

Abstract
Thoracic aortic aneurysms and dissections (TAAD) cause significant morbidity and mortality, but the genetic origins of TAAD remain largely unknown. In a genome-wide analysis of 418 sporadic TAAD cases, we identified 47 copy number variant (CNV) regions that were enriched in or unique to TAAD patients compared to population controls. Gene ontology, expression profiling, and network analysis showed that genes within TAAD CNVs regulate smooth muscle cell adhesion or contractility and interact with the smooth muscle-specific isoforms of α-actin and β-myosin, which are known to cause familial TAAD when altered. Enrichment of these gene functions in rare CNVs was replicated in independent cohorts with sporadic TAAD (STAAD, n = 387) and inherited TAAD (FTAAD, n = 88). The overall prevalence of rare CNVs (23%) was significantly increased in FTAAD compared with STAAD patients (Fisher's exact test, p = 0.03). Our findings suggest that rare CNVs disrupting smooth muscle adhesion or contraction contribute to both sporadic and familial disease.
Introduction
Thoracic aortic aneurysms and dissections (TAAD) cause more than 8000 deaths in the United States every year.1 Progressive enlargement of the aorta is usually asymptomatic until a catastrophic event occurs, typically an acute aortic dissection leading to pericardial tamponade, stroke, acute aortic regurgitation, or other complications.2 The pathological hallmark of TAAD is “medial degeneration,” previously termed cystic medical necrosis.3 The medial layer of the aorta is normally composed of laminar smooth muscle cells (SMCs) surrounded by a tightly packed meshwork of elastin and collagen that provides tensile strength and flexibility. Medial degeneration corresponds to progressive depletion of elastin fibers from the media and predisposes to dissection or rupture.4 The annual risk of sudden death from an enlarged thoracic aneurysm as a result of an acute aortic dissection is more than 10%.5 Timely surgical repair of aneurysms can prevent death. However, there are few recognized risk factors for the development of thoracic aneurysms and no reliable biomarkers that might be used for screening. The aortic segment affected, age of onset, and rate of enlargement of aneurysms are heterogeneous.6 Eighty percent of TAAD patients do not disclose a family history of vascular disease (sporadic thoracic aortic aneurysms, or STAAD).7 Because of these challenges, the genetic causes of STAAD have remained largely unknown. Prospective identification of patients at risk for TAAD via a genetic strategy will be critical to prevent sudden deaths from this treatable disease.
Twenty percent of TAAD patients have an affected relative (familial thoracic aortic aneurysm and dissection, or FTAAD), and the phenotype is primarily inherited in an autosomal-dominant manner characterized by variable expression and incomplete penetrance.8 Large pedigrees with multiple affected members have been assembled to allow the genetic mapping of seven distinct loci for FTAAD on 5q13-q14, 16p13.12-p13.13, 11q23.3-q24, 3p24-p25, 9q22, 10q22-q24, and 15q24-q26 (MIM 607087, 132900, 607086, 6103080, 608967, 611788, and unassigned). Four of these genes have been identified: ACTA2 (MIM 102620, 10q23), MYH11 (MIM 160745, 16p13), TGFBR1 (MIM 190181, 9q22), and TGFBR2 (MIM 190182, 3p22).9–12 ACTA2 and MYH11 encode the smooth muscle-specific isoforms of actin and myosin, which are primary components of the contractile apparatus. The TGF-β pathway regulates the expression of contractile proteins by vascular SMCs.13 Together, these genes account for less than 20% of the familial cases, suggesting that additional locus heterogeneity could be extensive. Similarly, genetic syndromes that predispose to the development of thoracic aneurysms are also caused by mutations of SMC adhesive or cytoskeletal proteins such as fibrillin-1 (FBN1 [MIM 134797]) in Marfan syndrome (MIM 154700) and filamin-A (FLNA [MIM 300017]) in patients with X-linked periventricular nodular heterotopia (MIM 300049) and thoracic aortic disease.14 Both proteins perform an essential function in vascular development by anchoring actin fibers to focal adhesions on SMC membranes.15 Based on these observations, we hypothesized that disruption of vascular SMC contractility by mutation of contractile proteins or impairment of SMC adhesion predisposes to TAAD.16
Unanswered questions concerning the pathogenesis of TAAD include how mutations in SMC contractile proteins lead to aortic pathology and why the phenotypic manifestations of identical mutations are so variable. We propose that variations in currently unidentified modifying genes may exert a profound influence on an individual's susceptibility to aneurysm disease. These genetic variants almost certainly interact with environmental factors such as age, gender, hypertension, and tobacco use to determine an individual's total risk for TAAD.17 According to this model, gene products that relay environmental signals to regulate smooth muscle functions are ideal candidates to modify the pathogenesis of TAAD.
Copy number variations (CNVs) have been shown to confer increased risk for common multifactorial diseases such as autism (MIM 209850) and schizophrenia (MIM 181500), as well as congenital cardiovascular disorders such as tetralogy of Fallot (TOF [MIM 187500]).18–20 These findings are consistent with a genetic model wherein any of a large number of individually rare copy number mutations contribute to disease causation or predisposition.21 We therefore hypothesized that individually rare structural variants could contribute to disease risk in TAAD and that such variants are likely enriched within genes involved in vascular SMC contractility.
Subjects and Methods
TAAD and Control Cohorts
The study protocol was approved by the institutional review boards at Baylor College of Medicine and University of Texas Health Science Center at Houston (UTHSCH). After obtaining written informed consent, we enrolled non-Hispanic patients of European descent with sporadic ascending aortic aneurysms or thoracic aortic dissections (STAAD) who were evaluated at the Texas Heart Institute at St. Luke's Episcopal Hospital and Methodist Hospital (BCM cohort) or Memorial Hermann Hospital (UTHSCH cohort). BCM cases included 418 unrelated individuals, 269 males and 149 females, with a mean age of 63 years (32–86 years). The replication cohort consisted of 387 unrelated individuals enrolled at UTHSCH. In all patients, the diagnosis was confirmed by echocardiography, computed tomography, magnetic resonance imaging, or aortography. We excluded patients who were less than 31 years of age because of the higher prevalence of Mendelian disorders in this age group. We also excluded patients with aortic lesions associated with trauma, infection, aortitis, syndromic forms of TAAD, or connective tissue disorders, patients with a first-degree relative with thoracic aortic aneurysm or dissection, patients with an isolated intramural hematoma, penetrating aortic ulcer, or pseudoaneurysm, and patients who received packed red blood cell, whole blood, or platelet transfusions within 72 hr of blood collection.
The familial TAAD cohort (FTAAD) consisted of 88 affected probands from unrelated families with multiple members with TAAD who did not have a genetic mutation or syndrome identified as the cause of the inherited TAAD. The proband and family members were considered to be affected if they had dissection of the thoracic aorta, surgical repair of an ascending aneurysm, or dilatation of the ascending aorta greater than 4.5 cm based on echocardiography images of the aortic diameter at the sinuses of Valsalva and/or the ascending aorta.
The primary controls for the study were Illumina genotypes of 6809 subjects obtained from the Database of Genotypes and Phenotypes (dbGAP). The characteristics of the five control cohorts and genotypes are listed in Table S1 available online. Our analysis was confined to unrelated individuals of European descent over 31 years of age from each data set. Phenotypic data relevant to TAAD were not available from any of the multiple control samples used.
DNA Extraction and Genotyping
DNA was extracted from saliva, whole blood, or buffy coat samples collected from each patient at enrollment and stored at −80°C until DNA extraction. The quantity of double-stranded DNA was measured with PicoGreen (Invitrogen). Samples with a DNA concentration below 40 μg/ml were concentrated with a Speed Vac system; samples with a DNA concentration above 150 μg/ml were normalized to 100 μg/ml. Purity of the DNA samples was assessed with a NanoDrop spectrophotometer (Thermo Scientific).
Genotyping for the study was performed with the BeadStation system with the Human CNV370-Quad BeadChip (Illumina) for STAAD samples and the Human 660W-Quad BeadChip (Illumina) for FTAAD samples. Briefly, genomic DNA was amplified and fragmented prior to hybridization to the genotyping chip. A single base pair extension reaction was then performed, followed by staining with detectable fluorescent dye labels. The resulting products were imaged with the BeadArray Reader (Illumina). Quality standards for labeling, single base extension, hybridization, stringency, and nonspecific binding were verified. Additional quality control thresholds included standard deviation of normalized intensity values <0.35, inferred European ancestry by comparison to HapMap CEU (Utah residents with ancestry from northern and western Europe) samples (with multidimensional scaling implemented in PLINK), and >97% call rate of attempted SNPs. Replicates, unexpected duplicates, or samples with mismatched gender, excess heterozygosity, excess homozygosity, or missing genotypes were excluded from further analysis.
Data Analysis
After allele detection and genotype calling were performed with Genome Studio software (Illumina), B allele frequencies and logR ratios were exported as text files for PennCNV analysis. CNVPartition was run as a plug-in within the Genome Studio browser with the following settings: confidence threshold 50, minimum number of probes 5. Sample-level quality control analysis was performed with PennCNV software. Samples were excluded from further analysis if any of the following criteria were met: standard deviation of logR ratios > 0.35, B allele frequency drift > 0.1, waviness factor > 0.05, number of CNVs identified > 2 standard deviations above the mean of each data set. CNVs in pericentromeric and immunoglobulin regions were also excluded. Because of technical limitations with the FTAAD data set, CNV detection was restricted to 311,803 SNPs that are common to the Illumina 370K and 660K platforms, and association tests were limited to CNVs with a minimum of ten SNPs. Numbers of excluded genotypes were not significantly different between data sets.
