

Abstract
This mini-symposium aims to provide an integrated perspective on recent developments in optogenetics. Research in this emerging field combines optical methods with targeted expression of genetically encoded, protein-based probes to achieve experimental manipulation and measurement of neural systems with superior temporal and spatial resolution. The essential components of the optogenetic toolbox consist of two kinds of molecular devices: actuators and reporters, which respectively enable light-mediated control or monitoring of molecular processes. The first generation of genetically encoded calcium reporters, fluorescent proteins, and neural activators has already had a great impact on neuroscience. Now, a second generation of voltage reporters, neural silencers, and functionally extended fluorescent proteins hold great promise for continuing this revolution. In this review, we will evaluate and highlight the limitations of presently available optogenic tools and discuss where these technologies and their applications are headed in the future.
Introduction
It is our everyday experience that light can both provide informative signals (e.g., vision) and control behavior (e.g., traffic lights). Neuroscientists have applied optical methods since the days of Santiago Ramón y Cajal, with each new generation of researchers contributing more powerful tools ranging from simple stains to highly specific molecular probes to strategies for cellular manipulation with high temporal and spatial resolution. During the last decade, the combination of optical and genetic—also known as “optogenetic”—methods has greatly increased the effective spatial resolution and specificity of probing and manipulation. The core tools of optogenetics are targetable, genetically encoded actuator and reporter proteins that allow the use of light to either control or report on the activity of molecular processes in specific cell populations within networks of heterologous cell types. Controlling and reporting are the two branches of a comprehensive optogenetic approach (Fig. 1). The reporter branch involves engineered biosensors derived from the fusion of fluorescent protein (FP) reporters to “detector” proteins, which convert physiological signals into changes in fluorescence output (for a recent comprehensive review, see Chudakov et al., 2010). The actuator branch emerged from pioneering work combining genetics and stimulation via optical uncaging techniques (Lima and Miesenböck, 2005) and now appears to be converging toward a steadily improving family of light-activated channels and pumps (for a recent review, see Gradinaru et al., 2010).
Figure 1.
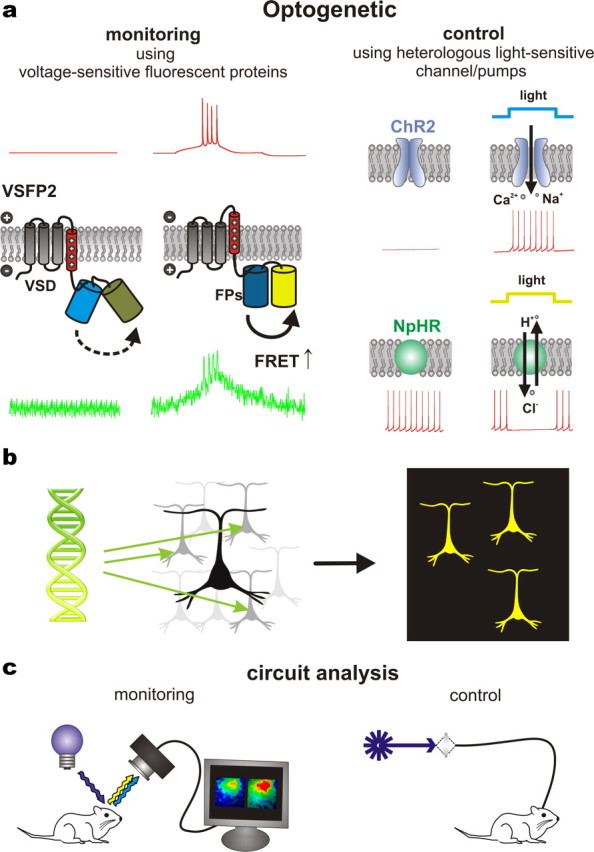
Genetically encoded, targetable actuator and reporter proteins that allow the use of light to either control or report molecular processes in specific cell populations within networks of heterologous cell types. a, Optical reporting and control are exemplified respectively by a voltage-sensitive fluorescent protein (VSFP2, left) and a light-sensitive ion channel (ChR2, top right) or ion pump [Np-halorhodopsin (NpHR), bottom right] that are directly gated by photoabsorption. Membrane potential traces are schematically indicated in red. VSFP2 reports action potential firing via an optical signal (green trace), while light is used to induce (ChR2) or inhibit (NpHR) action potential generation via the activity of actuator proteins. b, Optogenetic reporters and actuators can be selectively expressed in specific sets of neurons using genetic techniques. c, Light may be delivered (and collected) either by simple epi-illumination optics or more precisely targeted configurations, such as via fiber optics. Combinations of both methods can be used to investigate causality between the activity of specific neuronal populations and behavior.
The first six chapters of this summary present the topics covered by the six speakers at the mini-symposium. We begin with recent progress in ongoing efforts to generate improved fluorescent proteins that span the visual spectrum and have now begun to enter the near-infrared range. We then proceed to the reporter branch, and evaluate the most promising voltage-sensitive and calcium-sensitive FP probes. Similar analysis is provided for the various actuators that trigger and inhibit neuronal activity, and for novel approaches to the optical control of intracellular signal transduction. We conclude the report with a discussion of how these technologies and their applications are likely to evolve in the future.
Expanding the range of fluorescent proteins
Monomeric FPs serve as the basic components for the engineering of optical reporters of voltage, ions, and biochemical activity. The ever-increasing need for simultaneous imaging of multiple cellular parameters has driven the expansion of the FP color palette across the entire visible spectrum—and even beyond it. Following on the engineering of bright monomeric FPs based on Aequorea victoria-enhanced green fluorescent protein (EGFP) and Discosoma DsRed to obtain derivatives that cover the optical region from blue to red (Shaner et al., 2005), further expansion and improvements have been achieved at both ends of the spectrum.
