

Abstract
TGF-β plays a crucial role in immune regulation. It has been reported that pro–TGF-β, latency-associated peptide (LAP), latent TGF-β and/or active TGF-β (LAP/TGF-β) is localized on the cell surface of Foxp3+ regulatory T cells. However, the molecular mechanism(s) of how LAP/TGF-β is anchored on the cell membrane is unknown. In this study, we show that forced expression of human TGF-β1 gene by retrovirus transduction into P3U1 mouse myeloma cells, and other cell types including murine CD4+CD25− T cells, makes these cells surface LAP/TGF-β-positive. The surface LAP/TGF-β contains high-glycosylated, furin-processed latent TGF-β, which is different from the low-glycosylated, furin-unprocessed intracellular form or the high-glycosylated, furin-unprocessed secreted form. Furthermore, surface LAP/TGF-β forms a complex with the molecular chaperone glucose-regulated protein 78 (GRP78, also known as BiP), and knockdown of GRP78 reduced the expression levels of surface LAP/TGF-β. GRP78, however, is not involved in GARP-mediated surface LAP/TGF-β. Our results suggest that GRP78 provides an additional surface localization mechanism for LAP/TGF-β, which may play an important role in controlling TGF-β activity.
Transforming growth factor-β plays a crucial role in immune regulation; it acts as an immunosuppressant, T regulatory cell (Treg)-inducer, or Th17-inducer depending on the context (1). The mechanisms by which TGF-β is synthesized and expressed by immune cells are not well understood. TGF-β is first synthesized as prepro–TGF-β peptide. It quickly forms a dimer (pro–TGF-β) connected by disulfide bondings in the endoplasmic reticulum, and pro–TGF-β becomes highly glycosylated in the Golgi complex. Pro–TGF-β is cleaved by furin (2) before secretion and becomes latent TGF-β that is a noncovalently associated complex of the latency-associated peptide (LAP) dimer and TGF-β peptide dimer (mature TGF-β) (3) (Fig. 1). Latent TGF-β does not have biological activity, and needs a further activation process after secretion to be able to bind TGF-β receptors, such as proteolytic removal of LAP to release mature TGF-β, or a conformational change so that TGF-β is exposed to the surface of the latent TGF-β complex (4, 5). Thus, each processing step must be clarified to understand how TGF-β activity is controlled.
FIGURE 1.
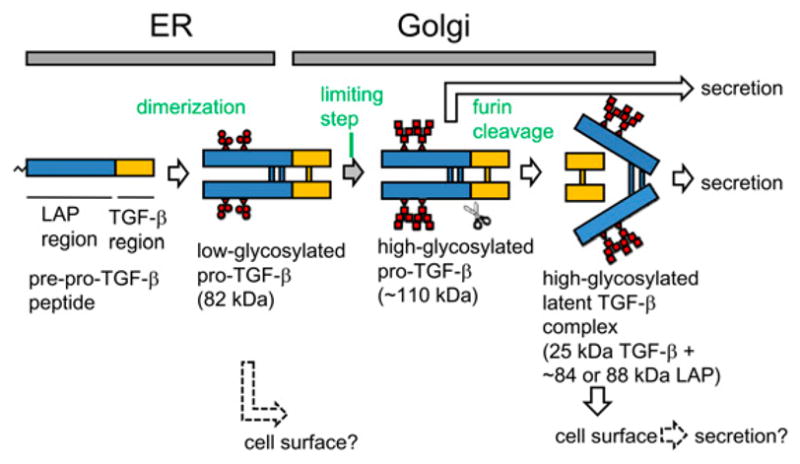
Schematic intracellular processing and transport of LAP/TGF-β. Low-glycosylated (immature high-mannose type) pro–TGF-β is a major intracellular form, whereas high-glycosylated (highly branched type), furin-processed latent TGF-β is secreted rapidly. In artificially overexpression systems, furin-unprocessed pro–TGF-β is also secreted. The molecular mass is based on our SDS-PAGE under nonreducing conditions. Although the illustration depicts LAP/TGF-β as soluble forms, some of them may be anchored on membranes and transported to the cell surface.
Nakamura et al. (6) first reported that pro–TGF-β, LAP, latent TGF-β, and/or mature TGF-β (hereafter referred as LAP/TGF-β) is anchored on CD4+CD25+ Treg surface. They proposed that the surface TGF-β is presented to TGF-β receptors on target effector T cells by cell–cell contact and this is an important mechanism of the Treg-mediated suppression. Since then, other laboratories, including ours, described the existence of surface LAP/TGF-β (7–10). However, it is still a matter of debate because surface LAP/TGF-β is not always observed (11), and the TGF-β effects on Treg-mediated suppression have been challenged (11).
One of the reasons for the controversial issues about surface LAP/TGF-β relates to the fact that we do not have reliable systems where we can constantly observe surface LAP/TGF-β to conduct biochemical analysis. Unless the molecular mechanisms of the surface anchoring of LAP/TGF-β are revealed, it is difficult to make a comprehensive view of the idea of surface LAP/TGF-β.
In this study, we report that simple overexpression of the TGF-β gene makes cells surface LAP/TGF-β positive. Taking advantage of the system, we were able to obtain a large number of surface LAP/TGF-β+ cells, and we found that surface LAP/TGF-β forms a complex with the molecular chaperone glucose-regulated protein 78 (GRP78, also known as BiP). Surface LAP/TGF-β–bound GRP78 has a slightly higher molecular mass than canonical GRP78, suggesting the presence of special glycosylation. Surface LAP/TGF-β contains high-glycosylated, furin-processed latent TGF-β, which is different from the major intracellular pool of low-glycosylated unprocessed pro–TGF-β or the secreted form of high-glycosylated unprocessed pro–TGF-β.