Base coverage and clustering of CNV intervals was determined with the Galaxy Web server. Genes that overlap with TAAD-associated CNVs were identified with the regional association test function in PLINK.22 CNV regions called by both PennCNV and CNVPartition were identified with the overlap function for rare CNVs in PLINK. CNVs that were present in more than 1% of samples were defined as copy number polymorphisms and excluded from comparative analysis. CNVs less than 50 kb in length were also excluded from case-control association tests. Expression profiles for CNV genes were determined with GeneHub-GEPIS.23 For functional analysis, gene lists were entered into the IPA Web server (Ingenuity Systems) and the MetaCore Web server (GeneGo, Bioinformatics Software). The significance of associations was scored with p values derived from two-tailed Fisher's exact tests.
Confirmatory Studies
We validated a subset of suspected CNVs via comparative genomic hybridization (CGH) with custom Agilent oligonucleotide microarrays.18 Array design and hybridizations were performed according to the manufacturer's recommended protocols. Briefly, 400 ng of genomic DNA was digested with restriction enzymes AluI and RsaI and fluorescently labeled with the Agilent DNA Labeling kit. The test samples were labeled with cyanine 5-dUTP, and the reference sample was labeled with cyanine 3-dUTP. Labeled DNA was denatured and preannealed with Cot-1 DNA and Agilent blocking reagent prior to hybridization for 24 hr in a 658C Agilent hybridization oven. Standard wash procedures were followed. Arrays were scanned at 5 μm resolution with an Agilent scanner, and image analysis was performed with default CGH settings of Feature Extraction software 9.1.1.1 (Agilent Technologies).
Primers and probes used to detect CNVs with real-time quantitative PCR were designed with PrimerExpress software (Applied Biosystems). Primers were purchased from Applied Biosystems, Integrated DNA Technologies, and Eurofin MWG Operon. Reactions were run on the ABI 7900 real-time PCR system (Applied Biosystems) with the following cycling parameters: one cycle at 95°C for 10 min, followed by 40 cycles at 95°C for 15 s and 60°C for 60 s. Each reaction was carried out in a total volume of 20 μl containing 1× Taqman Universal PCR Master mix, 900 nM of each primer, 250 nM probe, and 10 ng of genomic DNA. Copy number was determined by Copy Caller 1.0 software (Applied Biosystems) with a probe set within the RnaseP gene as a reference.
Gene Ontology and Network Analysis
Functional analysis was performed with the GeneGo MetaCore and Ingenuity Pathways Analysis (IPA) databases. Tables of genes that were found to be enriched within TAAD-associated CNVs in the BCM, UTHSCH, and FTAAD cohorts were imported into the MetaCore or IPA Web servers as individual experiments. All types of copy num-ber alterations were included together in our analysis. The significance of biological pathways was estimated through a variation of Fisher's exact test, as implemented in MetaCore and IPA, and was adjusted for multiple testing via Benjamini-Hochberg analysis, with a false discovery rate of less than 0.05.
Ingenuity Pathways Analysis
Gene networks were algorithmically generated based on their connectivity to the Ingenuity knowledge base. A maximum of 35 genes from the input data set was allowed in each network. Network scores were based on the hypergeometric distribution and were calculated via right-tailed Fisher's exact test. To present most of the relevant network data on the same figure, we used the Merge Networks feature. The resulting figure (Figure 2) is derived from known interactions between nodes from the top two networks of each data set.
Figure 2.
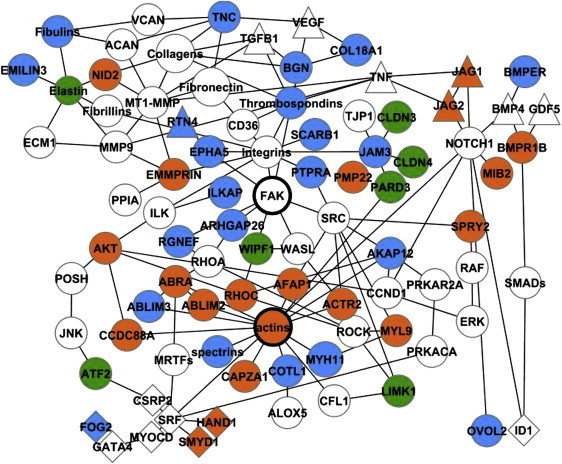
Network of Interacting TAAD CNV Genes
Genes within TAAD-associated CNVs are colored green (FTAAD), orange (BCM), or blue (UTHSCH); lines indicate functional or physical interactions between gene products; not all genes in the network are shown. All CNVs represented in this network are rare and occur in one or none of 4922 control individuals. The following abbreviations are used: ABRA, actin-binding Rho-activating protein; ABLIM, actin-binding LIM protein family; ACAN, aggrecan; ACTR2, actin-related protein 2 homolog; AFAP1, actin filament-associated protein 1; AKAP12, A kinase anchor protein12; ALOX5, 5-lipoxygenase; ARHGAP26, GTPase regulator associated with FAK; ATF2, activating transcription factor 2; BGN, biglycan; BMP4, bone morphogenetic protein 4; BMPR1B, bone morphogenetic protein receptor 1B; BMPER, BMP-binding endothelial cell precursor-derived regulator; EPHA5, ephrin type A receptor 5; EMMPRIN, extracellular matrix metalloproteinase inducer; CAPZA1, capping protein muscle Z-line, alpha 1; CCDC88A, coiled-coil domain containing 88A; CD36, thrombospondin receptor; CLDN, claudins; COL18A1, collagen type 18, alpha 1 and endostatin; CSRP2, cysteine-rich protein 2; FAK, focal adhesion kinase; FOG2, friend of GATA2; GDF5, growth differentiation factor 5; HAND1, heart and neural crest derivatives expressed 1; ID1, inhibitor of DNA binding 1; ILKAP, integrin-linked kinase-associated phosphatase; JAG, jagged homolog; JAM3, junctional adhesion molecule 3; JNK, JUN N-terminal kinase; LIMK1, LIM domain kinase 1; MIB2, mindbomb homolog 2; MMP9, matrix metalloproteinase 9; MT1-MMP, matrix metalloproteinase 14; MRTFs, myocardin- related transcription factors; MYH11, myosin heavy chain, smooth muscle isoform; MYL9, myosin regulatory light chain 2, smooth muscle isoform; MYOCD, myocardin; NID2, nidogen 2; OVOL2, Ovo-like 2; PARD3, partitioning defective 3 homolog; PMP22, peripheral myelin protein 22; POSH, plenty of SH3s; PRKAR2A, cAMP-dependent protein kinase, regulatory subunit alpha 2; PPIA, cyclophilin A; PRKACA, cAMP-dependent protein kinase catalytic subunit alpha; PTPRA, protein tyrosine phosphatase, receptor type, alpha polypeptide; RGNEF, Rho guanine nucleotide exchange factor; RHOC, Ras homolog gene family, member C; ROCK, Rho-associated, coiled-coil domain-containing protein kinase; SMAD, mothers against decapentaplegic homolog; SMYD1, set and MYND domain-containing 1; SPRY2, sprouty homolog 2; SRF, serum response factor; TJP1, tight junction protein ZO-1; TNC, tenascin C; VCAN, versican; WASL, Wiskott-Aldrich syndrome gene-like protein; WIPF1, WAS/WASL-interacting protein family member 1. Figure was derived from IPA analysis (version 8.1) and created with Cytoscape (version 2.6.3).
The significance of associations between genes within TAAD CNVs and canonical pathways was measured (1) as a ratio of the number of genes from the data set that map to the pathway divided by total number of genes within the canonical pathway and (2) as a p value calculated via Fischer's exact test, representing the probability that the association between the genes and the canonical pathway is explained by chance alone.
MetaCore Analysis
In MetaCore, we performed enrichment analysis of four independent ontologies: Gene Ontology (GO) biological processes, canonical pathways, disease categories, and GeneGo cellular processes, which are based on experimentally validated interactions. Calculation of hypergeometric p values was similar to IPA. Networks were generated via Dijkstara's shortest path algorithm, which connected nodes from the TAAD experimental data sets by the shortest directed paths with manually curated interactions from the MetaCore database.
Results
CNV Discovery in TAAD Cases
The methods used for CNV discovery in cases and controls with two independent algorithms are outlined in Figure 1. We also performed CNV analysis of 88 unrelated affected probands from families with at least two relatives affected with TAAD (FTAAD cohort). DNA samples from these individuals were collected and genotyped via identical methods. The characteristics of the study populations are shown in Table 1.
Figure 1.
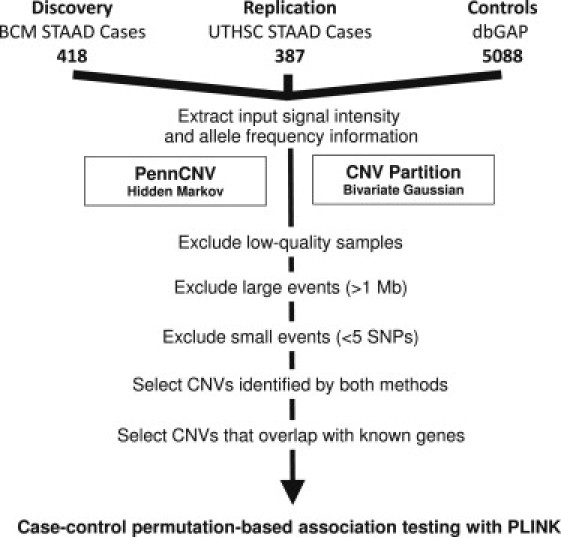
Flow Diagram of CNV Analysis
The following abbreviations are used: STAAD, sporadic thoracic aortic aneurysms and dissections; BCM, Baylor College of Medicine; UTHSCH, University of Texas Health Science Center; dbGAP, Database of Genotypes and Phenotypes. Samples that did not meet quality thresholds were identified and excluded via functions implemented in the PennCNV package. Rare events larger than 1 Mb were analyzed separately.
Table 1.