Until recently, monomeric red FPs have yielded inferior brightness and photostability relative to the widely used EGFP, with a tendency to aggregate following prolonged expression. However, proteins derived recently from the bulb tip anemone Entacmaea show significant improvement over existing red FPs in various aspects. The proteins TagRFP and mRuby offer the brightest fluorescence in the orange wavelengths near 600 nm, with overall brightness values (the product of extinction coefficient and quantum yield) that are respectively 150% (Merzlyak et al., 2007) and 120% (Kredel et al., 2009) of that of EGFP. TagRFP-T, a variant of TagRFP, is as bright as EGFP but is also more photostable under arc lamp or single-photon laser stimulation (Shaner et al., 2008). A red-shifted TagRFP variant with improved monomeric character, mKate2, has shown excellent performance in a variety of protein fusions while exhibiting 74% of the brightness of EGFP (Shcherbo et al., 2009).
Recent efforts have succeeded in further extending the wavelength range of FPs. The TagRFP derivative mNeptune shows peak excitation at 600 nm and peak emission in the far red, at 650 nm (Lin et al., 2009). mNeptune can be excited with a 633 nm laser line, enabling the creation of reporters that can be imaged in a channel distinct from that used for green and orange FPs. It may also be a suitable Förster resonance energy transfer (FRET) acceptor for orange FPs, allowing dual FRET imaging in conjunction with a cyan-and-yellow FRET pair. Ongoing efforts to improve mNeptune have resulted in the development of brighter, and more monomeric and robustly folding variants.
Beyond the realm of autocatalytic FPs, directed evolution of bacteriophytochrome domains has recently given rise to the infrared fluorescent protein IFP1.4 (Shu et al., 2009), which is excited at 680 nm and emits at 700 nm, allowing for the use of yet another optical channel. Importantly, IFP1.4 incorporates biliverdin as its chromophore, which typically must be supplied exogenously for maximum brightness.
There have been significant improvements in FPs at the blue end of the spectrum as well. While excitation by violet light should be performed sparingly to avoid phototoxicity, blue and cyan FPs can provide an additional channel for optical reporters that may prove useful for multiplexing. Two bright blue FPs, EBFP2 and mTagBFP, have been engineered from different precursors and use different chromophore structures (Ai et al., 2007; Subach et al., 2008). mTagBFP is brighter than EBFP2 (98 vs 53% of EGFP), but EBFP2 is more photostable and, being derived from EGFP, should be a straightforward replacement for Aequorea-derived FPs in existing reporters. In addition, an improved form of ECFP, mTurquoise (Goedhart et al., 2010), exhibits 75% of the brightness of EGFP. Collectively, these developments allow the simultaneous imaging of four reporters using blue/cyan, green/yellow, orange, and far-red FPs, with the potential for a fifth channel if IFP1.4 is included.
Finally, a pair of bright orange photoactivatable (PA) FPs has been recently developed. PA-mCherry and PA-TagRFP are nonfluorescent in their preactivated state, but upon photoactivation convert to an orange fluorescent form with peak emission at 595 nm and brightness reaching 25 and 75% of EGFP, respectively (Subach et al., 2010). Potential applications in living neurons could include visualization of protein movement and single-molecule super-resolution microscopy in two channels in combination with PA-EGFP.
Genetically targeted fluorescent protein voltage probes
Practically all fluorescent protein-based probes follow a common design principle, involving the molecular fusion of a protein that senses a parameter of interest (e.g., calcium, membrane voltage, cyclic nucleotides) with an FP-based reporter. Voltage-gated ion channels have been extensively studied and were therefore the obvious choice for the first generation of FP-based voltage probes (for recent reviews, see Baker et al., 2008; Perron et al., 2009a). Two of the early voltage sensor designs (Flash and SPARC) were based on the molecular insertion of an FP into a potassium channel subunit or a sodium channel (Siegel and Isacoff, 1997; Ataka and Pieribone, 2002). Concurrently, the Knöpfel laboratory conceptualized a design for voltage-sensitive FPs (VSFPs) based on the fusion of an isolated voltage sensor domain to an FP reporter construct (Sakai et al., 2001). This work led to the development of first-generation VSFP1s, which can function in non-neuronal mammalian cells. Unfortunately, only a small fraction of these first-generation reporters (Flash, SPARC, and VSFP1) localized to the plasma membranes of neurons, hindering their application for the monitoring of activity in neuronal networks.
More recently, the derivation of probes with highly efficient membrane targeting based on the voltage-sensing domain (VSD) of Ciona intestinalis voltage sensor-containing phosphatase has helped to catapult this field of research forward (Dimitrov et al., 2007). The initial prototypes of these second-generation VSFP2 probes proved capable of reporting membrane potential transients following the kinetics of synaptic potentials and action potentials recorded from mammalian neurons (Dimitrov et al., 2007). However, additional tuning for response speed and dynamic range, as well as implementation of appropriate gene transfer strategies, were required to convincingly demonstrate voltage reporting in neurons embedded within intact tissue (for details, see Akemann et al., 2010).
While the second-generation VSFP2s incorporate a FRET pair, the early identification of a FRET-independent component gave rise to a series of VSFP3 probes (Lundby et al., 2008; Perron et al., 2009b). Because VSFP3s contain a single FP, it became a straightforward process to generate variants for essentially any color represented by an FP. These probes offer the advantages of broad coverage of the color spectrum and fast kinetics, but their dynamic range is much smaller than that of VSFP2s. VSFP3s are the first genetically encoded probes for membrane voltage with which optical recordings from patch-clamped neurons have been reported (Perron et al., 2009b).
For imaging under in vivo recording conditions, VSFP2-type probes are advantageous because ratiometric measurements allow separation of hemodynamic and movement-related signals from voltage signals. Recently, VSFP2s were expressed in pyramidal cells of the mouse somatosensory cortex to demonstrate for the first time that these probes are capable of reporting cortical electrical responses to single sensory stimuli in vivo (Akemann et al., 2010). After passing this benchmark, we expect that the ability of VSFPs to report faster signals, along with genetic targeting, will allow new approaches to the study of the dynamic interaction of assemblies of neurons. This will facilitate the investigation of fundamental questions of information processing in the brain, such as the circuit operations involved in sensing our environment and generation of body movements, but will also be applicable to directly visualize cognitive functions.