Materials and Methods
Abs
Anti-human LAP mAb clone 27232 and anti–TGF-β mAb clone 9016 were obtained from R&D Systems (Minneapolis, MN). Anti-human LAP mAbs TW4-2F8 (mouse IgG1) and TW4-4E5 (mouse IgG1), and anti–TGF-β mAb TW4-9E7 (mouse IgG1) were made by immunizing BALB/c mice with purified human recombinant LAP (R&D Systems) emulcified with TiterMax (Sigma-Aldrich, St. Louis, MO), and boosting with P3U1–TGF-β cells. These inhouse anti-LAP mAbs and anti–TGF-β mAb were con-firmed to bind purified recombinant human LAP (R&D Systems) or purified recombinant TGF-β (R&D Systems), respectively (Supplemental Fig. 1). Goat anti-GRP78 was purchased from R&D Systems. Anti-mouse CD3 and anti-mouse CD28 were from BD Biosciences (San Diego, CA). Anti–β-actin was from Santa Cruz Biotechnologies (Santa Cruz, CA). PE-labeled anti-mouse GARP (clone YGIC86) was from eBioscience (San Diego, CA).
Cells and retroviral transduction
P3U1 is a subline of NS0 mouse myeloma cell line and was originally obtained from American Type Culture Collection (ATCC) (Manassas, VA). Retroviral vector pMCs-IRES-GFP (12), ecotropic retroviral packaging cell line Plat-E (13) and pantropic retroviral packaging cell line Plat-GP (13) were kindly provided by Dr. Kitamura (Tokyo University, Tokyo, Japan). Human TGF-β gene (TGFB1) was cloned into pMCs-IRES-GFP vector or modified pMCs vector lacking IRES-GFP, and the retroviral supernatant was produced by Plat-E. P3U1 cells or mouse CD4+25− T cells from BALB/c mice (The Jackson Laboratories, Bar Harbor, ME) preactivated with plate-bound anti-CD3 and anti-CD28 for 30 h were infected with human TGF-β or control ecotropic retrovirus by centrifugation at 3000 rpm for 1 h. The human TGF-β–transduced P3U1 (P3U1–TGF-β) cells were subjected to single-cell cloning to obtain stable clones. TA3 cells (Dr. L. Glimcher, Harvard School of Public Health, Boston, MA) and Jurkat cells (ATCC) were first transduced with the ecotropic receptor (Slc7a1)-expressing vector using glycoprotein of the vesicular stomatitis virus (VSV-G) envelope, then transduced with human TGF-β retrovirus as above. NRK cells (ATCC) were directly transduced with human TGF-β retrovirus using VSV-G envelope. Two hundred ninety-three T cells were first transfected with pcDNA3.1-ecotropic receptor vector using TransIt-293 transfection reagent (Mius Bio, Madison, WI), and then transduced with human TGF-β ecotropic retrovirus. P3U1–TGF-β clone No. 32 cells were transduced with pMCs-mouse GARP-IRES-Thy1.1 vector and cloned.
FACS staining
P3U1–TGF-β cells were first treated with anti-CD16/CD32 (BD Biosciences) to block Fc receptors, and stained with anti-LAP mAb or anti–TGF-β mAb, followed by allophycocyanin-labeled goat anti-mouse Ig (BD Biosciences). The cells were analyzed by FACS Calibur (BD Biosciences) and plotted by Cellquest (BD Biosciences) or FlowJo (TreeStar, Ashland, OR) software.
Surface LAP immunoprecipitation
To identify molecular form(s) of surface LAP/TGF-β, P3U1–TGF-β clone No. 16 cells were surface labeled with anti-human LAP mAb TW4-2F8 after washing with PBS and treatment with anti-CD16/CD32 mAb. Unbound anti-LAP mAbs were removed by washing with PBS three times, and the cells were lysed with 1% Triton X-100, 0.25% deoxycholate-Tris buffered saline (pH 7.4) containing EDTA and protease inhibitor mixture (Thermo-Pierce, Rockford, IL). The lysate was clarified by centrifugation at 13,000 rpm for 10 min, and was subjected to immunoprecipitation with anti-mouse IgG-coated magnetic beads (BioMag Plus [Polysciences, Warrington, PA]). The bound fraction was eluted by heating at 70°C for 10 min in 1× LDS buffer without reducing reagents. The elute was run on SDS-PAGE under nonreducing conditions using Nupage 7% Tris-Acetate Gel (Invitrogen, Carlsbad, CA). After transfer to a polyvinylidene difluoride membrane, Western blotting for LAP or anti–TGF-β was done with goat anti–LAP-biotin (R&D Systems, BAF246), followed by streptavidin-HRP (Invitrogen), or chicken anti–TGF-β (R&D Systems, AF-101-NA), followed by donkey anti-chicken IgY-HRP (Jackson ImmunoResearch Laboratories, West Grove, PA), respectively. The membranes blotted with one Ab were reblotted with the other to check whether each band contained LAP epitope and/or TGF-β epitope.
Protein identification of LAP/TGF-β–associated molecules
P3U1–TGF-β clone No. 16 cells, or control untransduced P3U1 cells were first surface-biotinylated by sulfo-LS-biotin-NHS (Pierce) according to the manufacturer’s instruction. The cells were lysed with 1% Triton X-100-PBS containing EDTA and protease inhibitor mixture (Pierce). The lysate was immunoprecipitated with magnetic beads (Dynabeads MyOne, tosyl-activated, Invitrogen) covalently coupled with anti-human LAP mAb TW4-4E5 or with isotype control mAb MOPC21. The bead-bound fraction was eluted 1× LDS sample buffer (Invitrogen) by heating at 70°C for 10 min. The biotinylated proteins were further purified by streptavidin-coated magnetic beads (Streptavidin Dynabeads, Invitrogen) and eluted with 1× LDS sample buffer containing 100 mM biotin by heating at 100°C for 10 min. The fractions were run on SDS-PAGE using Nupage 4–12% Bis-Tris gel and MOPS buffer (Invitrogen). After silver staining, a band specific on P3U1–TGF-β cells was cut and liquid chromatography/mass spectrometry (LC/MS) analysis was conducted by 4800 Plus proteomics analyzer (Applied Biosystems, Foster City, CA).