Clinical Characteristics of TAAD Study Groups
|
Sporadic TAAD |
p Value | FTAAD (88)c | ||
|---|---|---|---|---|
| BCM (418) | UTHSCH (387) | |||
| Female | 36% | 35% | NS | 26% |
| Age | 63 (12) | 63 (12) | NS | 45 (14) |
| BMI | 27.8 (5.2) | 28.0 (6.1) | NS | 27.5 (5.5) |
| Smoking | 62% | 60% | NS | NA |
| Hypertension | 67% | 85% | <0.01 | NA |
| Diabetes | 5% | 7% | NS | 6% |
| Maximum aortic diametera | 5.9 (1.2) | 5.6 (1.0) | NS | 5.5 (1.8) |
| Bicuspid aortic valve | 15% | 23% | <0.01 | 15% |
| Dissectionb | 47% | 49% | NS | 44% |
| Stanford type A | 33% | 26% | 0.04 | 36% |
| Stanford type B | 15% | 23% | <0.01 | 8% |
| Aortic rupture | 2% | 4% | NS | 3% |
| Underwent aortic repair | 94% | 93% | NS | 57% |
| Involvement of ascending aorta | 89% | 83% | NS | 94% |
| Annuloaortic ectasia or root replacement | 24% | 33% | NS | 34% |
The following abbreviations are used: BCM, Baylor College of Medicine cohort; UTHSCH, University of Texas Health Science Center at Houston cohort; FTAAD, familial TAAD; p, probability value derived from Fisher's exact test; BMI, body mass index (kg/m2); NS, not significant. Categorical variables are shown as percentages, and continuous variables are shown as mean (standard deviation).
Aortic diameters were available for 397 BCM patients and 234 UTHSCH patients.
Four patients had both Stanford type A and type B dissections.
With the exception of age, gender, and aortic diagnosis (dissection, Stanford class, aortic rupture, repair, involvement of ascending aorta), values were calculated from 68 available records.
In a genome-wide analysis of 392 BCM STAAD patients (after exclusion of low-quality samples), CNVPartition detected 1924 CNVs (760 copy gains and 1164 copy losses), with an average of 5 CNVs per individual (0–21) and a mean length of 294,000 bp (646–18,724,786 bp). PennCNV detected 8103 CNVs (3346 copy gains and 4757 copy losses), with an average of 20 CNVs per individual (1–93) and an average length of 73,000 bp (597–4145007 bp). A total of 2063 CNVs in 123 separate chromosomal regions containing 380 Mb or 13% of the genome were detected by both algorithms. The results of our CNV analysis are summarized in Table 2.
Table 2.
Summary of CNV Discovery
|
TAAD |
Controls | |||
|---|---|---|---|---|
| BCM | UT | Familial | ||
| Number of individuals | 392 | 366 | 87 | 4922 |
| Microarray platform | 370Q | 370Q | 660W | Multiple |
| Number of CNVs: PennCNV | 8103 | 8576 | 664a | 146423 |
| Number of CNVs: CNVP | 1924 | 2086 | 1764 | 107282 |
| Number of CNVs: Overlap | 1025 | 1038 | 93 | 26168 |
| Number of CNVs: Duplications only | 562 | 493 | 49 | 11180 |
| Number of CNVs: >500 kb | 193 | 148 | 12 | 2803 |
| Number of CNVs: Rare CNV regions | 47 | 57 | 20 | ND |
| Number of CNVs: Percentage of individuals | 11 | 15 | 23 | ND |
| Number of genes overlapped by rare CNVs | 186 | 210 | 84 | ND |
Number denotes number of unrelated individuals in each data set with usable CNV data; PennCNV denotes number of autosomal CNVs detected by PennCNV algorithm; CNVP denotes number of autosomal CNVs detected by CNVPartition algorithm; Overlap denotes number of autosomal CNVs detected by both PennCNV and CNVPartition; ND denotes not determined. Rare CNV regions are a subset of the overlapping CNVs identified by both algorithms.
Called with 311,803 SNPs common to 370K and 660K platforms.
To validate our findings, we amplified nine predicted rare CNV regions (CNVRs) (involving ABLIM3 [MIM 611305], AKAP13 [MIM 604698], BMPR1B [MIM 603248], JAG1 [MIM 601920], MYH11, PHLPPL [MIM 611066], RHOC [MIM 165380], RGNEF [MIM 612790], and SLFN12 [MIM unassigned]) from genomic DNA via real-time PCR. Nine genomic DNA samples with eight predicted rare CNVRs (involving ACTB [MIM 102630], THBS4 [MIM 600715], SPTBN1 [MIM 182790], SPTBN4 [MIM 606214], SLFN12, MYH11, TGFBR3 [MIM 600742], and DFNB31 [MIM 607084]) were also hybridized to a custom CGH oligonucleotide array that was designed to interrogate predicted CNV regions based on our preliminary results. All of the predicted CNVs were successfully verified. In pairs of duplicated samples, the rate of concordance for CNVs determined by the same algorithm was 90%. Intersection of CNV calls from two different algorithms enabled us to derive a high-confidence data set for subsequent analysis.
To identify risk CNVs for STAAD, we analyzed the burden of large CNVs in cases and controls. In 392 BCM patients, we identified 63 copy gains or losses larger than 500 kb (43 duplications, 20 deletions). For comparison, control genotypes from unaffected adults were selected from five publicly available cohorts in dbGAP (Table S1). Multidimensional scaling (as implemented in PLINK) with HapMap reference populations (CEU, Japanese in Tokyo, Japan [JPT], and Yoruba in Ibadan, Nigeria [YRI]) was used to select 5088 ethnically matched controls of European descent from 6809 dbGAP controls. Identical genotype calling, quality control, and CNV detection methods were used in STAAD cases and controls. In the controls, PennCNV and CNVPartition jointly detected 26,168 CNVs in 558 separate chromosomal regions. The overall prevalence of duplications and deletions, as well as the percentage of CNVs that are located within gene boundaries, was significantly increased in STAAD patients (p < 0.001). Forty-three large CNVs (68%) discovered in the STAAD cases were also observed in at least one control individual, whereas 20 large CNVs were unique to STAAD cases and were not present in controls. These included a 12 Mb deletion in one individual that overlaps the critical region defined by linkage analysis for the FTAAD locus FAA1 in 11q23.3.24 In two other individuals, we identified 800 kb and 1.2 Mb duplications of 16p13.1 involving MYH11, which is also mutated in FTAAD.10,11 None of the individuals with large CNVs were found to have associated defects, such as patent ductus arteriosus or bicuspid aortic valves, or other extravascular anomalies.
To discover CNVRs that are enriched in STAAD patients, we performed association tests with the segmental CNV data restricted to CNVs less than 1 Mb in length via permutation methods implemented in PLINK. We identified 31 CNVRs involving 120 genes that were observed in cases, but not in controls, as well as 14 additional CNVRs involving 66 genes that are more prevalent in cases compared to controls. The enriched CNVRs are rare (prevalence of less than 0.1%) or not present at all in control individuals. Five of these CNVRs were found in more than one individual (Table 3). Sixteen of 37 copy gains are predicted to lead to three intact copies of all affected genes, with the exception of CNVs that disrupt one allele of ABRA (MIM 609747), ADAMTS2 (MIM 604539), BMPR1B (MIM 603248), CALB2 (MIM 114051), CAPZA1 (MIM 601580), CDRT4 (MIM unassigned), COX10 (MIM 602125), CTNNA3 (MIM 607667), DNAJA5 (MIM unassigned), DPP10 (MIM 608209), FHOD3 (MIM 609691), GARS (MIM 600287), LRCH3 (MIM unassigned), MTUS (MIM 609589), PBLD (MIM 612189), PHLPPL (MIM 611066), RASGRP3 (MIM 609531), RASSF2 (MIM 609492), RNF24 (MIM 612489), RNF150 (MIM unassigned), RREB1 (MIM 602209), SH3TC1 (MIM unassigned), TAFA2 (MIM unassigned), TBC1D9 (MIM unassigned), TBC1D14 (MIM unassigned), and TSSC1 (MIM 608998).
Table 3.
