with fever, which were sometimes triggered by viral infection (Table 1). Previous studies in mouse embryonic fibroblasts (MEFs) suggested that FADD plays a crucial role in type I interferon (IFN) -dependent antiviral immunity.15,29 We therefore tested the hypothesis that the unusually severe illnesses associated with viral infections in FADD-deficient patients might be due to an impaired type I IFN-dependent antiviral response, by assessing the antiviral effect of IFN-α in vesicular stomatitis virus (VSV)-infected P3 and IV.1 EBV-B cells. STAT1-deficient EBV-B cells, lacking a key type-I-IFN-responsive pathway,29–31 were included as controls. Prior treatment with IFN-α protected both P3 and IV.1 cells from VSV-induced cell death until 36 hr after infection (Figure 3A). However, IFN-α-treated P3 cells eventually succumbed to VSV infection, whereas IFN-α-treated IV.1 cells remained protected until 60 hr after infection (Figures 3A and 3B). These results suggest that the IFN-α-responsive pathway is intact but that the positive feedback of IFN signaling is impaired in FADD-deficient cells. Interferon regulatory factor 7 (IRF7), induced by autocrine type I IFNs, is known to play a critical role in the positive feedback of IFN signaling, by inducing IFN-α gene family members. We found that the induction of IRF7 in response to VSV infection was impaired in P3 cells, despite levels of IFN-β induction similar to those in IV.1 cells (Figure 3C). These data provide a molecular basis for the increase in susceptibility to viral diseases in FADD-deficient patients.
Figure 3.
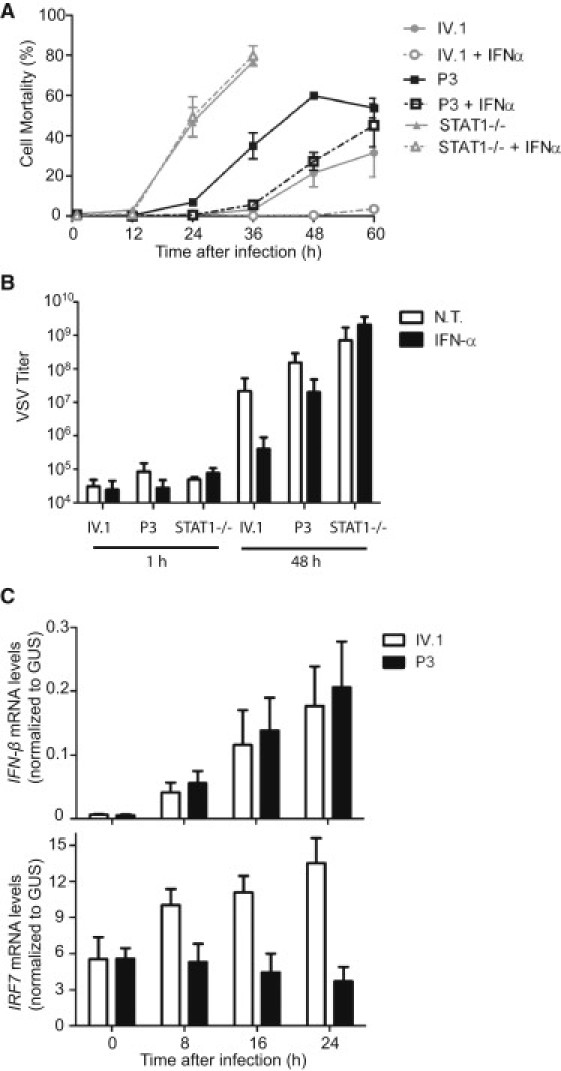
Impaired Antiviral Immunity
(A) EBV-B cells from P3, IV.1, and a STAT1-deficient patient were infected with VSV at a multiplicity of infection (MOI) of 1, with or without prior treatment with 100 IU/ml of IFN-α for 24 hr. Cell mortality was assessed by measuring the amount of LDH released into the medium. For STAT1-deficient cells, results are shown only until 36 hr after infection because of high levels of spontaneous LDH release at later time points. A mean of three independent experiments is shown. Error bars indicate the SEM.