GRP78 knockdown
The 293T cells (Clontech, Mountain View, CA) were transfected with a short hairpin RNA (shRNA) lentiviral vector against GRP78 (Open Biosystems, Huntsville, AL, RMM3981-9577763) or control PLKO.1 vector mixed with psPAX2 lentiviral packaging plasmid (Addgene, Cambridge, MA) and pCMV-VSV-G envelope plasmid (Cell Biolabs, San Diego, CA), and viral supernatant was made. P3U1–TGF-β clone No. 32 cells were infected with the virus by spinfection. Three days postinfection, GRP78 knockdown was checked by Western blot, and surface LAP/TGF-β levels were analyzed by FACS.
Results
Retroviral transduction of human TGF-β into cell lines and murine T cells induces surface LAP/TGF-β
To study molecular mechanisms of surface localization of LAP/TGF-β (Fig. 1), we needed to have a reproducible method to obtain surface LAP/TGF-β+ cells. P3U1 mouse myeloma cell line is a fast-growing, nonadherent cell line that is as well suited for retroviral transduction. Untransduced P3U1 cells are negative for surface staining with anti-human LAP mAb 27232 or anti-TGF-β mAb 9016. However, after retroviral transduction of human TGF-β1, the cells became surface positive for anti-LAP mAb and anti–TGF-β mAb (Fig. 2A). Furthermore, our inhouse anti-LAP mAb TW4-2F8 and anti–TGF-β mAb TW4-9E7 showed similar staining (Fig. 2A). This indicates that the cells have intrinsic molecular mechanisms to anchor LAP/TGF-β on the cell surface. Next, we asked whether surface LAP/TGF-β induction is unique to P3U1 cells. For these experiments, murine CD4+CD25− T cells were similarly transduced with human TGF-β, and we also found that mouse CD4+ CD25− T cells became surface LAP/TGF-β positive (Fig. 2B). We also tested the human embryonic kidney 293T cell line, the TA3 murine B hybridoma cell line, the Jurkat human leukemic cell line, the NRK rat fibroblastic cell line, and the 3T3 mouse embryonic fibroblast cell line, and they too became surface LAP/TGF-β positive after transduction with human TGF-β (Supplemental Fig. 2). These results demonstrate that multiple cell types can express surface LAP/TGF-β when TGF-β is overexpressed.
FIGURE 2.
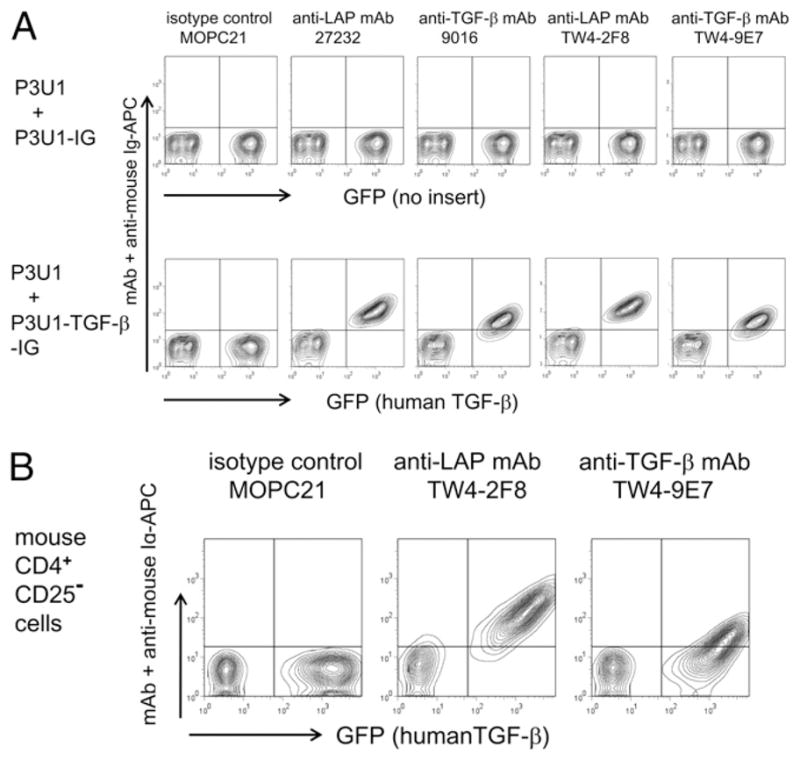
Surface LAP/TGF-β induction by forced expression of human TGF-β1. A, P3U1 cells were retrovirally transduced with TGFB1 or control vector, and were stained with commercially available or inhouse anti-human LAP mAb or anti–TGF-β mAb. Untransduced P3U1 cells were mixed as controls. B, mouse CD4+CD25− cells were preactivated with anti-CD3/anti-CD28 for 30 h and retrovirally transduced with TGFB1. Two days postinfection, the cells were stained with inhouse anti-human LAP mAb or anti–TGF-β mAb.