Rare CNV Regions in BCM TAAD Patients
| Chr. | Band | Start | Length | Gene(s) | Copy | Cases | Controls |
|---|---|---|---|---|---|---|---|
| 1 | p36.33 | 1142494 | 475171 | DVL1, CDC2L2, CDC2L1, MIB2, MMP23B, SSU72, ATAD3A, ATAD3B, ATAD3C, VWA1, CCNL2, AURKAIP1, MXRA8, SCNN1D, ACAP3, CPSF3L, PUSL1, UBE2J2, B3GALT6, SDF4, GLTPD1, TAS1R3, MRPL20 | Gainb | 2 | 1 |
| 2 | q14.1 | 116135534 | 531094 | DPP10a | Gainc | 2 | 0 |
| 4 | q22.3 | 95973863 | 372128 | BMPR1Ba | Loss/Gainc | 2 (U) | 0 |
| 4 | q31.21 | 141875917 | 129684 | RNF150,aTBC1D9a | Loss/Gainc | 2 | 0 |
| 16 | p13.11 | 14929488 | 1509330 | MYH11, ABCC6, NOMO2, NDE1, ABCC1, MPV17L, C16ORF45, C16ORF63, KIAA0430 | Gain | 2 (U,F) | 5 |
| 2 | p25.3 | 3342118 | 419473 | ALLC, COLEC11, RPS7, ADI1, RNASEH1, TTC15, TSSC1a | Gain | 1 | 0 |
| 1 | p13.2 | 111664830 | 94386 | C1ORF88, OVGP1d | Loss | 1 | 1 |
| 1 | p13.2 | 113015620 | 449830 | RHOC, SLC16A1, CAPZA1,aFAM19A3, PPM1J | Gain | 1 | 0 |
| 1 | q24.2 | 167524929 | 225256 | BLZF1, C1orf114, SLC19A2, F5 | Loss | 1 | 1 |
| 2 | p22.3 | 32208994 | 363359 | YIPF4, NLRC4, SPAST,dSLC30A6 | Gain | 1 | 0 |
| 2 | p22.3 | 33532429 | 66034 | RASGRP3a | Gain | 1 | 1 |
| 2 | p22.1 | 38371019 | 376145 | HNRPLL, ATL2 | Gain | 1 | 0 |
| 2 | p14 | 65256382 | 121219 | ACTR2 | Gain | 1 | 0 |
| 2 | p11.2 | 88684402 | 246926 | SMYD1, FABP1, THNSL2, EIF2AK3, RPIA | Loss | 1 | 0 |
| 2 | q33.3 | 207353709 | 198248 | CPO, FASTKD2 | Loss | 1 | 0 |
| 3 | q29 | 199053957 | 252802 | IQCG, LRCH3 | Gain | 1 | 1 |
| 4 | p16.1 | 6969581 | 1289129 | AFAP1, ABLIM2, SH3TC1,aSORCS2, TBC1D14,aGRPEL1, CCDC96, TADA2B | Gain | 1 | 0 |
| 4 | p14 | 37251646 | 184002 | RELL1, c4orf19 | Gain | 1 | 0 |
| 5 | p15.33 | 143199 | 254392 | SDHA, CCDC127, PLEKHG4Bd | Gain | 1 | 1 |
| 5 | p13.2 | 34984289 | 106107 | AGXT2, DNAJA5, PRLR | Gain | 1 | 0 |
| 5 | q33.1 | 148274871 | 566620 | ABLIM3, AFAP1L1, SH3TC2, HTR4, GRPEL2, PCYOX1L, IL17B | Gain | 1 (U) | 0 |
| 5 | q33.2 | 153778073 | 203294 | SAP30L, HAND1 | Gain | 1 | 0 |
| 5 | q35.3 | 178661437 | 204439 | ADAMTS2a | Gain | 1 | 1 |
| 6 | p24.3 | 7137785 | 135061 | RREB1,aSSR1, CAGE1a | Gain | 1 | 1 |
| 7 | p22.1 | 5441935 | 142828 | ACTB, FBXL18 | Gain | 1 | 0 |
| 7 | p15.1 | 30623992 | 507188 | GHRHR, AQP1, ADCYAP1R1, INMT, FAM188B, CRHR2, GARSa | Gain | 1 | 0 |
| 8 | p22 | 17741656 | 153872 | PCM1, FGL1, MTUS1d | Gain | 1 | 0 |
| 8 | q23.1 | 107847500 | 39522 | ABRAa | Gain | 1 | 0 |
| 10 | q21.2 | 63577609 | 75066 | RTKN2 | Loss | 1 | 0 |
| 10 | q21.3 | 68672493 | 1073082 | MYPN,dSIRT1, CTNNA3,aHERC4, PBLD,a,dDNAJC12, ATOH7 | Gain | 1 | 1 |
| 10 | q23.31 | 90129215 | 158693 | C10ORF59 | Loss | 1 | 0 |
| 12 | q14.1 | 60119327 | 729214 | TAFA2a | Gain | 1 | 0 |
| 13 | q31.1 | 79702503 | 154705 | SPRY2 | Gain | 1 | 0 |
| 14 | q22.1 | 51343413 | 391832 | NID2, GNG2,dC14orf166 | Gain | 1 | 0 |
| 14 | q32.33 | 104688087 | 65485 | JAG2, NUDT14, BRF1 | Loss | 1 | 1 |
| 16 | q22.3 | 69965031 | 295906 | CALB2,aPHLPPL,aZNF23, ZNF19, TAT, MARVELD3, CHST4 | Gain | 1 | 0 |
| 17 | p13.1 | 7162432 | 58562 | NEURL4,aCENTB1, KCTD11,dTMEM95d | Gain | 1 | 0 |
| 17 | p12 | 14045669 | 1353365 | COX10,a,dCDRT15,dHS3ST3B1,dPMP22, TEKT3, CDRT4a | Gain | 1 (U) | 1 |
| 17 | q11.2 | 25958123 | 504562 | CRLF3, ATAD5, ADAP2, RNF135, NF1a | Gain | 1 | 0 |
| 18 | q12.2 | 32105471 | 269288 | FHOD3a | Gain | 1 | 1 |
| 18 | q23 | 73094966 | 704483 | GALR1 | Gain | 1 (F) | 1 |
| 19 | p13.3 | 532070 | 94443 | EMMPRIN, HCN2, POLRMT, FGF22, RNF126 | Loss | 1 | 0 |
| 19 | q13.2 | 45748633 | 171836 | SPTBN4, LTBP4, SHKBP1, NUMBL, ITPKC, AKT2, MAP3K10, CNTD2, TTC9B, C19ORF47, PRX, PLD3, HIPK4, BLVRB, SERTAD1, SERTAD3, ADCK4 | Loss | 1 (U) | 0 |
| 20 | p13 | 3923107 | 104045 | RNF24a | Gain | 1 | 0 |
| 20 | p13 | 4726754 | 47646 | RASSF2a | Gain | 1 | 0 |
| 20 | p12.2 | 8044130 | 5278047 | PLCB1, PLCB4, PAK7, SNAP25, MKKS, JAG1, TASP1, BTBD3, ANKRD5, ISM1, SPTCL3 | Loss | 1 | 0 |
| 20 | q13.11 | 41356169 | 184228 | SFRS6 | Gain | 1 | 0 |
Chr. denotes chromosome; Start denotes beginning of CNV based on build 36.1 of the reference genome; Length denotes size of CNV in base pairs; Type denotes duplication (gain) or deletion (loss); Controls denotes prevalence of CNV in control population (4922 individuals). Known aneurysm-associated genes are shown in bold type; genes that are expressed at above-average levels in SMCs are underlined; presence of CNVRs in UTHSCH (U) or familial (F) groups is indicated by parentheses.
Genes that are potentially disrupted by duplications.
p = 0.02.
p = 0.006.
Genes that overlap with 1–2 additional control CNVs.
Gene Ontology Analysis
To determine whether specific cellular functions are disrupted by STAAD-associated CNVs, we performed gene ontology and pathway analysis of the 186 genes within CNVRs that were enriched in BCM STAAD patients (Table 4). We found that 27 of these genes encode proteins that are involved in the regulation of the actin-based cytoskeleton (GO terms “Cell Assembly and Organization” and “Cell Morphology,” p = 5.2 × 10−3): ACTR2 (MIM 604221), AFAP1 (MIM 608252), AFAP1L1 (MIM unassigned), ABLIM2 (MIM 612544), ABLIM3 (MIM 611305), HTR4 (MIM 602164), RHOC, CAPZA1, DVL1 (MIM 601365), MYH11, MYPN (MIM 608517), FHOD3, PDLIM5 (MIM 605904), SNAP25 (MIM 600322), NDE1 (MIM 609449), ABCC1 (MIM 158343), SPRY2 (MIM 602466), POMZP3 (MIM 600587), PAK7 (MIM 608038), JAG1, BLZF1 (MIM 608692), SPTBN4, ABRA, CRHR2 (MIM 602034), AQP1 (MIM 107776), ADCYAP1R1 (MIM 102981), and HCN2 (MIM 602780). MetaCore analysis of these genes also identified pathways that mediate myofibril assembly (p = 5.9 × 10−8), elastic fiber assembly (p = 4.9 × 10−7), cytoskeleton remodeling (p = 1.7 × 10−4), and cell adhesion (p = 1.2 × 10−3) as the most significantly enriched GO processes. MetaCore and IPA also showed that these gene products interact together in canonical Rho GTPase signal transduction pathways (“Actin Cytoskeleton Signaling,” p = 5 × 10−4, and “Regulation of Actin-based Motility by Rho,” p = 2.6 × 10−3). We also analyzed CNVs that are more prevalent in control individuals compared with TAAD cases, but we did not find significant enrichment for cytoskeletal remodeling or other discrete intracellular processes. The most highly enriched GO terms for control CNVs were ribosomal RNA (2.9 × 10−11), enzyme interaction (1.7 × 10−8), and cytokine receptor binding (2.5 × 10−8).
Table 4.