(B) EBV-B cells from P3, IV.1, and a STAT1-deficient patient were either left untreated (NT, bars in white) or treated with 100 IU/ml IFN-α for 24 hr (IFN-α, bars in black), prior to infection with VSV at a MOI of 0.1, and were harvested 48 hr after infection. VSV titers (TCID50, ml) were determined on Vero cells. A mean of three independent experiments is shown. Error bars indicate the SEM.
(C) EBV-B cells from P3 or IV.1 were infected with VSV at a MOI of 10. Cells were harvested at the indicated time points, and total RNA was extracted. IFN-β and IRF7 mRNA levels were determined by quantitative RT-PCR. Threshold cycles normalized with respect to those of GUS (ΔCT) are plotted as 2−ΔCT. A mean of three independent experiments is shown. Error bars indicate the SEM.
By contrast, we can only speculate about the mechanisms underlying the developmental abnormalities within this kindred. P3 displayed pulmonary atresia (MIM 178370) and P4 had a left-sided superior vena cava (SVC) draining into the left atrium, both of which are rare as sporadic defects and were probably caused by FADD insufficiency. Indeed, mice with a null mutation in FADD are not viable because of embryonic defects, including cardiac abnormalities, such as thinning of the ventricular myocardium.32 Moreover, FADD expression in mice, at embryonic day 11.5, is detected not only in the myocardium but also in the brain, liver, and developing vertebrae.32 Spinal anatomy appeared normal in all FADD-deficient patients, but hepatic and cerebral dysfunctions were clearly key elements of the disease phenotype. Cerebral atrophy was observed in P1, P3, and P4 at a very early age and appeared to be progressive in P4 (scan images in Figure S4), despite continuing neurodevelopment (Table 1). This, and the mild portal fibrosis observed on liver biopsy in two patients (Figures 4J–4L), should be considered as sequelae of FADD deficiency. Finally, FADD seems to be important for the development of a functional spleen in humans, as demonstrated by the presence of Howell-Jolly bodies—the hallmark of a lack of splenic function—in the blood smears of P3 and P4.7,8 In P2, a limited postmortem examination of lymph nodes, spleen, and thymus revealed no histological features of ALPS or other lymphoid tissue abnormalities (Figure 4). However, the spleen showed an expanded, congested red pulp containing increased neutrophils, consistent with sepsis. Moreover, the normal cord-sinus architecture of the red pulp was not readily apparent; reticulin deposition appeared disordered, normal sinusoidal “barrel hoop” fibers were not identified, and macrophages appeared haphazardly arranged (Figures 4C–4I). Examination of spleen histology in other affected individuals is unlikely to be possible, thus precluding firm conclusions regarding the relationship between human FADD deficiency and disordered red pulp microarchitecture. The prominence but incomplete penetrance of cardiac, cerebral, hepatic, and splenic abnormalities in the clinical phenotype strongly suggests the existence of organ-specific effects of FADD deficiency worthy of further study in suitably designed mouse models.
Figure 4.
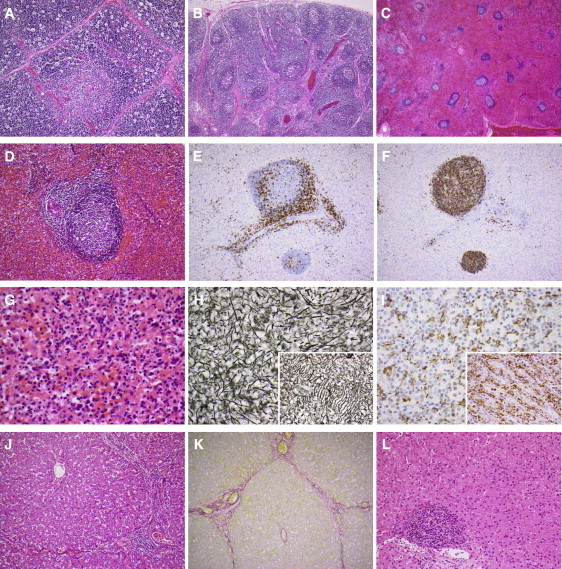
Histopathology of P2 and P4
(A–K) postmortem histopathology of P2 at 14 months.