Molecular form of surface LAP/TGF-β on P3U1–TGF-β cells
TGF-β is sequentially processed intracellularly before secretion (Fig. 1). To identify how surface LAP/TGF-β is processed, we attempted to separate surface LAP/TGF-β. Thus, P3U1–TGF-β clone No. 16 cells, which is the clone expressing the highest surface LAP/TGF-β, were first surface labeled with anti-LAP mAb TW4-2F8, then lysed with Triton X-100–containing buffer. After clarifying by centrifugation, the lysate was immunoprecipitated with anti-mouse IgG-coated magnetic beads. The surface immunoprecipitated LAP/TGF-β was run on SDS-PAGE under nonreducing conditions along with the anti-LAP immunoprecipitate from the whole-cell lysate and with cell culture supernatant containing secreted LAP/TGF-β. As shown in Fig. 3A, lane 1, anti-LAP identified smear bands at 110 kDa and 84 kDa in the culture supernatant. Anti–TGF-β also detected the 110 kDa band but not the 84 kDa band (Fig. 3B, lane 1). Based on their sizes (14, 15), the 110 kDa band is consistent with the high-glycosylated form (highly branched type) of pro–TGF-β (furin-unprocessed), and the 84 kDa is consistent with the high-glycosylated form of the LAP (furin-processed). We believe the reason why pro–TGF-β was the major form of secreted LAP/TGF-β in this system is that overexpression of TGF-β exceeded the capacity of endogenous furin proprotein convertase activity in the P3U1–TGF-β clone No. 16 cells, because a less expressing c P3U1–TGF-β clone No. 32 cells had a higher ratio of the 84 kDa LAP to 110 kDa pro–TGF-β in the culture supernatant than clone No. 16 (data not shown). In contrast, we found that most of the intracellular form of TGF-β was a 82 kDa form detectable both with anti-LAP (Fig. 3A, lane 3) and with anti–TGF-β Abs (Fig. 3B, lane 3). This 82 kDa form is considered as low-glycosylated (immature high-mannose type) pro–TGF-β (furin-unprocessed) as has been reported (14, 15). Surface LAP/TGF-β showed 88 kDa and 82 kDa bands by anti-LAP detection (Fig. 3A, lane 2), and a single 82 kDa band by anti–TGF-β detection (Fig. 3B, lane 2). The 88 kDa band is considered as high-glycosylated LAP (furin-processed), and the 82 kDa band is considered as low-glycosylated pro–TGF-β (furin-unprocessed). Anti–TGF-β blotting also showed the 25 kDa mature TGF-β band in the surface anti–LAP-immunoprecipitated sample (Fig. 3C), suggesting that LAP and mature TGF-β existed as the latent TGF-β complex on the surface. Although the surface anti–LAP-immunoprecipitated sample also contained the 82 kDa low-glycosylated unprocessed pro–TGF-β, this could be contamination from the whole-cell lysate considering the divalency of the anti-LAP mAb and possibility of exchange with whole lysate-derived LAP/TGF-β during the immunoprecipitation process. These results suggest that at least some of surface LAP/TGF-β is the high-glycosylated, furin-processed form that is distinct from the secreted form of TGF-β (high-glycosylated pro–TGF-β) or the intracellular form of TGF-β (low-glycosylated pro–TGF-β). In summary, this demonstrates that surface LAP/TGF-β has a unique route for processing and localization.
FIGURE 3.

Molecular forms of surface LAP/TGF-β. P3U1–TGF-β clone No. 16 cells were surface labeled with mouse anti-human LAP mAb, lysed, and immunoprecipitated with anti-mouse IgG magnetic beads. The elute was run on SDS-PAGE (A–C; lane 2) along with cell culture supernatant (A–C; lane 1) and anti-LAP immunoprecipitate from whole cell lysate (A–C; lane 3), and immunoblotted with goat anti-LAP (A) or chicken anti–TGF-β (B, C). The arrows indicate positions of the anti-LAP reacted bands.
GRP78 is associated with surface LAP/TGF-β
We then investigated how surface LAP/TGF-β is anchored on the cell membrane. First, we checked known LAP/TGF-β binding proteins, for example, insulin-like growth factor II/mannose 6-phosphate receptor (IGFIIR/M6PR) and αV integrin. IGFIIR/M6PR binds mannose-6-phosphate on N-linked carbohydrate chains of LAP/TGF-β (16), and αV integrin binds RGD sequence on LAP (17). We made mutant human TGF-β constructs; TGF-β (N82Q, N136Q) (18, 19) in which Asn82 and Asn136 are changed to Gln, and TGF-β (RGD→RGE) (20) in which the RGD sequence is disrupted to RGE. We found that retroviral transduction of TGF-β (N82Q, N136Q) or TGF-β (RGD→RGE) induced surface expression of LAP/TGF-β (Supplemental Fig. 3). These results indicate that IGFIIR/M6PR or αV integrin are not involved in the surface localization of LAP/TGF-β of P3U1–TGF-β cells.
To identify surface LAP/TGF-β associatedmolecules, P3U1–TGF-β No. 16 cells or parent P3U1 cells were first surface biotinylated, and then lysed and immunoprecipitated with anti-LAP mAb TW4-4E5, which we found works for immunoprecipitation (Supplemental Fig. 4). The anti-LAP immunoprecipitated fractions were further separated with streptavidin-coupled magnetic beads, and bound fractions were run on SDS-PAGE under reducing conditions, and then visualized by streptavidin-HRP blotting (Fig. 4A), or by silver staining (Fig. 4B). We found that only a ~80 kDa band was identified as specific to P3U1–TGF-β cells in the silver staining (Fig. 4B, arrow), which was also seen in the streptavidin-HRP blotting (Fig. 4A, arrow). The 80 kDa band was cut and subjected to LC/MS analysis. One peptide corresponding to GRP78 was identified. To determine whether GRP78 was truly immunoprecipitated with anti-LAP mAb from P3U1–TGF-β cells, anti-LAP immunoprecipitated samples from P3U1–TGF-β cells or P3U1 parent cells were checked by Western blotting with anti-GRP78 Ab. We found that there were 75 and 80 kDa bands of GRP78 in the anti-LAP immunoprecipitated sample from P3U1–TGF-β cells, but not from P3U1 parent cells (Fig. 5A). We also tested streptavidin-bound, and -unbound fractions of anti-LAP immunoprecipitated samples from surface biotinylated P3U1–TGF-β cells. Interestingly, the 75 kDa GRP78 band was associated with the streptavidin-unbound (nonsurface) fraction, whereas the 80 kDa band was found exclusively in the streptavidin-bound (surface) fraction (Fig. 5B). The 75 kDa band most likely represents canonical GRP78, whereas the 80 kDa band would correspond to glycosylated GRP78 that is reported on the cell surface of many cancer cells (21). In summary, these results demonstrate that LAP/TGF-β is associated with GRP78 both intracellularly and on the surface, but that the forms of GRP78 at the two locations are different.