Genetically encoded calcium indicators for optical recording of brain activity
Genetically encoded calcium indicators (GECIs) can be used to monitor calcium transients in living cells and organisms. In general, GECIs consist of a calcium-binding domain (e.g., calmodulin or troponin C) fused to one or two FPs (Fig. 2a–c). In single-FP GECIs, the fluorescence intensity of a circularly permuted FP is modulated by calcium binding-dependent changes in the chromophore environment (Baird et al., 1999; Nagai et al., 2001; Nakai et al., 2001). In two-FP GECIs, such as cameleon, calcium binding allosterically modulates the relative donor–acceptor emission spectra through a distance- and orientation-dependent change in FRET (Miyawaki et al., 1997; Heim and Griesbeck, 2004). In many cases, a conformational actuator, such as the M13 peptide, is included in the fusion protein to enhance the conformational change and fluorescence modulation. GECIs have been iteratively improved and are becoming useful for imaging neural activity in vivo. The calmodulin-based FRET sensor D3cpV (Palmer et al., 2006) has been used to detect single action potentials (APs) in organotypic mouse brain slices and in vivo in layer 2/3 somatosensory cortical neurons (Wallace et al., 2008). Recently, another sensor derived from cameleon, YC3.60 (Nagai et al., 2004), was reported to allow in vivo detection of single APs with a sensitivity comparable to D3cpV, but with faster kinetics and minimal saturation while reporting bursts of up to 10 APs (Lütcke et al., 2010). Likewise, the troponin C-based FRET sensor TN-XXL has been used for chronic in vivo activity imaging in mice and flies (Mank et al., 2008). The single-FP sensor GCaMP3 achieves high expression and sensitivity with no apparent cytotoxicity or behavioral phenotype in Caenorhabditis elegans and Drosophila (Tian et al., 2009). Long-term overexpression of GCaMP3 in the rodent cortex sometimes led to a cytomorbid phenotype characterized by filled cells and nuclei, decreased sensor response, and lower excitability; similar outcomes were observed with other GECIs tested (Tian et al., 2009). The precise cause of this cytomorbidity is not yet known. Lowering GECI expression level may resolve this problem as no adverse effects were apparent in transgenic mice expressing GCamP2 (Díez-García et al., 2007; Fletcher et al., 2009).
Figure 2.
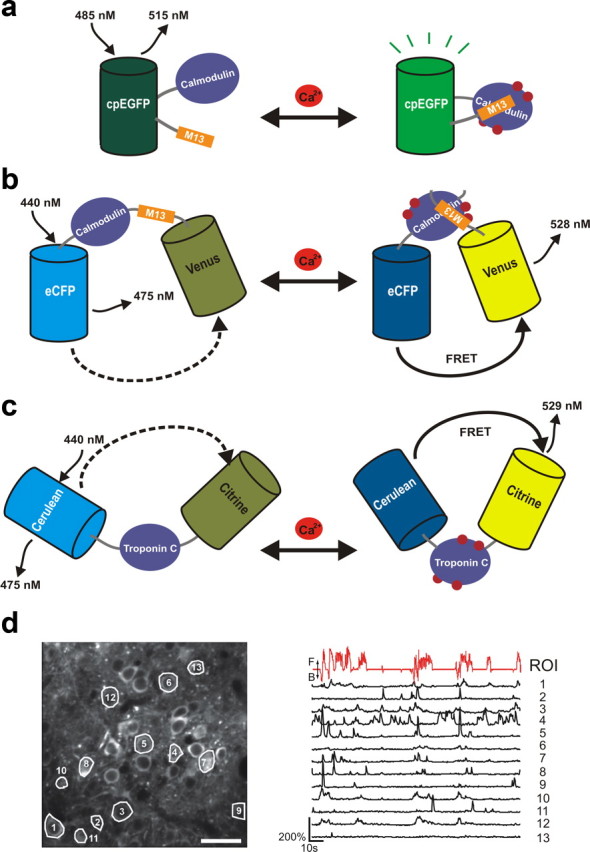
Schematic representation of the three major GECI classes. a, Schematic of the single-FP GECI sensing mechanism. Upon calcium binding, conformational changes in the CaM/M13 complex induce fluorescence changes in the circularly permuted GFP (cpEGFP). b, The cameleon family of FRET-based GECIs. A calcium-dependent increase in FRET between a cyan fluorescent protein (CFP) and yellow fluorescent protein (YFP) FRET pair is coupled to the binding of calmodulin to the M13 peptide. c, Troponin C-based FRET GECIs. Binding of calcium to troponin C induces conformational changes and an increase in FRET between CFP and YFP. d, Imaging neural activity with GCaMP3 using a two-photon microscope in awake, behaving mice. GCaMP3 expression in layer 2/3 neurons of the primary motor cortex at 72 d postinjection (scale bar, 30 mm) and ΔF/F traces of individual cells (black lines). Relative treadmill movement indicated by red line. F, forward; B, backward; eCFP, enhanced CFP; ROI, region of interest. Adapted from Tian et al., 2009.
Experiments with GCaMP3 have demonstrated the detection of single action potentials in pyramidal neurons in acute mouse brain slices, and long-term imaging studies with GCaMP3 revealed large fluorescence changes in the motor cortices of behaving mice over the course of months, with the capacity to detect trains of 3–20 APs with linearity (Fig. 2d) (Tian et al., 2009).