(A) Normal thymic architecture; increased cortical tingible body macrophages consistent with a stress response (P2, hematoxylin and eosin [H&E] stain, original magnification × 100).
(B) Normal-appearing reactive lymph node with follicular hyperplasia (P2, H&E stain, original magnification × 40).
(C) Low-power view of the spleen showing expanded, congested red pulp with relatively reduced white pulp (P2, H&E stain, original magnification × 20).
(D) White pulp nodule containing a follicle with a reactive germinal centre, but no marginal zone hyperplasia, arising from a periarteriolar lymphoid sheath (P2, H&E stain, original magnification × 200).
(E) Immunohistochemistry for CD3 shows a normal distribution of CD3-positive T cells in a white pulp nodule (P2, Ventana Benchmark XT immunostainer, monoclonal antibody LN10, original magnification × 100).
(F) Immunohistochemistry for CD20 shows a normal distribution of CD20-positive B cells in a white pulp nodule (P2, Ventana Benchmark XT immunostainer, monoclonal antibody L26, original magnification × 100).
(G) Congested, disorganized red pulp containing increased neutrophils consistent with sepsis (P2, H&E stain, original magnification × 400).
(H) Disorganized reticulin staining pattern of splenic red pulp. Inset: “barrel hoop” arrangement of sinusoidal reticulin fibers in a normal control spleen (P2, Gordon and Sweet's reticulin stain, original magnification × 400).
(I) Immunohistochemistry for CD68 shows a haphazard arrangement of macrophages in the splenic red pulp. Inset: orderly distribution of cordal macrophages in a normal control spleen (P2, Ventana Benchmark XT immunostainer, monoclonal antibody PG-M1, original magnification × 400).
(J) Liver showing portal-portal linkage and expansion of portal tracts by a moderate chronic inflammatory cell infiltrate without interface activity (P2, H&E stain, original magnification × 100).
(K) Sirius red staining of liver shows collagen deposition (red) within portal-portal bridges. Elastin could also be demonstrated (not shown), consistent with established portal-portal fibrosis (P2, Sirius red and Fast green stain, original magnification × 100).
(L) Histopathology of P4 at 22 months. The liver shows mild expansion of portal tracts by a chronic inflammatory cell infiltrate but no bridging fibrosis (H&E stain, original magnification × 200).
In conclusion, we have described the clinical features of an autosomal-recessive inherited disorder with biological features of ALPS, susceptibility to bacterial and viral infections, and developmental abnormalities. Furthermore, we have determined its genetic etiology, which results in FADD deficiency in the affected children, by genome-wide linkage analysis and whole-exome sequencing. Finally, we have dissected the molecular mechanisms of disease for several aspects of this phenotype, including bacterial susceptibility (functional hyposplenism), viral susceptibility (impaired IFN immunity), and features of ALPS (impaired Fas-dependent apoptosis). Other phenotypes, particularly those of a developmental nature, remain unexplained but are probably due to the disruption of Fas-independent pathways, given that they were not documented in Fas-deficient patients. These unanticipated findings highlight the power of exome sequencing used in combination with genome-wide linkage analysis within consanguineous families.9–11,33 We anticipate rapid advances in molecular medicine,5,9 facilitated by this strategy, in the coming years.