FIGURE 4.

Identification of surface LAP/TGF-β–associated proteins. A, P3U1–TGF-β clone No. 16 cells or parent P3U1 cells were first surface labeled with biotin. The cell lysates were immunoprecipitated with anti-LAP mAb. The elutes further separated with SA magnetic beads, and blotted with SA-HRP. SA-bound fractions (lanes 1, 2) and -unbound fractions (lanes 3, 4) from P3U1 cells (lanes 1, 3) or P3U1–TGF-β cells (lanes 2, 4). B, Silver staining of the SA-bound fractions from P3U1 cells (lane 1) or from P3U1–TGF-β cells (lane 2). The band at the arrow was cut and subjected to LC/MS analysis. SA, streptavidin.
FIGURE 5.

GRP78 association with LAP/TGF-β. A, Cell lysate from P3U1–TGF-β clone No. 16 cells was immunoprecipitated with anti-LAP mAb (lane 1) or isotype control (IC) (lane 2), and immunoblotted with anti-GRP78. B, Cell lysates from surface biotinylated P3U1–TGF-β cells or parent P3U1 cells were immunoprecipitated with anti-LAP, and then were further separated with SA-magnetic beads. SA-bound fractions (lanes 1, 3) and -unbound fractions (lanes 2, 4) from P3U1–TGF-β cells (lanes 1, 2) or P3U1 cells (lanes 3, 4) were immunoblotted with anti-GRP78.
Knockdown of GRP78 reduces surface LAP/TGF-β levels
To test the importance of GRP78 for surface localization of LAP/TGF-β, we attempted to knockdown GRP78 in P3U1–TGF-β cells by lentiviral short hairpin RNA (shRNA) transduction. P3U1–TGF-β clone No. 32 was used for the knockdown experiments as a representative of TGF-β–transduced P3U1 cells because this clone has a typical level of surface LAP expression and a similar FACS scatter profile as gross cultures of P3U1–TGF-β cells prior to cloning. Our preliminary tests using P3U1 cells transduced with GRP78-IRES-Thy1.1 as reporter cells indicated that P3U1 cells could be infected at nearly 100% efficiency and that the GRP78 shRNA had the ability to strongly knockdown GRP78 mRNA, as judged by the reduction of Thy1.1 expression by the foreign promoter driven GRP78-IRES-Thy1.1 to ~5% of the initial (Supplemental Fig. 5). However, we observed only ~50% reduction of the GRP78 protein level in GRP78 shRNA-transduced P3U1–TGF-β No. 32 cells (Fig. 6A). We postulate that the cells have a feedback circuit that serves to maintain the GRP78 protein level either by upregulation of the endogenous GRP78 transcription or by reducing the protein degradation. Nevertheless, the surface LAP/TGF-β levels were also reduced by ~50% (Fig. 6B). We also used another clone, P3U1–TGF-β No. 16, and observed a decrease of surface LAP/TGF-β levels although the GRP78 knockdown level and consequent surface LAP/TGF-β reduction were less apparent (data not shown). Despite GRP78 knockdown was not complete, these results suggest that GRP78 is directly linked to LAP/TGF-β and important for the surface localization.
FIGURE 6.

Knockdown of GRP78 reduced surface LAP/TGF-β. A, P3U1–TGF-β clone No. 32 cells were transduced with control- or GRP78-shRNA lentivirus, and serial dilutions of cell lysates were immunoblotted with anti-GRP78 (left) or anti–β-actin (right). B, The cells were surface stained with anti-LAP mAb (left) or anti–TGF-β (right) and analyzed by FACS. Note that 80 kDa form of GRP78 is quite minor in whole-cell lysate, and it cannot be seen unless a film is overexposed.
GRP78 is independent of GARP-mediated surface LAP expression
Recently, it was reported that human FOXP3+ Tregs uniquely express LAP on the surface after activation (22, 23), and GARP is the LAP-anchoring molecule on human Tregs (24, 25). GARP was not detectable by FACS staining (Fig. 7A) or RT-PCR (data not shown) in P3U1–TGF-β cells, and thus it is not likely that GARP is involved in surface localization of LAP/TGF-β in our P3U1–TGF-β cells. We then asked whether GRP78 is involved in GARP-mediated surface LAP/TGF-β expression. We attempted GRP78 knockdown in murine CD4+CD25+ Tregs with the lentiviral shRNA used for P3U1–TGF-β cells in our studies. Although we achieved reasonable infection efficiency of the lentiviral shRNA and obtained good cell survival after puromycin selection, we did not observe detectable knockdown of GRP78 by Western blot, or reduction of surface LAP staining in CD4+CD25+ Tregs (data not shown). This may be because of counteraction of the knockdown pressure as seen in P3U1–TGF-β cells. As an alternative way to address this question, we transduced P3U1–TGF-β cells with GARP. Consistent with previous reports (24, 25), GARP transduction into P3U1–TGF-β cells proportionally enhanced surface LAP expression (Fig. 7A). Lentiviral GRP78 knockdown did not alter surface LAP expression on P3U1–TGF-β-GARP cells (Fig. 7B, upper panel), although GRP78 knockdown was confirmed by Western blot (Fig. 7B, lower panel), and parallel GRP78 knockdown of P3U1–TGF-β cells reduced surface LAP expression (Fig. 7B, upper panel). This suggests that GRP78-mediated surface LAP machinery is independent of GARP-anchored surface LAP.
FIGURE 7.