As each biological system or cell type employs different calcium-signaling mechanisms, a GECI with high sensitivity in one setting may not be the best fit under different conditions—a fact that needs to be considered when developing and choosing GECIs for neurophysiological applications. GECI developers need to establish a multistep screening paradigm, including a primary screen that enables characterization of brightness, calcium sensitivity, dynamic range, and kinetics with high throughput while capturing as many aspects of intact preparations (e.g., calcium buffering and cytopathicity) as possible. Advanced validation can be achieved by performing two-photon imaging of GECI responses to back-propagating action potentials in acute brain slices, which recapitulates many aspects of in vivo functional imaging (e.g., allowing for long-term expression and observation of developmental effects), while allowing moderate throughput (in conjunction with in utero electroporation). Because of greater imaging depth, motion artifacts, and hemodynamics, in vivo imaging has more challenging signal-to-noise requirements, and before deployment it is necessary to identify the best candidates based on testing in an in vivo preparation. Neuroscientists should ideally select the most appropriate probe for their specific application by comparing several GECIs under identical conditions (e.g., expression cassette, expression time and level, cell type, and stimuli) in their system. Careful experiments will also be required to determine the effects of GECI expression on cells and circuits.
Ongoing and future neurophysiological applications of GECIs will take advantage of their ability to target specific cell populations while allowing for optical detection of action potential firing at the single cell level. These features are particularly well suited for the identification of neuronal assemblies that are recruited during specific circuit operations.
Quieting neural activity with light
The last few years have seen rapid innovation in the arena of light-driven neural silencers. The first generation of such optogenetic tools included rat rhodopsin 4 (RO4), which, when expressed in neurons, can couple with endogenous GIRK potassium channels via endogenous Gi/o G-proteins to enable light-induced neural hyperpolarization (Li et al., 2005). It should be noted, however, that RO4 exhibits relatively slow current onset and offset kinetics due to the slow nature of the signaling cascades involved. On the other hand, the light-driven inward chloride pump halorhodopsin from Natronomonas pharaonis (Halo/NpHR) mediates much more rapid hyperpolarization in neurons upon illumination with yellow light, with onset and offset in tens of milliseconds (Han and Boyden, 2007; Zhang et al., 2007). Although useful as a proof of concept or as a tool for confronting specific neuroscience questions, the light-driven currents mediated by natural halorhodopsins are still small compared with the currents needed to effectively silence neurons in the awake mammalian brain. Furthermore, the N. pharaonis halorhodopsin has a tendency to form aggregates when expressed at high levels in neurons (Gradinaru et al., 2008; Zhao et al., 2008) and enters into an electrogenically inactive state that persists for tens of minutes when illuminated for extended periods of time (Han and Boyden, 2007; Chow et al., 2010), although this phenomenon can be reversed using pulses of blue light. Improved neural silencers have subsequently been generated via diverse methodologies. One approach is to boost halorhodopsin expression levels by appending sequences from other mammalian proteins at the N and C termini that boost trafficking of halorhodopsin to the cell membrane (Gradinaru et al., 2008; Zhao et al., 2008). This results in higher peak currents but presumably would not improve recovery from the inactive state or other potential side effects of expressing halorhodopsins in cells (Tønnesen et al., 2009).
A genomic screen searching for novel, naturally occurring, light-driven neural silencers revealed that the green light-driven outward proton pump archaerhodopsin-3 from Halorubrum sodomense (Arch) could safely and effectively mediate light-driven hyperpolarizing currents exceeding 900 pA—sufficient to achieve near-total neural silencing in an awake, behaving mouse (Fig. 3a). Arch-mediated proton pumping is well tolerated in neurons because the pH fluctuations triggered in response to illumination are self-limiting to levels comparable to those mediated by channelrhodopsins (ChRs) (Nagel et al., 2003; Tønnesen et al., 2009) or natural neural activity. The same genomic screen also yielded other novel neural silencing reagents with varying spectral activation properties, such that neurons expressing different reagents can be inhibited by different colors of light (Fig. 3b) (Chow et al., 2010). Ongoing innovations in this area include the identification of more light-sensitive reagents with faster kinetics by mining new genomes, as shown in a recent study demonstrating transient optical silencing of the activity of neurons in the rhesus monkey brain to near-zero activity levels (Han et al., 2010), and the characterization of new, multi-color-responsive sets of neural silencers (Klapoetke et al., 2010).
Figure 3.

Quieting neural activity with light. a, Neural activity in a representative neuron expressing Arch (i) before, during, and after 5 s of illumination with yellow light, shown as a spike raster plot in which each spike is represented by a dot. This is displayed above a histogram that shows spike firing rate for each 20-ms-duration period throughout the light delivery process, averaged across trials. The bottom (ii) depicts the averaged instantaneous firing rate in a population of Arch-expressing neurons before, during and after yellow light illumination (black line, mean; gray lines, mean ± SE; data from 13 neurons). b, Multicolor silencing of two neural populations, enabled by ion pumps of different classes that are selectively induced by blue (Mac) or red (Halo) light. i, Action spectra of Mac versus Halo; rectangles indicate filter bandwidths used for multicolor silencing in vitro. Blue light illumination was achieved via a 470 + 20 nm filter at 5.3 mW/mm2, and red light illumination was achieved via a 630 + 15 nm filter at 2.1 mW/mm2. Action potentials evoked by current injection into patch-clamped cultured neurons transfected with Halo (ii) were selectively silenced by the red light but not the blue light, while the opposite was true for neurons expressing Mac (iii). Adapted from Chow et al., 2010.