Gene Functional Categories Enriched in BCM TAAD CNVs
| Category | UTHSCH | FTAAD | Genes in CNVs | p Value |
|---|---|---|---|---|
| Cardiovascular disease | + | + | CRHR2, SH3TC2, SCNN1D, SNAP25, BMPR1B, ZFPM2, F5, SORCS2, PLCB1, JAG1, FHOD3, ITPKC, MYH11 | 0.024 |
| Cardiovascular system development and function | + | + | CHST4, ZFPM2, ABRA, SMYD1, HCN2, RHOC, DVL1, IL17B, HAND1, SMYD1 | 0.023 |
| Cell morphology | − | + | CRHR2, HTR4, RHOC, DVL1, AQP1, MYH11, ADCYAP1R1, HCN2, SPRY2, JAG1, BLZF1, ACTB, CAPZA1, AFAP1, AFAP1L1, ABRA, ABLIM2, ABLIM3, MYPN | 0.026 |
| Cell-to-cell signaling and interaction | + | + | RHOC, DVL1, BMPR1B, PLCB4, EMMPRIN, SPRY2, TAS1R3, PLCB1, PAK7, JAG1, ACTN1, CCNL2, SDF4, IL17B, FGF22 | 0.026 |
| Cellular assembly and organization | + | + | ACTR2, HTR4, RHOC, DVL1, MYH11, SNAP25, NDE1, ABCC1, SPRY2, POMZP3, FABP1, PAK7, JAG1, BLZF1, ACTN1, AFAP1, AFP1L1, ACTB, CTNNA3, SPTBN4, PDLIM5, FHOD3 | 0.026 |
| Cellular compromise | + | − | HTR4, RHOC, ABCC1, DVL1, PLCB1, AQP1, PAK7, PHLPPL | 0.027 |
| Cellular development | + | + | CCNL2, TNFRSF4, ZFPM2, JAG2, HTR4, SMYD1, SPRY2, FABP1, MYH11, JAG1 | 0.024 |
| Cellular growth and proliferation | + | + | CCNL2, BMPR1B, TNFRSF4, ADCYAP1R1, RHOC, SPRY2, GHRHR, FABP1, UGT2B17, MYH11, JAG1, AFAP1, TSSC1, CSNK1A1, JAG2, MTUS1 | 0.023 |
| Cellular movement | + | + | EMMPRIN, SPRY2, TAS1R3, MYH11, MANEA, ACTB, MYPN, ABRA, ABLIM2, ABLIM3 | 0.024 |
| Connective tissue development and function | + | + | CRHR2, BMPR1B, EMMPRIN, ABCC1, MANEA, NID2, UGDH, MMP23B, CPO | 0.024 |
| Genetic disorder | + | + | SDF4, CRHR2, SH3TC2, HTR4, ABCC6, F5, PLCB1, CAPZA1, RHOC, TBC1D9, ACTB, ALLC, TAT, SNAP25, BMPR1B, PLCB4, ZFPM2, ADCYAP1R1, EMMPRIN, NDE1, PCM1, ANKRD5, SORCS2, PAK7, SLC19A2, MOV10, FAM19A2, JAG1, TASP1, FHOD3, DPP10 | 0.021 |
| Inflammatory response | + | + | TNFRSF4, EMMPRIN, ABCC1, FABP1, AQP1, PLCB1, MYH11, IL17B, ITPKC | 0.028 |
| Molecular transport | + | + | ABCC6, EMMPRIN, SLC16A1, ABCC1, DVL1, KLB, AQP1, FABP1, SLC19A2, SNAP25, SLC30A6, SORCS2 | 0.024 |
| Organ development | + | + | BMPR1B, JAG2, ZFPM2, ADCYAP1R1, ABRA, NDE1, SMYD1, SPRY2, DVL1, GHRHR, FABP1, JAG1, DPP10, MKKS | 0.023 |
| Skeletal and muscular system development and function | + | + | BMPR1B, JAG2, SMYD1, HCN2, MYH11, JAG1, IL17B, HAND1 | 0.023 |
| Small molecule biochemistrŷ | + | + | B3GALT6, DVL1, SNAP25, ADI1, THNSL2, ABCC6, UGDH, EMMPRIN, ABCC1, SLC16A1, KLB, UGT2B17, FABP1, PLCB1, SLC19A2, LIAS | 0.024 |
Category denotes GO molecular processes; + or − indicate whether or not the same processes are also enriched for genes in UTHSCH or FTAAD CNVs; Genes in CNVs denotes genes within BCM CNVs that map to GO processes; p value denotes estimated genome-wide significance derived by permutation.
CNV Discovery in Replication Data Set
We then evaluated these findings in an independent cohort of 387 individuals with STAAD recruited from UTHSCH. Their demographic characteristics are well matched with BCM individuals except for a higher incidence of hypertension and more frequent involvement of the distal aorta (Table 1). CNV detection and analysis were performed identically for the two data sets. In 366 UTHSCH STAAD patients, CNVPartition and PennCNV identified 5302 and 8576 copy gain or copy loss events, respectively. A total of 1038 CNVs were detected by both algorithms. We identified 56 rare CNVRs (46 gains, 11 losses) that were associated with STAAD in case-control analysis, including 40 CNVRs involving 161 genes that are unique to STAAD and not present in controls, as well as 17 additional CNVRs involving 49 genes that are significantly more prevalent in cases compared to controls (Table S2). Nine of the top ten predicted functional categories for genes within BCM and UTHSCH CNVs were shared (Table 4). Five CNVRs involving 31 genes (13%) in 4q22.3, 5q33.1, 16p13.1, 17p12, and 19q13.2 were present in both groups (annotated in Table S2). The CNVR in 16p13.1 involves the MYH11 gene (AAT4). The other recurrent CNVRs do not involve loci previously suspected to play a role in TAAD.
Genes within UTHSCH CNVs are also predicted to interact within Rho GTPase-mediated actin cytoskeleton-based pathways. “Smooth Muscle Contraction” (p = 1.0 × 10−7) and “Muscle Thick Filament Assembly” (p = 6.9 × 10−7) were the two most highly enriched GO processes. IPA showed that the 210 genes within UTHSCH CNVs are enriched for the functional categories “Cardiovascular System Development and Function” (20 genes, p = 1.8 × 10−4), “Molecular Transport” (13 genes, p = 5.8 × 10−5), and “Tissue Development” (27 genes, p = 3.2 × 10−4). In the development category, MetaCore specifically identified gene networks regulating angiogenesis (p = 2 × 10−5) or blood vessel morphogenesis (p = 0.01) among the most highly enriched functions. These results suggest that disruption of connective tissue structure or smooth muscle contractility perturbs vascular integrity and predisposes to TAAD.
CNV Discovery in Familial TAAD Data Set
We also performed CNV analysis of 88 probands of European ancestry from families with at least two family members with TAAD but without an identified causative gene mutation or syndrome (FTAAD cohort) recruited at UTHSCH. On average, these patients were significantly younger at diagnosis than STAAD patients (45 years). However, gender distribution, body mass index, and the prevalence of aortic dissection were not significantly different between cohorts. In case-control analysis, we identified 20 unique or rare CNVRs (15 gains, 5 losses) involving 84 genes in 87 FTAAD patients (Table S3). In comparison with STAAD, we found that the overall prevalence of rare CNVs in FTAAD patients (23%) was significantly increased (Fisher's exact test, p = 0.03). Fourteen of the 20 CNVRs were unique to FTAAD. Four of these regions were also present in individuals with sporadic disease, including a 3 Mb duplication of 16p13.1 involving MYH11, which is the only event that is present in all three data sets. None of the genes within the other recurrent CNVs were previously implicated in vascular disorders. In one individual, we also identified a 1.4 Mb duplication of the Williams-Beuren syndrome region in 7q11.23 that includes the elastin gene (ELN [MIM 130160]). Using qPCR, we tested relatives of affected probands in three families for three different CNVRs and found that they do not segregate with TAAD (Figure S1). However, one CNVR (the ELN duplication) appears to be mosaic in the peripheral blood of the proband. These findings are consistent with prior studies showing variable severity and reduced penetrance of FTAAD.
MetaCore analysis showed that networks regulating muscle contraction (p = 9.2 × 10−6), cell adhesion (p = 3.1 × 10−3), and the actin cytoskeleton (p = 5 × 10−3) are highly enriched within FTAAD CNVs. The largest IPA functional categories were derived from actin, myosin, and Rho GTPase-mediated networks for “Cellular Movement” (10 genes, p = 9.4 × 10−5) and “Molecular Transport” (10 genes, p = 2.3 × 10−4), which were also enriched in the BCM data set (Table 4). In summary, analysis of GO annotation enrichment of the CNV genes was consistent among these three independent cohorts and strongly supports our hypothesis that disruption of actin-based motility is a common and specific element of sporadic and familial TAAD.
Further analysis with IPA showed that 50 genes within BCM, UTHSCH, and FTAAD CNVs interact in a common network that includes components of the extracellular matrix, intercellular junctions, the actin cytoskeleton, and nuclear transcription factors (Figure 2). Signal transduction from extracellular matrix receptors to Rho kinase (ROCK1 [MIM 601702]) and the serum response factor-myocardin transcriptional complex regulates SMC contractility and the expression of contractile proteins. Several genes within this network, including NOTCH1 (MIM 190198), JAG1, MMP9 (MIM 120361), ALOX5 (MIM 152390), JNK1 (MIM 601158), fibulins, and PPIA (MIM 123840), have been implicated in the pathogenesis of aortic aneurysms. The canonical pathway “Cytoskeleton Remodeling: Regulation of Cytoskeleton by Rho GTPases” and the major functional categories “Muscle Filament Assembly,” “Elastic Fiber Assembly,” and “Muscle Development” are shared by the BCM, UTHSCH, and FTAAD data sets (Figure 3). We also found evidence for functional interaction between genes in the TAAD network and ACTA2, the smooth muscle α-actin gene that is mutated in FTAAD. Comparison of vascular SMC gene expression profiles from explanted murine Acta2-deficient and wild-type aortic SMCs showed that highly upregulated transcripts in ACTA2 null cells were significantly enriched for genes within TAAD-associated CNVs: MYH11 (8.4-fold), CDH13 (MIM 601364, 8.1-fold), ELN (8.1-fold), COL18A1 (MIM 120328, 7.0-fold), INMT (MIM 604854, 6.5-fold), AQP1 (MIM 107776, 6.5-fold), COLEC11 (MIM 612502, 3.9-fold), AKAP12 (2.8-fold), MMP23 (MIM 603320, 2.7-fold), and FOXD1 (MIM 601091, 2.5-fold) (Fisher's exact test, p = 0.012). In addition, BMPER (MIM 608699), EFEMP1 (MIM 601548), TNC (MIM 187380), and FOG2 (MIM 603693) were significantly downregulated in Acta2-deficient SMCs (D.M.M., unpublished data).25 These findings implicate a network of genes linking dynamic changes in the extracellular matrix, cell adhesion, and vascular smooth muscle contractility to the pathogenesis of TAAD.
Figure 3.
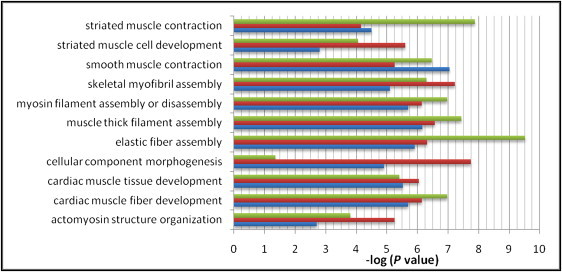
Gene Ontology Processes in BCM, UTHSC, and FTAAD Cohorts
Negative logarithms of p values are plotted on the x axis. Green: FTAAD; orange: BCM; blue: UTHSCH. Graph was generated with Microsoft Excel with data from GeneGo MetaCore pathway analysis software. P values are for enrichment of the specific GO terms. For calculation of p values, please refer to Subjects and Methods.