Acknowledgments
We thank the patients' family for their trust and all members of our laboratories for helpful discussions. We thank Capucine Picard for her expert analysis of the lymphocyte subsets of the patients. We thank Louise Tee for technical assistance and SNP genotyping and Shanaz Pasha for patient ascertainment. We thank Magorzata Dobaczewska for the production of proteins in bacteria and Andrey Bobkov (protein facility at SBMRI) for DSC measurement. The Laboratory of Human Genetics of Infectious Diseases is supported by grants from INSERM, University Paris Descartes, The Rockefeller University Center for Clinical Translational Science (grant number 5UL1RR024143-03), The Rockefeller University, and St. Giles Foundation. The laboratory of S.H. is supported by an MRC grant (G0701897). This work was also supported by a R01AA017238 grant to S.J.R., by an ANR grant (ANR-08-GENO-015-01) to F.R.L., and by the WellChild foundation. M.B. is supported by a fellowship from the Cancer Research Institute.
Supplemental Data
Web Resources
The URLs for the data presented here are as follows:
1000 Genomes Project, http://www.1000genomes.org
HomozygosityMapper, http://www.homozygositymapper.org/
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
Picard Tools, http://picard.sourceforge.net/
SeattleSeq SNP Annotation, http://gvs.gs.washington.edu/SeattleSeqAnnotation/
References
- 1.Del-Rey M., Ruiz-Contreras J., Bosque A., Calleja S., Gomez-Rial J., Roldan E., Morales P., Serrano A., Anel A., Paz-Artal E., Allende L.M. A homozygous Fas ligand gene mutation in a patient causes a new type of autoimmune lymphoproliferative syndrome. Blood. 2006;108:1306–1312. doi: 10.1182/blood-2006-04-015776. [DOI] [PubMed] [Google Scholar]
- 2.Fisher G.H., Rosenberg F.J., Straus S.E., Dale J.K., Middleton L.A., Lin A.Y., Strober W., Lenardo M.J., Puck J.M. Dominant interfering Fas gene mutations impair apoptosis in a human autoimmune lymphoproliferative syndrome. Cell. 1995;81:935–946. doi: 10.1016/0092-8674(95)90013-6. [DOI] [PubMed] [Google Scholar]
- 3.Rieux-Laucat F., Le Deist F., Hivroz C., Roberts I.A., Debatin K.M., Fischer A., de Villartay J.P. Mutations in Fas associated with human lymphoproliferative syndrome and autoimmunity. Science. 1995;268:1347–1349. doi: 10.1126/science.7539157. [DOI] [PubMed] [Google Scholar]
- 4.Su H.C., Lenardo M.J. Genetic defects of apoptosis and primary immunodeficiency. Immunol. Allergy Clin. North Am. 2008;28:329–351, ix. doi: 10.1016/j.iac.2008.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Casanova J.L., Abel L. Primary immunodeficiencies: a field in its infancy. Science. 2007;317:617–619. doi: 10.1126/science.1142963. [DOI] [PubMed] [Google Scholar]
- 6.Brigden M.L. Detection, education and management of the asplenic or hyposplenic patient. Am. Fam. Physician. 2001;63:499–506, 508. [PubMed] [Google Scholar]
- 7.Doll D.C., List A.F., Yarbro J.W. Functional hyposplenism. South. Med. J. 1987;80:999–1006. doi: 10.1097/00007611-198708000-00016. [DOI] [PubMed] [Google Scholar]
- 8.Katcher A.L. Familial asplenia, other malformations, and sudden death. Pediatrics. 1980;65:633–635. [PubMed] [Google Scholar]