GRP78 is not involved in GARP-mediated surface LAP expression. A, GARP-transduction into P3U1–TGF-β cells proportionally increased surface LAP expression. P3U1–TGF-β clone No. 32 cells were retrovirally transduced with pMCs-mouse GARP-IRES-Thy1.1 vector and stained with anti-LAP TW4-2F8 and anti-mouse GARP mAb. B, GRP78 knockdown in P3U1–TGF-β-GARP cells did not decrease surface LAP expression. Upper panel, Cloned GARP-transduced P3U1–TGF-β No. 32 cells (P3U1–TGF-β-GARP clone No. 16 cells) or parent P3U1–TGF-β No. 32 cells were transduced with control- or GRP78-shRNA lentivirus, and surface LAP expression was analyzed by FACS. Note that the allophycocyanin (FL4) detector amp gain was intentionally set lower than the one used for Fig. 7A to make signals within the range. Lower panel, GRP78 knockdown was confirmed by Western blot.
Discussion
The concept of surface LAP/TGF-β was first reported in mouse CD4+CD25+ Tregs (6), although there has been controversy regarding the existence and the role of surface LAP/TGF-β (11). We reported a unique subset of CD4+CD25−LAP+ Tregs (10) and the induction of CD4+CD25−LAP+ Tregs following of oral administration of anti-CD3 Ab (26). Biochemical analysis of how LAP/TGF-β is anchored on the cell membrane has been difficult because surface LAP/TGF-β seems to be observed only under special conditions.
We found that simple overexpession of human TGF-β induced surface LAP/TGF-β on P3U1 mouse myeloma cells and other cell types, including mouse CD4+CD25− T cells. This suggests that the surface localization mechanism is common to many cell types rather than depending on a cell type-specific anchoring molecule. The surface LAP/TGF-β on the P3U1–TGF-β cells exists at least in part as high-glycosylated, furin-processed latent TGF-β. We found no detectable high-glycosylated, furin-unprocessed form (high-glycosylated pro–TGF-β) on the surface that, however, is abundant in the cell culture supernatant. Surface LAP/TGF-β is also different from the major intracellular form, low-glycosylated pro–TGF-β. The fact that surface LAP/TGF-β has a special processing feature different from other compartments suggests that surface LAP/TGF-β is not transported merely by miss-sorting from intracellular organella or recaptured after it is secreted, but has biological machineries to be transported to the surface.
We do not know whether surface localization of LAP/TGF-β is a part of the process of secretion, or it is independent from the conventional secretory pathway. Considering that surface LAP/TGF-β does not contain high-glycosylated pro–TGF-β that is abundant in the culture supernatant, we speculate that the membrane-anchored pathway is independent from the conventional secretory pathway. However, it is possible that in our overexpression system, LAP/TGF-β may overwhelm the capacity of the membrane-anchored transportation and most newly synthesized TGF-β goes into the fluid phase secretion pathway. In physiological conditions, TGF-β may reside primarily in the membrane-anchored transport system.
We found from a proteomics approach that GRP78 is associated with surface LAP/TGF-β. GRP78 is a member of Hsp70 family, and it acts as a molecular chaperone for emerging membrane or secretory proteins in an ATP-dependent manner in the endoplasmic reticulum (27). Because LAP/TGF-β associates with GRP78 not only on the cell surface but also intracellularly, it is possible that LAP/TGF-β and GRP78 make a complex in the endoplasmic reticulum. In early studies, it was shown that LAP/TGF-β is secreted very slowly (15). LAP/TGF-β is retained in the intracellular compartment for days as a low-glycosylated, furin-unprocessed form in nonoverexpressing cells. It thus seems that it is the intrinsic nature of TGF-β to stay in the intracellular compartment, presumably because it is captured by (a) sequestering protein(s). GRP78 may be one of such sequestering proteins, and at the same time GRP78 could be a transporter of LAP/TGF-β to the cell surface depending on the molecular forms of GRP78, such as glycosylation. GRP78 is mainly localized in the endoplasmic reticulum, but it is also reported to be detected on the cell surface (28). Cell surface GRP78 is N- and O-glycosylated, and shows a higher molecular mass (~82 kDa) in SDS-PAGE than in-tracellular canonical GRP78 (~78 kDa) in cancer cells (21). Our results show that LAP/TGF-β–bound forms of GRP78 are both of higher molecular mass (80 kDa) and of lower molecular mass (75 kDa) in our SDS-PAGE system, which would correspond to the glycosylated GRP78 and the canonical GRP78, respectively. Interestingly, surface LAP/TGF-β–bound GRP78 is exclusively in the higher molecular mass form, whereas nonsurface (intracellular) LAP/TGF-β–bound GRP78 is in the lower molecular mass form. It may be that glycosylation of GRP78 is the key for GRP78-LAP/TGF-β surface localization. Although GRP78 mainly exists as a soluble protein, it can also take a transmembrane configuration (29). How the topology of GRP78 is controlled is not known. We still do not know whether LAP/TGF-β–bound GRP78 is the trans-membrane form, or it is a soluble form and needs to be bound to another transmembrane protein to localize on the cell surface.
Recently, it was reported that GARP is the LAP-anchoring molecule on the surface of human FOXP3+ Tregs (24, 25). GRP78 knockdown did not change the surface LAP expression level of P3U1–TGF-β-GARP cells, suggesting that GRP78 is not involved in GARP-mediated surface LAP/TGF-β expression. Although it is not known whether GARP is the sole anchoring molecule of surface LAP on Foxp3+ Tregs, it appears that GRP78 is not a dominant factor controlling the surface LAP on Foxp3+ Tregs. Our results demonstrate that multiple anchoring molecular mechanisms for surface LAP/TGF-β exist. Thus, we have identified a previously unknown molecule that serves this function, GRP78. We recently reported that a small population of human CD4+ T cells express surface LAP without activation (30), and these LAP+ cells do not express FOXP3 or GARP (R. Gandhi, unpublished observations), suggesting a role for other molecules in surface LAP/TGF-β expression. Further studies are now needed to understand the importance of GRP78 in vivo for regulating TGF-β activity at this moment. Although GRP78 knockout mice are embryonic lethal (31), Cre-mediated conditional GRP78 knockout mice have been reported (32), and, thus, cell- or organ-specific conditional knockout animals would be helpful to address these issues.