Discussion
TAAD is a multifactorial disease with a significant genetic component, as illustrated by the fact that patients have similarly affected family members in 20% of cases, and the genes identified for familial TAAD demonstrate reduced penetrance and variable severity.7,8 We found rare copy number variants in 13% of sporadic TAAD cases in both the discovery and replication cohorts, as well as significantly higher numbers of CNVs (23%) in FTAAD cases. Although the statistical association between any single rare variant and TAAD does not attain genome-wide significance, we observed recurrent rare events in cases that are not present in 5088 controls (Figure S1). TAAD-associated CNVs were enriched for genes that regulate cell adhesion or the actin cytoskeleton through direct interactions with contractile units containing the smooth muscle isoforms of actin and myosin. Mutations in genes encoding these proteins are known to cause FTAAD. Our findings support a common underlying mechanism for the pathogenesis of TAAD whereby any one of multiple, individually rare variants predispose to the disorder by disruption of SMC function, specifically cell adhesion and contraction.
The discovery of mutations of SMC-specific actin or myosin isoforms in FTAAD cases has implicated disruption of aortic SMC contractile functions in the development of aneurysms.16 SMC contractile units are distinctly different from sarcomeres in cardiac or skeletal muscle. Actin and myosin filaments are anchored to the SMC membrane at dense plaques found at focal adhesions that contain integrin receptors. These integrin receptors, in turn, bind to extracellular ligands, including fibrillin in microfibrils and collagen fibers, which form a supportive lattice around SMCs in the aortic media. Fibrillin-containing microfibrils provide the link between SMCs and elastic fibers in elastic arteries, including the aorta.26 In the cytoplasm, integrins are linked to the actin cytoskeleton by filamins, dystrophins, and other actin-binding proteins. Therefore, interactions between contractile units in SMCs, dense plaques, microfibrils, and elastic fibers are necessary for SMCs to generate contractile force and are required to coordinate contractile and elastic tensions in response to mechanical stresses imposed on the vessel wall.27 Disruption of any component is predicted to compromise SMC contractility and activate stress and stretch pathways in SMCs.16
Our findings indicate that TAAD-associated CNV genes are enriched in genes involved in an interacting network that regulates vascular SMC adhesion and contractility. The network includes the ligands jagged-1 and jagged-2 (JAG1 and JAG2 [MIM 602570]), sprouty-2 (SPRY2), reticulon-4 (RTN4 [MIM 604475]), bone morphogenetic protein-binding endothelial precursor-derived regulator (BMPER), the elastin-associated growth factor endostatin (a proteolytic fragment of the COL18A1 gene product), and VG5Q (AGGF1 [MIM 608464]), which modify the proliferation and differentiation of vascular cells during angiogenesis or recovery from vascular injury.28–30 The adhesive glycoproteins tenascin C (TNC), biglycan (BGN [MIM 301870]), nidogen-2 (NID2 [MIM 605399]), fibulin-3 (EFEMP1), elastin microfibril interface-located protein-3 (EMILIN3 [MIM 608929]), and thrombospondins THBS1 (MIM 188060), THBS4, and THSD7A (MIM 612249) facilitate interactions between collagen or elastin fibers in the tunica media and integrin receptors on SMCs.31–34 Their tissue distributions and functions are similar to fibrillin-1, and mutations of FBN1 encoding fibrillin-1 cause TAAD in patients with Marfan syndrome.35 Elevated expression of tenascin C is consistently found in ascending aortic aneurysms.36 We also found rare CNVs that affect the integrin-associated membrane proteins tetraspanin 1 (TSPAN1 [MIM 613170]), ephrin receptor A5 (EPHA5 [MIM 600004]), extracellular matrix metalloproteinase inducer (EMMPRIN [MIM 109480]), protein tyrosine phosphatase A (PTPRA [MIM 176884]), integrin-linked kinase associated phosphatase (ILKAP [MIM 601646]), junctional adhesion molecule C (JAM3 [MIM 606871]), and scavenger receptor type B1 (SCARB1 [601040]). These proteins interact with integrins and regulate cell migration or adhesion in response to ligand binding or deformation of the cell membrane.37–39 Integrins are linked to the actin cytoskeleton and SMC contractile units by spectrins, alpha-actinins, and other actin-binding proteins, including myopallidin (MYPN), formins (FHOD3), coactosin-like protein (COTL1 [MIM 606748]), actin-related proteins (ACTR2), actin-binding LIM domain proteins (ABLIM2 and ABLIM3), and actin filament-associated proteins (AFAP1 and AFAP1L1).40,41 These genes are highly expressed in SMCs and are also disrupted by rare CNVs in TAAD patients. Based on these results, we conclude that TAAD-associated CNVs systematically disrupt interactions between vascular SMCs and the extracellular matrix.
TGF-β ligands and receptors were not enriched in our data set, although the canonical TGF-β pathway was significantly overrepresented in STAAD patients. Loss of one copy of the TGF-β inhibitors LTBP4 (MIM 604710) or WWP1 (MIM 602307) because of CNVs in three TAAD patients would be expected to increase TGF-β activity.42,43 Alterations of the TGF-β pathway promote the progression of aneurysm disease in Marfan syndrome, with implications for a similar pathogenic role in TAAD.44
Abnormal organization of myofibrils was found in aortic SMCs from FTAAD patients with ACTA2 mutations.9,10 Depletion of elastin, collagens, fibulins, or fibrillin-1 in the ECM or uncoupling of SMCs from the ECM cause similar pathological changes and aortic disease. We hypothesize that the rare, sporadically occurring CNVs identified in TAAD patients also compromise SMC contractility by direct effects on contractile proteins or disruption of SMC adhesion to the ECM via focal adhesions. Although these predictions need to be verified with functional studies in cell and animal models, it is instructive that CNVs and familial mutations affect a common pathway that is critical for SMC function.
It is interesting to note that TAAD-associated CNVs contain several genes known to predispose to arterial aneurysms. The AGGF1 gene encoding the secreted angiogenic factor VG5Q is mutated in Klippel-Trenaunay-Weber syndrome (MIM 149000), which is associated with hemangiomas and large arterial aneurysms. Mutations of JAG1 cause Alagille syndrome (MIM 188450), a developmental disorder with arterial aneurysms in 10% of cases, as well as multiple extravascular manifestations. A large deletion involving JAG1 was also reported in a single individual with isolated sporadic tetralogy of Fallot, which is associated with the late development of aortic root dilation and ascending aneurysms in 12%–20% of patients after surgical repair.20,45 Mutations in the transcription factor FOG2 (also known as ZFPM2) have also been described in tetralogy of Fallot and are associated with cardiovascular developmental defects in mice.46,47 ELN mutations cause thoracic aortic aneurysms in association with autosomal-dominant cutis laxa (MIM 123700), and larger deletions that include ELN contribute to aortic and pulmonary artery defects in individuals with Williams-Beuren syndrome (MIM 194050). However, the association between microduplications of this region and TAAD is currently unknown.48 MYH11 is mutated in 1%–2% of FTAAD cases with a distinctive phenotype of TAAD associated with a patent ductus arteriosus (AAT4 [MIM 132900]), but similarly the association of duplications of this region and TAAD has not been identified.10,49 In addition, ACTB, SLC2A11 (MIM 610367), and EFEMP1 interact with or are paralogs of genes that are implicated in other aneurysm disorders. TAAD patients with these and other rare variants do not possess distinguishing clinical features or extravascular abnormalities.
In addition to the 16p13.1 duplications involving MYH11, unique CNVs in TAAD patients contain genes with known vascular functions. Duplications of galanin receptor 1 (GALR1 [MIM 600377]) are present in BCM and FTAAD patients. Galanin is a peptide hormone with potent vasodilatory effects that modulates smooth muscle contractility.50 EMILIN-3 is a secreted glycoprotein that interacts with elastin in the arterial wall.32 Mutation of a closely related paralog (EMILIN1 [MIM 130660]) causes aortic defects with connective tissue abnormalities in mice.51 We identified recurrent disrupting duplications of EMILIN3 in UTHSCH and FTAAD patients. Inhibition of bone morphogenetic protein and Notch signal transduction by BMPER and OVOL2 (MIM unassigned), respectively, is required for normal vascular development in mice and zebrafish,29,52 and deficiency of biglycan (BGN) causes spontaneous thoracic aortic dissections in mice.53 Alterations in the expression of these genes may increase the vulnerability of the aorta to mechanical or oxidative stress and predispose to aneurysm formation.
TAAD patients harbor a remarkable number of rare CNVs that are enriched for genes known to be involved in TAAD or other vascular disorders. We identified a total of 84 CNVRs, as well as larger chromosomal aberrations that are unique to TAAD patients and not present in 5088 controls. The CNV-associated gene set identified in individuals with TAAD contains excess duplications and deletions of protein-encoding genes compared with controls. Because 13% of STAAD patients harbor these rare variants, CNVs may contribute to a greater proportion of TAAD cases than single gene mutations. Limitations of our study include the confounding and unpredictable influence of multiple Illumina platforms on CNV discovery and the absence of familial samples that might be used to adjudicate the CNV findings. The pathway analysis is based on curated knowledgebases that may not include biologically relevant interactions, and this may lead to bias toward detecting well-defined pathways. Our results also do not include CNVs that were detected by a single algorithm or genes that are adjacent to CNV boundaries and are, therefore, likely to underestimate the overall impact of CNVs in TAAD. The burden of rare CNVs that we discovered in TAAD is consistent with what has been described in autism and in tetralogy of Fallot, a comparable cardiovascular developmental disorder that predisposes to aneurysm formation.20 Our findings provide strong support for the involvement of multiple rare genic CNVs, both genome-wide and at specific loci in TAAD, and have implications for other adult-onset cardiovascular disorders.