- 9.Byun M., Abhyankar A., Lelarge V., Plancoulaine S., Palanduz A., Telhan L., Boisson B., Picard C., Dewell S., Zhao C. Whole-exome sequencing-based discovery of STIM1 deficiency in a child with fatal classic Kaposi sarcoma. J. Exp. Med. 2010;207:2307–2312. doi: 10.1084/jem.20101597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ng S.B., Buckingham K.J., Lee C., Bigham A.W., Tabor H.K., Dent K.M., Huff C.D., Shannon P.T., Jabs E.W., Nickerson D.A. Exome sequencing identifies the cause of a mendelian disorder. Nat. Genet. 2010;42:30–35. doi: 10.1038/ng.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ng S.B., Turner E.H., Robertson P.D., Flygare S.D., Bigham A.W., Lee C., Shaffer T., Wong M., Bhattacharjee A., Eichler E.E. Targeted capture and massively parallel sequencing of 12 human exomes. Nature. 2009;461:272–276. doi: 10.1038/nature08250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., DePristo M.A. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chinnaiyan A.M., O'Rourke K., Tewari M., Dixit V.M. FADD, a novel death domain-containing protein, interacts with the death domain of Fas and initiates apoptosis. Cell. 1995;81:505–512. doi: 10.1016/0092-8674(95)90071-3. [DOI] [PubMed] [Google Scholar]
- 15.Balachandran S., Thomas E., Barber G.N. A FADD-dependent innate immune mechanism in mammalian cells. Nature. 2004;432:401–405. doi: 10.1038/nature03124. [DOI] [PubMed] [Google Scholar]
- 16.Osborn S.L., Diehl G., Han S.J., Xue L., Kurd N., Hsieh K., Cado D., Robey E.A., Winoto A. Fas-associated death domain (FADD) is a negative regulator of T-cell receptor-mediated necroptosis. Proc. Natl. Acad. Sci. USA. 2010;107:13034–13039. doi: 10.1073/pnas.1005997107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Walsh C.M., Bell B.D. T cell intrinsic roles of autophagy in promoting adaptive immunity. Curr. Opin. Immunol. 2010;22:321–325. doi: 10.1016/j.coi.2010.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tourneur L., Chiocchia G. FADD: a regulator of life and death. Trends Immunol. 2010;31:260–269. doi: 10.1016/j.it.2010.05.005. [DOI] [PubMed] [Google Scholar]
- 19.Scott F.L., Stec B., Pop C., Dobaczewska M.K., Lee J.J., Monosov E., Robinson H., Salvesen G.S., Schwarzenbacher R., Riedl S.J. The Fas-FADD death domain complex structure unravels signalling by receptor clustering. Nature. 2009;457:1019–1022. doi: 10.1038/nature07606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Peter M.E., Krammer P.H. The CD95(APO-1/Fas) DISC and beyond. Cell Death Differ. 2003;10:26–35. doi: 10.1038/sj.cdd.4401186. [DOI] [PubMed] [Google Scholar]
- 21.Taylor R.C., Cullen S.P., Martin S.J. Apoptosis: controlled demolition at the cellular level. Nat. Rev. Mol. Cell Biol. 2008;9:231–241. doi: 10.1038/nrm2312. [DOI] [PubMed] [Google Scholar]
- 22.Chun H.J., Zheng L., Ahmad M., Wang J., Speirs C.K., Siegel R.M., Dale J.K., Puck J., Davis J., Hall C.G. Pleiotropic defects in lymphocyte activation caused by caspase-8 mutations lead to human immunodeficiency. Nature. 2002;419:395–399. doi: 10.1038/nature01063. [DOI] [PubMed] [Google Scholar]
- 23.Wang J., Zheng L., Lobito A., Chan F.K., Dale J., Sneller M., Yao X., Puck J.M., Straus S.E., Lenardo M.J. Inherited human Caspase 10 mutations underlie defective lymphocyte and dendritic cell apoptosis in autoimmune lymphoproliferative syndrome type II. Cell. 1999;98:47–58. doi: 10.1016/S0092-8674(00)80605-4. [DOI] [PubMed] [Google Scholar]