Exciting neuronal activity by light
ChRs are algal light-gated channel proteins that nonselectively conduct cations when stimulated by visible light. These unique channels allow neuroscientists to optically depolarize transfected neurons, providing a valuable tool for studying neural circuitry. Both native (ChR2, VChR1) and engineered (ChIEF, ChETA, ChR2/C128X/D156A, ChR2/H134R) ChR variants can effectively depolarize transfected neurons in response to light (Boyden et al., 2005; Nagel et al., 2005; Zhang et al., 2008; Lin et al., 2009; Bamann et al., 2010; Gunaydin et al., 2010), although many of these ChRs remain incompletely characterized. The biophysical properties of a given ChR variant and the membrane properties of the transfected neurons (capacitance, resistance, and voltage-gated channel expression) determine the effectiveness and temporal precision of light-induced membrane depolarization. Altering the membrane properties of neurons of interest is generally undesirable or unfeasible, and therefore it would be beneficial to engineer optimized ChR variants. The key properties influencing the effectiveness of ChRs include channel conductance, ion selectivity, channel kinetics, desensitization, and recovery from desensitization, light sensitivity, spectral response, and membrane expression/trafficking. ChR2, the most commonly used ChR, has fast kinetics, responds strongly to blue-green light, and is highly expressed at neuronal membranes, but is also strongly desensitized by continuous or pulsed light, leading to inconsistent responses when such stimuli are used to depolarize the membrane (Boyden et al., 2005; Lin et al., 2009). Introduction of an H134R mutation modestly reduces the level of desensitization and increases light sensitivity at the expense of channel kinetics (Nagel et al., 2005; Lin et al., 2009). The use of ChETA (ChR2/E125T) does not alter the level of desensitization but increases channel kinetics at the expense of light sensitivity and reduced photocurrent (Gunaydin et al., 2010). The ChR variant ChIEF exhibits greatly reduced desensitization and identical kinetics and conductance to ChR2, making it the most consistent and ideal ChR for photostimulation experiments to date (Lin et al., 2009). The improved expression and membrane trafficking of ChIEF in mammalian cells are likely to result in toxicity when overexpressed in neuronal membranes, but modifications that hinder membrane trafficking or reduce expression levels of ChIEF should make it a more suitable neuroscientific tool than ChR2. ChR2 with C128X/D156A mutations displays enhanced light sensitivity but slow channel kinetics, and many additional properties of the C128X/D156A variants are not available in the existing literature (Schoenenberger et al., 2009; Bamann et al., 2010). VChR1 has a wide spectral response ranging from 400 to 600 nm but also has slow kinetics (Zhang et al., 2008), and its use as a neuroscientific tool is limited at present. More detailed comparisons and suggested uses of these various ChRs have been published in some recent reviews (Lin, 2010; Zhang et al., 2010).
Future engineering of ChRs for neuroscience applications should focus on the development of proteins that offer beneficial properties (e.g., higher conductance, narrower spectrum, red-shifted spectra, or faster kinetics) without sacrificing the advantages seen in existing ChRs, such as light sensitivity and conductance.
Control of intracellular signal transduction by light
Plant phytochromes and phytochrome-interacting factors (PIFs) undergo a light-gated dimerization cycle, associating strongly when stimulated by red light and dissociating when subsequently stimulated by infrared light (Quail, 2002). Given the ubiquity of protein–protein interactions as a mode of regulation via functional recruitment to macromolecular complexes or subcellular localization, the application of such light-gated protein interaction pairs to neuronal signaling networks could enable the optogenetic control of a wide range of biochemical processes important to adaptation, plasticity, and development of intact nervous systems. The phytochrome–PIF system was first demonstrated as an optogenetic tool in yeast to recruit a transcriptional transactivation domain to a DNA-binding domain to reversibly and quantitatively control transcription with the application of red and infrared light (Shimizu-Sato et al., 2002). This suggests the possibility of targeting the expression of other genetic tools by inducing transcription with light at specific times and places instead of controlling specificity with known promoters. The general principle of this system is illustrated in Figure 4. A phytochrome–PIF pair adapted for use in mammalian cells was shown to be capable of regulating monomeric G-protein signaling in a spatiotemporally programmable fashion based on light-directed membrane translocation of an upstream activator (Levskaya et al., 2009). This system was used to demonstrate the first optogenetic perturbation of cell morphology via spatially modulated red and infrared light. The principle drawback of using this system in vivo is the requirement of a chromophore, phycocyanobilin (PCB), which is non-native to mammalian tissue. However, this difficulty could be readily overcome by using exogenously supplied PCB or by engineering a biliverdin-binding phytochrome variant.
Figure 4.

Control of intracellular signal transduction by light. a, A plasma membrane-targeted phytochrome undergoes dimerization with a yellow fluorescent protein (YFP)-tagged PIF protein exposed to red light, which can be reversed by illumination with infrared light. b, This system can be used to translocate a targeted activator protein to the plasma membrane, enabling optogenetic control of a large number of biochemical signaling pathways.
Future directions
For all of the genetically encoded tools described above—both actuator and reporter proteins—advanced versions are already available and ready for use in neurobiological experiments. However, further progress in the development of these optogenetic resources is both highly desirable and feasible. We anticipate the rapid emergence of new probes with better signal-to-noise ratio and faster kinetics. More importantly, we eagerly await the development of good voltage and calcium probes that operate in the red or infrared spectrum. Such extension of the spectral range is also an important future direction for the improvement of membrane depolarizers and hyperpolarizers.
This current generation of available and emerging optogenetic techniques for the selective targeting of cell types and specific locations within the nervous system is only the beginning. The future will see more extensive exploitation of strategies for the expression of probes within particular subsets of functionally related neurons, including inducible promoters and conditional expression via the Cre/lox system, as well as methods of gene transfer using viral carriers, transsynaptic carriers, and transgenic techniques (Wickersham et al., 2007; Boldogkoi et al., 2009). Broader implementation of the established genetic toolbox will bring the neuroscience community closer to the goal of optically interrogating and controlling specific subsets of neurons to elucidate their specific functions in the execution or regulation of defined behaviors.
Footnotes
Work of the contributing laboratory is supported by the Howard Hughes Medical Institute and RIKEN. We thank members of our laboratories for discussion and support.