Acknowledgments
The authors are grateful to Chad Shaw for valuable discussions. Charles C. Miller provided additional clinical data for UTHSCH patients. The authors are extremely grateful to patients involved in this study. The following sources provided funding for these studies: P50HL083794-01 (D.M.M.), RO1 HL62594 (D.M.M.), the Vivian L. Smith Foundation (D.M.M.), the TexGen Foundation (D.M.M.), K08 HL080085 (S.A.L.), and the Thoracic Surgery Foundation for Research and Education (S.A.L.). D.M.M. is a Doris Duke Distinguished Clinical Scientist.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Galaxy, http://main.g2.bx.psu.edu
GeneHub-GEPIS: From Gene Integration to Expression Profiling, http://www.cgl.ucsf.edu/Research/genentech/genehub-gepis/
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
PLINK version 1.07, http://pngu.mgh.harvard.edu/purcell/plink/
UCSC Genome Browser, http://genome.ucsc.edu
References
- 1.Gillum R.F. Epidemiology of aortic aneurysm in the United States. J. Clin. Epidemiol. 1995;48:1289–1298. doi: 10.1016/0895-4356(95)00045-3. [DOI] [PubMed] [Google Scholar]
- 2.Tsai T.T., Fattori R., Trimarchi S., Isselbacher E., Myrmel T., Evangelista A., Hutchison S., Sechtem U., Cooper J.V., Smith D.E., International Registry of Acute Aortic Dissection Long-term survival in patients presenting with type B acute aortic dissection: Insights from the International Registry of Acute Aortic Dissection. Circulation. 2006;114:2226–2231. doi: 10.1161/CIRCULATIONAHA.106.622340. [DOI] [PubMed] [Google Scholar]
- 3.Ihling C., Szombathy T., Nampoothiri K., Haendeler J., Beyersdorf F., Uhl M., Zeiher A.M., Schaefer H.E. Cystic medial degeneration of the aorta is associated with p53 accumulation, Bax upregulation, apoptotic cell death, and cell proliferation. Heart. 1999;82:286–293. doi: 10.1136/hrt.82.3.286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nesi G., Anichini C., Tozzini S., Boddi V., Calamai G., Gori F. Pathology of the thoracic aorta: A morphologic review of 338 surgical specimens over a 7-year period. Cardiovasc. Pathol. 2009;18:134–139. doi: 10.1016/j.carpath.2008.04.001. [DOI] [PubMed] [Google Scholar]
- 5.Elefteriades J.A. Thoracic aortic aneurysm: Reading the enemy's playbook. Yale J. Biol. Med. 2008;81:175–186. [PMC free article] [PubMed] [Google Scholar]
- 6.Elefteriades J.A. Natural history of thoracic aortic aneurysms: Indications for surgery, and surgical versus nonsurgical risks. Ann. Thorac. Surg. 2002;74:S1877–S1880. doi: 10.1016/s0003-4975(02)04147-4. discussion S1892–S1898. [DOI] [PubMed] [Google Scholar]
- 7.Albornoz G., Coady M.A., Roberts M., Davies R.R., Tranquilli M., Rizzo J.A., Elefteriades J.A. Familial thoracic aortic aneurysms and dissections—incidence, modes of inheritance, and phenotypic patterns. Ann. Thorac. Surg. 2006;82:1400–1405. doi: 10.1016/j.athoracsur.2006.04.098. [DOI] [PubMed] [Google Scholar]
- 8.Milewicz D.M., Chen H., Park E.S., Petty E.M., Zaghi H., Shashidhar G., Willing M., Patel V. Reduced penetrance and variable expressivity of familial thoracic aortic aneurysms/dissections. Am. J. Cardiol. 1998;82:474–479. doi: 10.1016/s0002-9149(98)00364-6. [DOI] [PubMed] [Google Scholar]
- 9.Guo D.C., Pannu H., Tran-Fadulu V., Papke C.L., Yu R.K., Avidan N., Bourgeois S., Estrera A.L., Safi H.J., Sparks E. Mutations in smooth muscle alpha-actin (ACTA2) lead to thoracic aortic aneurysms and dissections. Nat. Genet. 2007;39:1488–1493. doi: 10.1038/ng.2007.6. [DOI] [PubMed] [Google Scholar]
- 10.Pannu H., Tran-Fadulu V., Papke C.L., Scherer S., Liu Y., Presley C., Guo D., Estrera A.L., Safi H.J., Brasier A.R. MYH11 mutations result in a distinct vascular pathology driven by insulin-like growth factor 1 and angiotensin II. Hum. Mol. Genet. 2007;16:2453–2462. doi: 10.1093/hmg/ddm201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhu L., Vranckx R., Khau Van Kien P., Lalande A., Boisset N., Mathieu F., Wegman M., Glancy L., Gasc J.M., Brunotte F. Mutations in myosin heavy chain 11 cause a syndrome associating thoracic aortic aneurysm/aortic dissection and patent ductus arteriosus. Nat. Genet. 2006;38:343–349. doi: 10.1038/ng1721. [DOI] [PubMed] [Google Scholar]
- 12.Tran-Fadulu V., Pannu H., Kim D.H., Vick G.W., 3rd, Lonsford C.M., Lafont A.L., Boccalandro C., Smart S., Peterson K.L., Hain J.Z. Analysis of multigenerational families with thoracic aortic aneurysms and dissections due to TGFBR1 or TGFBR2 mutations. J. Med. Genet. 2009;46:607–613. doi: 10.1136/jmg.2008.062844. [DOI] [PubMed] [Google Scholar]
- 13.Grainger D.J., Metcalfe J.C., Grace A.A., Mosedale D.E. Transforming growth factor-beta dynamically regulates vascular smooth muscle differentiation in vivo. J. Cell Sci. 1998;111:2977–2988. doi: 10.1242/jcs.111.19.2977. [DOI] [PubMed] [Google Scholar]
- 14.Sheen V.L., Jansen A., Chen M.H., Parrini E., Morgan T., Ravenscroft R., Ganesh V., Underwood T., Wiley J., Leventer R. Filamin A mutations cause periventricular heterotopia with Ehlers-Danlos syndrome. Neurology. 2005;64:254–262. doi: 10.1212/01.WNL.0000149512.79621.DF. [DOI] [PubMed] [Google Scholar]
- 15.Tu Y., Wu S., Shi X., Chen K., Wu C. Migfilin and Mig-2 link focal adhesions to filamin and the actin cytoskeleton and function in cell shape modulation. Cell. 2003;113:37–47. doi: 10.1016/s0092-8674(03)00163-6. [DOI] [PubMed] [Google Scholar]
- 16.Milewicz D.M., Guo D.C., Tran-Fadulu V., Lafont A.L., Papke C.L., Inamoto S., Kwartler C.S., Pannu H. Genetic basis of thoracic aortic aneurysms and dissections: Focus on smooth muscle cell contractile dysfunction. Annu. Rev. Genomics Hum. Genet. 2008;9:283–302. doi: 10.1146/annurev.genom.8.080706.092303. [DOI] [PubMed] [Google Scholar]
- 17.Patel H.J., Deeb G.M. Ascending and arch aorta: Pathology, natural history, and treatment. Circulation. 2008;118:188–195. doi: 10.1161/CIRCULATIONAHA.107.690933. [DOI] [PubMed] [Google Scholar]
- 18.Sebat J., Lakshmi B., Malhotra D., Troge J., Lese-Martin C., Walsh T., Yamrom B., Yoon S., Krasnitz A., Kendall J. Strong association of de novo copy number mutations with autism. Science. 2007;316:445–449. doi: 10.1126/science.1138659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kirov G., Grozeva D., Norton N., Ivanov D., Mantripragada K.K., Holmans P., Craddock N., Owen M.J., O'Donovan M.C., International Schizophrenia Consortium. Wellcome Trust Case Control Consortium Support for the involvement of large copy number variants in the pathogenesis of schizophrenia. Hum. Mol. Genet. 2009;18:1497–1503. doi: 10.1093/hmg/ddp043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Greenway S.C., Pereira A.C., Lin J.C., DePalma S.R., Israel S.J., Mesquita S.M., Ergul E., Conta J.H., Korn J.M., McCarroll S.A. De novo copy number variants identify new genes and loci in isolated sporadic tetralogy of Fallot. Nat. Genet. 2009;41:931–935. doi: 10.1038/ng.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schork N.J., Murray S.S., Frazer K.A., Topol E.J. Common vs. rare allele hypotheses for complex diseases. Curr. Opin. Genet. Dev. 2009;19:212–219. doi: 10.1016/j.gde.2009.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A., Bender D., Maller J., Sklar P., de Bakker P.I., Daly M.J., Sham P.C. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang Y., Luoh S.M., Hon L.S., Baertsch R., Wood W.I., Zhang Z. GeneHub-GEPIS: Digital expression profiling for normal and cancer tissues based on an integrated gene database. Nucleic Acids Res. 2007;35(Web Server issue) doi: 10.1093/nar/gkm381. W152–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vaughan C.J., Casey M., He J., Veugelers M., Henderson K., Guo D., Campagna R., Roman M.J., Milewicz D.M., Devereux R.B., Basson C.T. Identification of a chromosome 11q23.2-q24 locus for familial aortic aneurysm disease, a genetically heterogeneous disorder. Circulation. 2001;103:2469–2475. doi: 10.1161/01.cir.103.20.2469. [DOI] [PubMed] [Google Scholar]
- 25.Schildmeyer L.A., Braun R., Taffet G., Debiasi M., Burns A.E., Bradley A., Schwartz R.J. Impaired vascular contractility and blood pressure homeostasis in the smooth muscle alpha-actin null mouse. FASEB J. 2000;14:2213–2220. doi: 10.1096/fj.99-0927com. [DOI] [PubMed] [Google Scholar]