- 24.Bleesing J.J., Brown M.R., Straus S.E., Dale J.K., Siegel R.M., Johnson M., Lenardo M.J., Puck J.M., Fleisher T.A. Immunophenotypic profiles in families with autoimmune lymphoproliferative syndrome. Blood. 2001;98:2466–2473. doi: 10.1182/blood.v98.8.2466. [DOI] [PubMed] [Google Scholar]
- 25.Magerus-Chatinet A., Stolzenberg M.C., Loffredo M.S., Neven B., Schaffner C., Ducrot N., Arkwright P.D., Bader-Meunier B., Barbot J., Blanche S. FAS-L, IL-10, and double-negative CD4- CD8- TCR alpha/beta+ T cells are reliable markers of autoimmune lymphoproliferative syndrome (ALPS) associated with FAS loss of function. Blood. 2009;113:3027–3030. doi: 10.1182/blood-2008-09-179630. [DOI] [PubMed] [Google Scholar]
- 26.Wilson N.S., Dixit V., Ashkenazi A. Death receptor signal transducers: nodes of coordination in immune signaling networks. Nat. Immunol. 2009;10:348–355. doi: 10.1038/ni.1714. [DOI] [PubMed] [Google Scholar]
- 27.Mahlaoui N., Minard-Colin V., Picard C., Bolze A., Ku C.L., Tournilhac O., Gilbert-Dussardier B., Pautard B., Durand P., Devictor D. Isolated congenital asplenia: A French nationwide retrospective survey of 20 cases. J Pediatr. 2010 doi: 10.1016/j.jpeds.2010.07.027. [DOI] [PubMed] [Google Scholar]
- 28.Picard C., Puel A., Bustamante J., Ku C.L., Casanova J.L. Primary immunodeficiencies associated with pneumococcal disease. Curr. Opin. Allergy Clin. Immunol. 2003;3:451–459. doi: 10.1097/00130832-200312000-00006. [DOI] [PubMed] [Google Scholar]
- 29.Balachandran S., Venkataraman T., Fisher P.B., Barber G.N. Fas-associated death domain-containing protein-mediated antiviral innate immune signaling involves the regulation of Irf7. J. Immunol. 2007;178:2429–2439. doi: 10.4049/jimmunol.178.4.2429. [DOI] [PubMed] [Google Scholar]
- 30.Chapgier A., Wynn R.F., Jouanguy E., Filipe-Santos O., Zhang S., Feinberg J., Hawkins K., Casanova J.L., Arkwright P.D. Human complete Stat-1 deficiency is associated with defective type I and II IFN responses in vitro but immunity to some low virulence viruses in vivo. J. Immunol. 2006;176:5078–5083. doi: 10.4049/jimmunol.176.8.5078. [DOI] [PubMed] [Google Scholar]
- 31.Chapgier A., Kong X.F., Boisson-Dupuis S., Jouanguy E., Averbuch D., Feinberg J., Zhang S.Y., Bustamante J., Vogt G., Lejeune J. A partial form of recessive STAT1 deficiency in humans. J. Clin. Invest. 2009;119:1502–1514. doi: 10.1172/JCI37083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yeh W.C., Pompa J.L., McCurrach M.E., Shu H.B., Elia A.J., Shahinian A., Ng M., Wakeham A., Khoo W., Mitchell K. FADD: essential for embryo development and signaling from some, but not all, inducers of apoptosis. Science. 1998;279:1954–1958. doi: 10.1126/science.279.5358.1954. [DOI] [PubMed] [Google Scholar]
- 33.Walsh T., Shahin H., Elkan-Miller T., Lee M.K., Thornton A.M., Roeb W., Abu Rayyan A., Loulus S., Avraham K.B., King M.C., Kanaan M. Whole exome sequencing and homozygosity mapping identify mutation in the cell polarity protein GPSM2 as the cause of nonsyndromic hearing loss DFNB82. Am. J. Hum. Genet. 2010;87:90–94. doi: 10.1016/j.ajhg.2010.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.