References
- Ai HW, Shaner NC, Cheng Z, Tsien RY, Campbell RE. Exploration of new chromophore structures leads to the identification of improved blue fluorescent proteins. Biochemistry. 2007;46:5904–5910. doi: 10.1021/bi700199g. [DOI] [PubMed] [Google Scholar]
- Akemann W, Mutoh H, Perron A, Rossier J, Knöpfel T. Imaging brain electric signals with genetically targeted voltage-sensitive fluorescent proteins. Nat Methods. 2010;7:643–649. doi: 10.1038/nmeth.1479. [DOI] [PubMed] [Google Scholar]
- Ataka K, Pieribone VA. A genetically targetable fluorescent probe of channel gating with rapid kinetics. Biophys J. 2002;82:509–516. doi: 10.1016/S0006-3495(02)75415-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baird GS, Zacharias DA, Tsien RY. Circular permutation and receptor insertion within green fluorescent proteins. Proc Natl Acad Sci U S A. 1999;96:11241–11246. doi: 10.1073/pnas.96.20.11241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker BJ, Mutoh H, Dimitrov D, Akemann W, Perron A, Iwamoto Y, Jin L, Cohen LB, Isacoff EY, Pieribone VA, Hughes T, Knöpfel T. Genetically encoded fluorescent sensors of membrane potential. Brain Cell Biol. 2008;36:53–67. doi: 10.1007/s11068-008-9026-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamann C, Gueta R, Kleinlogel S, Nagel G, Bamberg E. Structural guidance of the photocycle of channelrhodopsin-2 by an interhelical hydrogen bond. Biochemistry. 2010;49:267–278. doi: 10.1021/bi901634p. [DOI] [PubMed] [Google Scholar]
- Boldogkoi Z, Balint K, Awatramani GB, Balya D, Busskamp V, Viney TJ, Lagali PS, Duebel J, Pásti E, Tombácz D, Tóth JS, Takács IF, Scherf BG, Roska B. Genetically timed, activity-sensor and rainbow transsynaptic viral tools. Nat Methods. 2009;6:127–130. doi: 10.1038/nmeth.1292. [DOI] [PubMed] [Google Scholar]
- Boyden ES, Zhang F, Bamberg E, Nagel G, Deisseroth K. Millisecond-timescale, genetically targeted optical control of neural activity. Nat Neurosci. 2005;8:1263–1268. doi: 10.1038/nn1525. [DOI] [PubMed] [Google Scholar]
- Chow BY, Han X, Dobry AS, Qian X, Chuong AS, Li M, Henninger MA, Belfort GM, Lin Y, Monahan PE, Boyden ES. High-performance genetically targetable optical neural silencing by light-driven proton pumps. Nature. 2010;463:98–102. doi: 10.1038/nature08652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chudakov DM, Matz MV, Lukyanov S, Lukyanov KA. Fluorescent proteins and their applications in imaging living cells and tissues. Physiol Rev. 2010;90:1103–1163. doi: 10.1152/physrev.00038.2009. [DOI] [PubMed] [Google Scholar]
- Díez-García J, Akemann W, Knöpfel T. In vivo calcium imaging from genetically specified target cells in mouse cerebellum. Neuroimage. 2007;34:859–869. doi: 10.1016/j.neuroimage.2006.10.021. [DOI] [PubMed] [Google Scholar]
- Dimitrov D, He Y, Mutoh H, Baker BJ, Cohen L, Akemann W, Knöpfel T. Engineering and characterization of an enhanced fluorescent protein voltage sensor. PLoS One. 2007;2:e440. doi: 10.1371/journal.pone.0000440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher ML, Masurkar AV, Xing J, Imamura F, Xiong W, Nagayama S, Mutoh H, Greer CA, Knöpfel T, Chen WR. Optical imaging of postsynaptic odor representation in the glomerular layer of the mouse olfactory bulb. J Neurophysiol. 2009;102:817–830. doi: 10.1152/jn.00020.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedhart J, van Weeren L, Hink MA, Vischer NO, Jalink K, Gadella TW., Jr Bright cyan fluorescent protein variants identified by fluorescence lifetime screening. Nat Methods. 2010;7:137–139. doi: 10.1038/nmeth.1415. [DOI] [PubMed] [Google Scholar]
- Gradinaru V, Thompson KR, Deisseroth K. eNpHR: a Natronomonas halorhodopsin enhanced for optogenetic applications. Brain Cell Biol. 2008;36:129–139. doi: 10.1007/s11068-008-9027-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gradinaru V, Zhang F, Ramakrishnan C, Mattis J, Prakash R, Diester I, Goshen I, Thompson KR, Deisseroth K. Molecular and cellular approaches for diversifying and extending optogenetics. Cell. 2010;141:154–165. doi: 10.1016/j.cell.2010.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunaydin LA, Yizhar O, Berndt A, Sohal VS, Deisseroth K, Hegemann P. Ultrafast optogenetic control. Nat Neurosci. 2010;13:387–392. doi: 10.1038/nn.2495. [DOI] [PubMed] [Google Scholar]
- Han X, Boyden ES. Multiple-color optical activation, silencing, and desynchronization of neural activity, with single-spike temporal resolution. PLoS One. 2007;2:e299. doi: 10.1371/journal.pone.0000299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han X, Chow BY, Yang A, Zhou H, Rajimehr R, Klapoetke NC, et al. Temporally precise optical neural silencing in the nonhuman primate brain. Soc Neurosci Abstr. 2010;36 106.6. [Google Scholar]
- Heim N, Griesbeck O. Genetically encoded indicators of cellular calcium dynamics based on troponin C and green fluorescent protein. J Biol Chem. 2004;279:14280–14286. doi: 10.1074/jbc.M312751200. [DOI] [PubMed] [Google Scholar]
- Klapoetke N, Chuong A, Chow B, Morimoto T, Han X, Boyden ES. Novel classes of optogenetic reagent derived from screening genomic and ecological diversity. Soc Neurosci Abstr. 2010;36 106.1. [Google Scholar]