Neuropilin-1 has also been reported as a LAP/TGF-β binding protein (33). However, Neuropilin-1 expression does not correlate with surface LAP/TGF-β in human Tregs (22), and we did not detect Neuropilin-1 by RT-PCR in our cells. The role of Neuropilin-1 as a surface LAP/TGF-β-anchoring protein will require further study.
In summary, we have found that overexpression of the human TGF-β gene induced surface LAP/TGF-β expression, and this surface LAP/TGF-β makes a complex with GRP78. This new system provides an avenue to conduct further biochemical analysis to identify molecular mechanisms of how surface LAP/TGF-β is regulated.
Supplementary Material
Acknowledgments
We thank Dr. Simon Dillon (Genomics Center and Dana Farber/Harvard Cancer Center, Beth Israel Deaconess Medical Center, Boston, MA) for LC/MS analysis.
This work was supported by National Institutes of Health Grants AI435801 and NS38037 (to H.L.W.).
Abbreviations used in this paper
- GRP78
glucose-regulated protein 78
- IGFIIR/M6PR
insulin-like growth factor II/mannose 6-phosphate receptor
- LAP
latency-associated peptide
- LC/MS
liquid chromatography/mass spectrometry
- SA
streptavidin
- shRNA
short hairpin RNA
- Treg
T regulatory cell
- VSV-G
glycoprotein of the vesicular stomatitis virus
Footnotes
The online version of this article contains supplemental material.
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Rubtsov YP, Rudensky AY. TGFβ signalling in control of T-cell-mediated self-reactivity. Nat Rev Immunol. 2007;7:443–453. doi: 10.1038/nri2095. [DOI] [PubMed] [Google Scholar]
- 2.Dubois CM, Blanchette F, Laprise MH, Leduc R, Grondin F, Seidah NG. Evidence that furin is an authentic transforming growth factor-β1-converting enzyme. Am J Pathol. 2001;158:305–316. doi: 10.1016/s0002-9440(10)63970-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Miyazono K, Ichijo H, Heldin CH. Transforming growth factor-β: latent forms, binding proteins and receptors. Growth Factors. 1993;8:11–22. doi: 10.3109/08977199309029130. [DOI] [PubMed] [Google Scholar]
- 4.Khalil N. TGF-β: from latent to active. Microbes Infect. 1999;1:1255–1263. doi: 10.1016/s1286-4579(99)00259-2. [DOI] [PubMed] [Google Scholar]
- 5.Annes JP, Munger JS, Rifkin DB. Making sense of latent TGFβ activation. J Cell Sci. 2003;116:217–224. doi: 10.1242/jcs.00229. [DOI] [PubMed] [Google Scholar]
- 6.Nakamura K, Kitani A, Strober W. Cell contact-dependent immunosuppression by CD4+CD25+ regulatory T cells is mediated by cell surface-bound transforming growth factor β. J Exp Med. 2001;194:629–644. doi: 10.1084/jem.194.5.629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Annunziato F, Cosmi L, Liotta F, Lazzeri E, Manetti R, Vanini V, Romagnani P, Maggi E, Romagnani S. Phenotype, localization, and mechanism of suppression of CD4+CD25+ human thymocytes. J Exp Med. 2002;196:379–387. doi: 10.1084/jem.20020110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N, McGrady G, Wahl SM. Conversion of peripheral CD4+CD25− naive T cells to CD4+ CD25+ regulatory T cells by TGF-β induction of transcription factor Foxp3. J Exp Med. 2003;198:1875–1886. doi: 10.1084/jem.20030152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Green EA, Gorelik L, McGregor CM, Tran EH, Flavell RA. CD4+ CD25+ T regulatory cells control anti-islet CD8+ T cells through TGF-β-TGF-β receptor interactions in type 1 diabetes. Proc Natl Acad Sci USA. 2003;100:10878–10883. doi: 10.1073/pnas.1834400100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Oida T, Zhang X, Goto M, Hachimura S, Totsuka M, Kaminogawa S, Weiner HL. CD4+CD25− T cells that express latency-associated peptide on the surface suppress CD4+CD45RBhigh-induced colitis by a TGF-β-dependent mechanism. J Immunol. 2003;170:2516–2522. doi: 10.4049/jimmunol.170.5.2516. [DOI] [PubMed] [Google Scholar]
- 11.Piccirillo CA, Letterio JJ, Thornton AM, McHugh RS, Mamura M, Mizuhara H, Shevach EM. CD4+CD25+ regulatory T cells can mediate suppressor function in the absence of transforming growth factor β 1 production and responsiveness. J Exp Med. 2002;196:237–246. doi: 10.1084/jem.20020590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kitamura T, Koshino Y, Shibata F, Oki T, Nakajima H, Nosaka T, Kumagai H. Retrovirus-mediated gene transfer and expression cloning: powerful tools in functional genomics. Exp Hematol. 2003;31:1007–1014. [PubMed] [Google Scholar]
- 13.Morita S, Kojima T, Kitamura T. Plat-E: an efficient and stable system for transient packaging of retroviruses. Gene Ther. 2000;7:1063–1066. doi: 10.1038/sj.gt.3301206. [DOI] [PubMed] [Google Scholar]
- 14.Miyazono K, Olofsson A, Colosetti P, Heldin CH. A role of the latent TGF-β 1-binding protein in the assembly and secretion of TGF-β 1. EMBO J. 1991;10:1091–1101. doi: 10.1002/j.1460-2075.1991.tb08049.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Miyazono K, Thyberg J, Heldin CH. Retention of the transforming growth factor-β 1 precursor in the Golgi complex in a latent endoglycosidase H-sensitive form. J Biol Chem. 1992;267:5668–5675. [PubMed] [Google Scholar]