- 26.Huang J., Davis E.C., Chapman S.L., Budatha M., Marmorstein L.Y., Word R.A., Yanagisawa H. Fibulin-4 deficiency results in ascending aortic aneurysms: A potential link between abnormal smooth muscle cell phenotype and aneurysm progression. Circ. Res. 2010;106:583–592. doi: 10.1161/CIRCRESAHA.109.207852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Davis E.C. Smooth muscle cell to elastic lamina connections in developing mouse aorta. Role in aortic medial organization. Lab. Invest. 1993;68:89–99. [PubMed] [Google Scholar]
- 28.Zhang C., Chaturvedi D., Jaggar L., Magnuson D., Lee J.M., Patel T.B. Regulation of vascular smooth muscle cell proliferation and migration by human sprouty 2. Arterioscler. Thromb. Vasc. Biol. 2005;25:533–538. doi: 10.1161/01.ATV.0000155461.50450.5a. [DOI] [PubMed] [Google Scholar]
- 29.Moser M., Binder O., Wu Y., Aitsebaomo J., Ren R., Bode C., Bautch V.L., Conlon F.L., Patterson C. BMPER, a novel endothelial cell precursor-derived protein, antagonizes bone morphogenetic protein signaling and endothelial cell differentiation. Mol. Cell. Biol. 2003;23:5664–5679. doi: 10.1128/MCB.23.16.5664-5679.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ergün S., Kilic N., Wurmbach J.H., Ebrahimnejad A., Fernando M., Sevinc S., Kilic E., Chalajour F., Fiedler W., Lauke H. Endostatin inhibits angiogenesis by stabilization of newly formed endothelial tubes. Angiogenesis. 2001;4:193–206. doi: 10.1023/a:1014027218980. [DOI] [PubMed] [Google Scholar]
- 31.Reinboth B., Hanssen E., Cleary E.G., Gibson M.A. Molecular interactions of biglycan and decorin with elastic fiber components: Biglycan forms a ternary complex with tropoelastin and microfibril-associated glycoprotein 1. J. Biol. Chem. 2002;277:3950–3957. doi: 10.1074/jbc.M109540200. [DOI] [PubMed] [Google Scholar]
- 32.Christian S., Ahorn H., Novatchkova M., Garin-Chesa P., Park J.E., Weber G., Eisenhaber F., Rettig W.J., Lenter M.C. Molecular cloning and characterization of EndoGlyx-1, an EMILIN-like multisubunit glycoprotein of vascular endothelium. J. Biol. Chem. 2001;276:48588–48595. doi: 10.1074/jbc.M106152200. [DOI] [PubMed] [Google Scholar]
- 33.Wang X.Q., Frazier W.A. The thrombospondin receptor CD47 (IAP) modulates and associates with alpha2 beta1 integrin in vascular smooth muscle cells. Mol. Biol. Cell. 1998;9:865–874. doi: 10.1091/mbc.9.4.865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang C.H., Su P.T., Du X.Y., Kuo M.W., Lin C.Y., Yang C.C., Chan H.S., Chang S.J., Kuo C., Seo K. Thrombospondin type I domain containing 7A (THSD7A) mediates endothelial cell migration and tube formation. J. Cell. Physiol. 2010;222:685–694. doi: 10.1002/jcp.21990. [DOI] [PubMed] [Google Scholar]
- 35.Booms P., Pregla R., Ney A., Barthel F., Reinhardt D.P., Pletschacher A., Mundlos S., Robinson P.N. RGD-containing fibrillin-1 fragments upregulate matrix metalloproteinase expression in cell culture: A potential factor in the pathogenesis of the Marfan syndrome. Hum. Genet. 2005;116:51–61. doi: 10.1007/s00439-004-1194-7. [DOI] [PubMed] [Google Scholar]
- 36.Majumdar R., Miller D.V., Ballman K.V., Unnikrishnan G., McKellar S.H., Sarkar G., Sreekumar R., Bolander M.E., Sundt T.M., 3rd Elevated expressions of osteopontin and tenascin C in ascending aortic aneurysms are associated with trileaflet aortic valves as compared with bicuspid aortic valves. Cardiovasc. Pathol. 2007;16:144–150. doi: 10.1016/j.carpath.2006.12.001. [DOI] [PubMed] [Google Scholar]
- 37.Zhu W., Saddar S., Seetharam D., Chambliss K.L., Longoria C., Silver D.L., Yuhanna I.S., Shaul P.W., Mineo C. The scavenger receptor class B type I adaptor protein PDZK1 maintains endothelial monolayer integrity. Circ. Res. 2008;102:480–487. doi: 10.1161/CIRCRESAHA.107.159079. [DOI] [PubMed] [Google Scholar]
- 38.Leung-Hagesteijn C., Mahendra A., Naruszewicz I., Hannigan G.E. Modulation of integrin signal transduction by ILKAP, a protein phosphatase 2C associating with the integrin-linked kinase, ILK1. EMBO J. 2001;20:2160–2170. doi: 10.1093/emboj/20.9.2160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li X., Stankovic M., Lee B.P., Aurrand-Lions M., Hahn C.N., Lu Y., Imhof B.A., Vadas M.A., Gamble J.R. JAM-C induces endothelial cell permeability through its association and regulation of beta3 integrins. Arterioscler. Thromb. Vasc. Biol. 2009;29:1200–1206. doi: 10.1161/ATVBAHA.109.189217. [DOI] [PubMed] [Google Scholar]
- 40.Bialkowska K., Saido T.C., Fox J.E. SH3 domain of spectrin participates in the activation of Rac in specialized calpain-induced integrin signaling complexes. J. Cell Sci. 2005;118:381–395. doi: 10.1242/jcs.01625. [DOI] [PubMed] [Google Scholar]
- 41.Kappert K., Furundzija V., Fritzsche J., Margeta C., Krüger J., Meyborg H., Fleck E., Stawowy P. Integrin cleavage regulates bidirectional signalling in vascular smooth muscle cells. Thromb. Haemost. 2010;103:556–563. doi: 10.1160/TH09-07-0478. [DOI] [PubMed] [Google Scholar]
- 42.Komuro A., Imamura T., Saitoh M., Yoshida Y., Yamori T., Miyazono K., Miyazawa K. Negative regulation of transforming growth factor-beta (TGF-beta) signaling by WW domain-containing protein 1 (WWP1) Oncogene. 2004;23:6914–6923. doi: 10.1038/sj.onc.1207885. [DOI] [PubMed] [Google Scholar]
- 43.Urban Z., Hucthagowder V., Schürmann N., Todorovic V., Zilberberg L., Choi J., Sens C., Brown C.W., Clark R.D., Holland K.E. Mutations in LTBP4 cause a syndrome of impaired pulmonary, gastrointestinal, genitourinary, musculoskeletal, and dermal development. Am. J. Hum. Genet. 2009;85:593–605. doi: 10.1016/j.ajhg.2009.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Choudhary B., Zhou J., Li P., Thomas S., Kaartinen V., Sucov H.M. Absence of TGFbeta signaling in embryonic vascular smooth muscle leads to reduced lysyl oxidase expression, impaired elastogenesis, and aneurysm. Genesis. 2009;47:115–121. doi: 10.1002/dvg.20466. [DOI] [PubMed] [Google Scholar]
- 45.Dimitrakakis G., Von Oppell U., Bosanquet D., Wilson D., Luckraz H. Aortic aneurysm formation five decades after tetralogy of Fallot repair. Ann. Thorac. Surg. 2009;88:1000–1001. doi: 10.1016/j.athoracsur.2009.02.029. [DOI] [PubMed] [Google Scholar]
- 46.Pizzuti A., Sarkozy A., Newton A.L., Conti E., Flex E., Digilio M.C., Amati F., Gianni D., Tandoi C., Marino B. Mutations of ZFPM2/FOG2 gene in sporadic cases of tetralogy of Fallot. Hum. Mutat. 2003;22:372–377. doi: 10.1002/humu.10261. [DOI] [PubMed] [Google Scholar]
- 47.Crispino J.D., Lodish M.B., Thurberg B.L., Litovsky S.H., Collins T., Molkentin J.D., Orkin S.H. Proper coronary vascular development and heart morphogenesis depend on interaction of GATA-4 with FOG cofactors. Genes Dev. 2001;15:839–844. doi: 10.1101/gad.875201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Beunders G., van de Kamp J.M., Veenhoven R.H., van Hagen J.M., Nieuwint A.W., Sistermans E.A. A triplication of the Williams-Beuren syndrome region in a patient with mental retardation, a severe expressive language delay, behavioural problems and dysmorphisms. J. Med. Genet. 2010;47:271–275. doi: 10.1136/jmg.2009.070490. [DOI] [PubMed] [Google Scholar]
- 49.Zhu L., Bonnet D., Boussion M., Vedie B., Sidi D., Jeunemaitre X. Investigation of the MYH11 gene in sporadic patients with an isolated persistently patent arterial duct. Cardiol. Young. 2007;17:666–672. doi: 10.1017/S1047951107001473. [DOI] [PubMed] [Google Scholar]
- 50.Schmidhuber S.M., Santic R., Tam C.W., Bauer J.W., Kofler B., Brain S.D. Galanin-like peptides exert potent vasoactive functions in vivo. J. Invest. Dermatol. 2007;127:716–721. doi: 10.1038/sj.jid.5700569. [DOI] [PubMed] [Google Scholar]
- 51.Zanetti M., Braghetta P., Sabatelli P., Mura I., Doliana R., Colombatti A., Volpin D., Bonaldo P., Bressan G.M. EMILIN-1 deficiency induces elastogenesis and vascular cell defects. Mol. Cell. Biol. 2004;24:638–650. doi: 10.1128/MCB.24.2.638-650.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Unezaki S., Horai R., Sudo K., Iwakura Y., Ito S. Ovol2/Movo, a homologue of Drosophila ovo, is required for angiogenesis, heart formation and placental development in mice. Genes Cells. 2007;12:773–785. doi: 10.1111/j.1365-2443.2007.01084.x. [DOI] [PubMed] [Google Scholar]
- 53.Heegaard A.M., Corsi A., Danielsen C.C., Nielsen K.L., Jorgensen H.L., Riminucci M., Young M.F., Bianco P. Biglycan deficiency causes spontaneous aortic dissection and rupture in mice. Circulation. 2007;115:2731–2738. doi: 10.1161/CIRCULATIONAHA.106.653980. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.