- Kredel S, Oswald F, Nienhaus K, Deuschle K, Röcker C, Wolff M, Heilker R, Nienhaus GU, Wiedenmann J. mRuby, a bright monomeric red fluorescent protein for labeling of subcellular structures. PLoS One. 2009;4:e4391. doi: 10.1371/journal.pone.0004391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levskaya A, Weiner OD, Lim WA, Voigt CA. Spatiotemporal control of cell signaling using a light-switchable protein interaction. Nature. 2009;461:997–1001. doi: 10.1038/nature08446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Gutierrez DV, Hanson MG, Han J, Mark MD, Chiel H, Hegemann P, Landmesser LT, Herlitze S. Fast noninvasive activation and inhibition of neural and network activity by vertebrate rhodopsin and green algae channelrhodopsin. Proc Natl Acad Sci U S A. 2005;102:17816–17821. doi: 10.1073/pnas.0509030102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lima SQ, Miesenböck G. Remote control of behavior through genetically targeted photostimulation of neurons. Cell. 2005;121:141–152. doi: 10.1016/j.cell.2005.02.004. [DOI] [PubMed] [Google Scholar]
- Lin JY. User's guide to channelrhodopsin variants: features, limitations and future developments. Exp Physiol. 2010 doi: 10.1113/expphysiol.2009.051961. Advance online publication. Retrieved September 19, 2010. PMID: 20621963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JY, Lin MZ, Steinbach P, Tsien RY. Characterization of engineered channelrhodopsin variants with improved properties and kinetics. Biophys J. 2009;96:1803–1814. doi: 10.1016/j.bpj.2008.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin MZ, McKeown MR, Ng HL, Aguilera TA, Shaner NC, Campbell RE, Adams SR, Gross LA, Ma W, Alber T, Tsien RY. Autofluorescent proteins with excitation in the optical window for intravital imaging in mammals. Chem Biol. 2009;16:1169–1179. doi: 10.1016/j.chembiol.2009.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundby A, Mutoh H, Dimitrov D, Akemann W, Knöpfel T. Engineering of a genetically encodable fluorescent voltage sensor exploiting fast Ci-VSP voltage-sensing movements. PLoS One. 2008;3:e2514. doi: 10.1371/journal.pone.0002514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lütcke H, Murayama M, Hahn T, Margolis DJ, Astori S, Zum Alten Borgloh SM, Göbel W, Yang Y, Tang W, Kügler S, Sprengel R, Nagai T, Miyawaki A, Larkum ME, Helmchen F, Hasan MT. Optical recording of neuronal activity with a genetically-encoded calcium indicator in anesthetized and freely moving mice. Front Neural Circuits. 2010;4:9. doi: 10.3389/fncir.2010.00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mank M, Santos AF, Direnberger S, Mrsic-Flogel TD, Hofer SB, Stein V, Hendel T, Reiff DF, Levelt C, Borst A, Bonhoeffer T, Hübener M, Griesbeck O. A genetically encoded calcium indicator for chronic in vivo two-photon imaging. Nat Methods. 2008;5:805–811. doi: 10.1038/nmeth.1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merzlyak EM, Goedhart J, Shcherbo D, Bulina ME, Shcheglov AS, Fradkov AF, Gaintzeva A, Lukyanov KA, Lukyanov S, Gadella TW, Chudakov DM. Bright monomeric red fluorescent protein with an extended fluorescence lifetime. Nat Methods. 2007;4:555–557. doi: 10.1038/nmeth1062. [DOI] [PubMed] [Google Scholar]
- Miyawaki A, Llopis J, Heim R, McCaffery JM, Adams JA, Ikura M, Tsien RY. Fluorescent indicators for Ca2+ based on green fluorescent proteins and calmodulin. Nature. 1997;388:882–887. doi: 10.1038/42264. [DOI] [PubMed] [Google Scholar]
- Nagai T, Sawano A, Park ES, Miyawaki A. Circularly permuted green fluorescent proteins engineered to sense Ca2+ Proc Natl Acad Sci U S A. 2001;98:3197–3202. doi: 10.1073/pnas.051636098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagai T, Yamada S, Tominaga T, Ichikawa M, Miyawaki A. Expanded dynamic range of fluorescent indicators for Ca(2+) by circularly permuted yellow fluorescent proteins. Proc Natl Acad Sci U S A. 2004;101:10554–10559. doi: 10.1073/pnas.0400417101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagel G, Szellas T, Huhn W, Kateriya S, Adeishvili N, Berthold P, Ollig D, Hegemann P, Bamberg E. Channelrhodopsin-2, a directly light-gated cation-selective membrane channel. Proc Natl Acad Sci U S A. 2003;100:13940–13945. doi: 10.1073/pnas.1936192100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagel G, Brauner M, Liewald JF, Adeishvili N, Bamberg E, Gottschalk A. Light activation of channelrhodopsin-2 in excitable cells of Caenorhabditis elegans triggers rapid behavioral responses. Curr Biol. 2005;15:2279–2284. doi: 10.1016/j.cub.2005.11.032. [DOI] [PubMed] [Google Scholar]
- Nakai J, Ohkura M, Imoto K. A high signal-to-noise Ca2+ probe composed of a single green fluorescent protein. Nat Biotechnol. 2001;19:137–141. doi: 10.1038/84397. [DOI] [PubMed] [Google Scholar]
- Palmer AE, Giacomello M, Kortemme T, Hires SA, Lev-Ram V, Baker D, Tsien RY. Ca2+ indicators based on computationally redesigned calmodulin-peptide pairs. Chem Biol. 2006;13:521–530. doi: 10.1016/j.chembiol.2006.03.007. [DOI] [PubMed] [Google Scholar]