- 16.Kovacina KS, Steele-Perkins G, Purchio AF, Lioubin M, Miyazono K, Heldin CH, Roth RA. Interactions of recombinant and platelet transforming growth factor-β 1 precursor with the insulin-like growth factor II/mannose 6-phosphate receptor. Biochem Biophys Res Commun. 1989;160:393–403. doi: 10.1016/0006-291x(89)91669-0. [DOI] [PubMed] [Google Scholar]
- 17.Munger JS, Harpel JG, Giancotti FG, Rifkin DB. Interactions between growth factors and integrins: latent forms of transforming growth factor-β are ligands for the integrin alphavbeta1. Mol Biol Cell. 1998;9:2627–2638. doi: 10.1091/mbc.9.9.2627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Purchio AF, Cooper JA, Brunner AM, Lioubin MN, Gentry LE, Kovacina KS, Roth RA, Marquardt H. Identification of mannose 6-phosphate in two asparagine-linked sugar chains of recombinant transforming growth factor-β 1 precursor. J Biol Chem. 1988;263:14211–14215. [PubMed] [Google Scholar]
- 19.Brunner AM, Lioubin MN, Marquardt H, Malacko AR, Wang WC, Shapiro RA, Neubauer M, Cook J, Madisen L, Purchio AF. Site-directed mutagenesis of glycosylation sites in the transforming growth factor-β 1 (TGFβ 1) and TGFβ 2 (414) precursors and of cysteine residues within mature TGFβ 1: effects on secretion and bioactivity. Mol Endocrinol. 1992;6:1691–1700. doi: 10.1210/mend.6.10.1448117. [DOI] [PubMed] [Google Scholar]
- 20.Yang Z, Mu Z, Dabovic B, Jurukovski V, Yu D, Sung J, Xiong X, Munger JS. Absence of integrin-mediated TGFβ 1 activation in vivo recapitulates the phenotype of TGFβ 1-null mice. J Cell Biol. 2007;176:787–793. doi: 10.1083/jcb.200611044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rauschert N, Brändlein S, Holzinger E, Hensel F, Müller-Hermelink HK, Vollmers HP. A new tumor-specific variant of GRP78 as target for antibody-based therapy. Lab Invest. 2008;88:375–386. doi: 10.1038/labinvest.2008.2. [DOI] [PubMed] [Google Scholar]
- 22.Tran DQ, Shevach EM. Therapeutic potential of FOXP3+ regulatory T cells and their interactions with dendritic cells. Hum Immunol. 2009;70:294–299. doi: 10.1016/j.humimm.2009.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tran DQ, Andersson J, Hardwick D, Bebris L, Illei GG, Shevach EM. Selective expression of latency-associated peptide (LAP) and IL-1 receptor type I/II (CD121a/CD121b) on activated human FOXP3+ regulatory T cells allows for their purification from expansion cultures. Blood. 2009;113:5125–5133. doi: 10.1182/blood-2009-01-199950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tran DQ, Andersson J, Wang R, Ramsey H, Unutmaz D, Shevach EM. GARP (LRRC32) is essential for the surface expression of latent TGF-β on platelets and activated FOXP3+ regulatory T cells. Proc Natl Acad Sci USA. 2009;106:13445–13450. doi: 10.1073/pnas.0901944106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stockis J, Colau D, Coulie PG, Lucas S. Membrane protein GARP is a receptor for latent TGF-β on the surface of activated human Treg. Eur J Immunol. 2009;39:3315–3322. doi: 10.1002/eji.200939684. [DOI] [PubMed] [Google Scholar]
- 26.Ochi H, Abraham M, Ishikawa H, Frenkel D, Yang K, Basso AS, Wu H, Chen ML, Gandhi R, Miller A, et al. Oral CD3-specific antibody suppresses autoimmune encephalomyelitis by inducing CD4+CD25−LAP+ T cells. Nat Med. 2006;12:627–635. doi: 10.1038/nm1408. [DOI] [PubMed] [Google Scholar]
- 27.Christis C, Lubsen NH, Braakman I. Protein folding includes oligomerization - examples from the endoplasmic reticulum and cytosol. FEBS J. 2008;275:4700–4727. doi: 10.1111/j.1742-4658.2008.06590.x. [DOI] [PubMed] [Google Scholar]
- 28.Gonzalez-Gronow M, Selim MA, Papalas J, Pizzo SV. GRP78: a multifunctional receptor on the cell surface. Antioxid Redox Signal. 2009;11:2299–2306. doi: 10.1089/ARS.2009.2568. [DOI] [PubMed] [Google Scholar]
- 29.Reddy RK, Mao C, Baumeister P, Austin RC, Kaufman RJ, Lee AS. Endoplasmic reticulum chaperone protein GRP78 protects cells from apoptosis induced by topoisomerase inhibitors: role of ATP binding site in suppression of caspase-7 activation. J Biol Chem. 2003;278:20915–20924. doi: 10.1074/jbc.M212328200. [DOI] [PubMed] [Google Scholar]
- 30.Gandhi R, Farez MF, Wang Y, Kozoriz D, Quintana FJ, Weiner HL. Cutting edge: human latency-associated peptide+ T cells: a novel regulatory T cell subset. J Immunol. 2010;184:4620–4624. doi: 10.4049/jimmunol.0903329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Luo S, Mao C, Lee B, Lee AS. GRP78/BiP is required for cell proliferation and protecting the inner cell mass from apoptosis during early mouse embryonic development. Mol Cell Biol. 2006;26:5688–5697. doi: 10.1128/MCB.00779-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fu Y, Wey S, Wang M, Ye R, Liao CP, Roy-Burman P, Lee AS. Pten null prostate tumorigenesis and AKT activation are blocked by targeted knockout of ER chaperone GRP78/BiP in prostate epithelium. Proc Natl Acad Sci USA. 2008;105:19444–19449. doi: 10.1073/pnas.0807691105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Glinka Y, Prud’homme GJ. Neuropilin-1 is a receptor for transforming growth factor β-1, activates its latent form, and promotes regulatory T cell activity. J Leukoc Biol. 2008;84:302–310. doi: 10.1189/jlb.0208090. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.