- Perron A, Mutoh H, Akemann W, Gautam SG, Dimitrov D, Iwamoto Y, Knöpfel T. Second and third generation voltage-sensitive fluorescent proteins for monitoring membrane potential. Front Mol Neurosci. 2009a;2:5. doi: 10.3389/neuro.02.005.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perron A, Mutoh H, Launey T, Knöpfel T. Red-shifted voltage-sensitive fluorescent proteins. Chem Biol. 2009b;16:1268–1277. doi: 10.1016/j.chembiol.2009.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quail PH. Phytochrome photosensory signalling networks. Nat Rev Mol Cell Biol. 2002;3:85–93. doi: 10.1038/nrm728. [DOI] [PubMed] [Google Scholar]
- Sakai R, Repunte-Canonigo V, Raj CD, Knöpfel T. Design and characterization of a DNA-encoded, voltage-sensitive fluorescent protein. Eur J Neurosci. 2001;13:2314–2318. doi: 10.1046/j.0953-816x.2001.01617.x. [DOI] [PubMed] [Google Scholar]
- Schoenenberger P, Gerosa D, Oertner TG. Temporal control of immediate early gene induction by light. PLoS One. 2009;4:e8185. doi: 10.1371/journal.pone.0008185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaner NC, Steinbach PA, Tsien RY. A guide to choosing fluorescent proteins. Nat Methods. 2005;2:905–909. doi: 10.1038/nmeth819. [DOI] [PubMed] [Google Scholar]
- Shaner NC, Lin MZ, McKeown MR, Steinbach PA, Hazelwood KL, Davidson MW, Tsien RY. Improving the photostability of bright monomeric orange and red fluorescent proteins. Nat Methods. 2008;5:545–551. doi: 10.1038/nmeth.1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shcherbo D, Murphy CS, Ermakova GV, Solovieva EA, Chepurnykh TV, Shcheglov AS, Verkhusha VV, Pletnev VZ, Hazelwood KL, Roche PM, Lukyanov S, Zaraisky AG, Davidson MW, Chudakov DM. Far-red fluorescent tags for protein imaging in living tissues. Biochem J. 2009;418:567–574. doi: 10.1042/BJ20081949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu-Sato S, Huq E, Tepperman JM, Quail PH. A light-switchable gene promoter system. Nat Biotechnol. 2002;20:1041–1044. doi: 10.1038/nbt734. [DOI] [PubMed] [Google Scholar]
- Shu X, Royant A, Lin MZ, Aguilera TA, Lev-Ram V, Steinbach PA, Tsien RY. Mammalian expression of infrared fluorescent proteins engineered from a bacterial phytochrome. Science. 2009;324:804–807. doi: 10.1126/science.1168683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel MS, Isacoff EY. A genetically encoded optical probe of membrane voltage. Neuron. 1997;19:735–741. doi: 10.1016/s0896-6273(00)80955-1. [DOI] [PubMed] [Google Scholar]
- Subach FV, Patterson GH, Renz M, Lippincott-Schwartz J, Verkhusha VV. Bright monomeric photoactivatable red fluorescent protein for two-color super-resolution sptPALM of live cells. J Am Chem Soc. 2010;132:6481–6491. doi: 10.1021/ja100906g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subach OM, Gundorov IS, Yoshimura M, Subach FV, Zhang J, Grüenwald D, Souslova EA, Chudakov DM, Verkhusha VV. Conversion of red fluorescent protein into a bright blue probe. Chem Biol. 2008;15:1116–1124. doi: 10.1016/j.chembiol.2008.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian L, Hires SA, Mao T, Huber D, Chiappe ME, Chalasani SH, Petreanu L, Akerboom J, McKinney SA, Schreiter ER, Bargmann CI, Jayaraman V, Svoboda K, Looger LL. Imaging neural activity in worms, flies and mice with improved GCaMP calcium indicators. Nat Methods. 2009;6:875–881. doi: 10.1038/nmeth.1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tønnesen J, Sørensen AT, Deisseroth K, Lundberg C, Kokaia M. Optogenetic control of epileptiform activity. Proc Natl Acad Sci U S A. 2009;106:12162–12167. doi: 10.1073/pnas.0901915106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace DJ, Meyer zum Alten Borgloh S, Astori S, Yang Y, Bausen M, Kügler S, Palmer AE, Tsien RY, Sprengel R, Kerr JN, Denk W, Hasan MT. Single-spike detection in vitro and in vivo with a genetic Ca2+ sensor. Nat Methods. 2008;5:797–804. doi: 10.1038/nmeth.1242. [DOI] [PubMed] [Google Scholar]
- Wickersham IR, Lyon DC, Barnard RJ, Mori T, Finke S, Conzelmann KK, Young JA, Callaway EM. Monosynaptic restriction of transsynaptic tracing from single, genetically targeted neurons. Neuron. 2007;53:639–647. doi: 10.1016/j.neuron.2007.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Wang LP, Brauner M, Liewald JF, Kay K, Watzke N, Wood PG, Bamberg E, Nagel G, Gottschalk A, Deisseroth K. Multimodal fast optical interrogation of neural circuitry. Nature. 2007;446:633–639. doi: 10.1038/nature05744. [DOI] [PubMed] [Google Scholar]
- Zhang F, Prigge M, Beyrière F, Tsunoda SP, Mattis J, Yizhar O, Hegemann P, Deisseroth K. Red-shifted optogenetic excitation: a tool for fast neural control derived from Volvox carteri. Nat Neurosci. 2008;11:631–633. doi: 10.1038/nn.2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Gradinaru V, Adamantidis AR, Durand R, Airan RD, de Lecea L, Deisseroth K. Optogenetic interrogation of neural circuits: technology for probing mammalian brain structures. Nat Protoc. 2010;5:439–456. doi: 10.1038/nprot.2009.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao S, Cunha C, Zhang F, Liu Q, Gloss B, Deisseroth K, Augustine GJ, Feng G. Improved expression of halorhodopsin for light-induced silencing of neuronal activity. Brain Cell Biol. 2008;36:141–154. doi: 10.1007/s11068-008-9034-7. [DOI] [PMC free article] [PubMed] [Google Scholar]