

Abstract
Chromosomal DNA polymerases are tethered to DNA by a circular sliding clamp for high processivity. However, lagging strand synthesis requires the polymerase to rapidly dissociate on finishing each Okazaki fragment. The Escherichia coli replicase contains a subunit (τ) that promotes separation of polymerase from its clamp on finishing DNA segments. This report reveals the mechanism of this process. We find that τ binds the C-terminal residues of the DNA polymerase. Surprisingly, this same C-terminal “tail” of the polymerase interacts with the β clamp, and τ competes with β for this sequence. Moreover, τ acts as a DNA sensor. On binding primed DNA, τ releases the polymerase tail, allowing polymerase to bind β for processive synthesis. But on sensing the DNA is complete (duplex), τ sequesters the polymerase tail from β, disengaging polymerase from DNA. Therefore, DNA sensing by τ switches the polymerase peptide tail on and off the clamp and coordinates the dynamic turnover of polymerase during lagging strand synthesis.
Replicases of cellular chromosomes are multiprotein machines that use a circular sliding clamp protein to attain a tight grip onto DNA for highly processive DNA synthesis (1, 2). In Escherichia coli, the processivity factor of DNA polymerase III holoenzyme, the β clamp, encircles DNA and slides along the duplex, tethering the holoenzyme to the template (3). The catalytic component within the holoenzyme is the heterotrimeric complex of α (the DNA polymerase), ε (proofreading 3′-5′ exonuclease), and θ, forming the “Pol III core” (polymerase III core) The holoenzyme contains two core polymerases that function with two β clamps for simultaneous synthesis of both leading and lagging strands of a chromosome (see Fig. 1). The strategy of using clamps generalizes to all cellular organisms. In eukaryotes, the sliding clamp is called PCNA (proliferating cell nuclear antigen) (4).
Fig. 1.

The processivity switch recycles DNA polymerase on the lagging strand. Shown in the figure is an outline of the cycle of Okazaki fragment synthesis in E. coli. (A) Architecture of the E. coli replication fork. The E. coli replisome consists of the DnaB hexameric helicase, primase, and DNA polymerase III holoenzyme. The holoenzyme contains two core polymerase/β clamp complexes for the two daughter strands and one γ complex clamp loader. The clamp loading subunits, γτ2δδ′, are arranged as a circular pentamer (χ and ψ subunits of γ complex are not shown in the figure). The τ subunits contain C-terminal extensions (τc) that bind to core and DnaB. (B) The lagging strand core finishes an Okazaki fragment, thereby triggering the τ processivity switch that ejects core from β.(C) The released core binds the new β clamp on the next primed site to start a new round of Okazaki fragment synthesis. (D Left) The polymerase C terminus binds the β clamp for high processivity. The τ switch protein recognizes that the DNA is incomplete and thus does not bind the polymerase C-tail. Only the τc of τ is shown, which extrudes up from the clamp loader (see A). (Right) The template has been fully converted to duplex DNA and τ subunit to bind the polymerase C terminus with high affinity. This action disengages core polymerase from β, allowing core to seek out a new β clamp on a RNA primed site (see A–C).
The two core polymerases within the E. coli replicase are held together by a single clamp loader complex (called γ complex), which hydrolyzes ATP to load β clamps onto DNA for both core polymerases. The clamp loader contains seven subunits (γτ2δδ′χψ), two of which (τ) bind directly to Pol III core (reviewed in ref. 5). The γ subunit and the N-terminal portions of the two τ subunits (identical to γ as explained below) bind δ and δ′, forming a circular pentamer that functions as a clamp loader (see Fig. 1) (6). The χ and ψ subunits are not required for clamp loading activity; they are involved in binding to single-stranded (SS) DNA-binding protein (SSB) and aid the polymerase-to-primase switch on the lagging strand (not shown in Fig. 1). The three ATP motor subunits (γ and two τ subunits) of the clamp loader are encoded by the same dnaX gene. The τ subunit (71 kDa) is the full-length product whereas γ (47 kDa) is truncated by a translational frameshift. Hence, τ contains an additional 24-kDa C-terminal domain relative to γ. This 24-kDa domain, referred to here as τc, is essential to E. coli and binds both DnaB helicase and Pol III core (7–11). As illustrated in Fig. 1 A, the C-terminal 24-kDa domains of the τ subunits organize the replisome by connecting two core polymerases to the clamp loader and by forming contacts to the hexameric DnaB helicase, which encircles the lagging strand and unwinds the parental duplex. The C-terminal 24-kDa domain of τ, referred to in this report as τc, is not required for actual clamp loading and seems to be connected to the rest of the protein by a flexible tether (5, 12).
The picture of a DNA polymerase held tightly to DNA by a protein ring fits nicely with action on the leading strand in which the core polymerase and β simply extend DNA continuously in one direction. However, a polymerase held tight to DNA by a protein ring is inconsistent with polymerase action on the lagging strand, where the direction of DNA synthesis is opposite that of the leading strand. As the helicase drives forward and unwinds the parental duplex, ssDNA template is generated on the lagging strand. Synthesis of the lagging strand is initiated by primase, a specialized RNA polymerase that produces short RNA primers that are subsequently extended by the lagging strand core polymerase (Okazaki fragments). Hence, as the lagging strand DNA polymerase extends the RNA primer, the duplex product is brought along with the replication fork machinery, producing a DNA loop (Fig. 1 A). As lagging strand synthesis proceeds, continued progression of the replication fork produces yet more ssDNA on the lagging strand, which must be primed and filled in. On completing each fragment, the polymerase must dissociate from the DNA (Fig. 1B) and cycle back to the next RNA primer to begin extension of another fragment (Fig. 1C). The cycle of synthesis and hopping between Okazaki fragments by Pol III repeats itself every 1–3 seconds, ≈3,000 times per chromosome division (13).
An early study on how the tightly bound Pol III holoenzyme could rapidly dissociate from one DNA and transfer to a new primed site showed that the enzyme was capable of this, despite its grip to DNA, provided two criteria were met. First, replication of the initial template must be completed and, second, a β clamp needed to be attached to the primed site that the holoenzyme was to transfer to (14). Having met these criteria, the polymerase transfers from finished DNA to another primed template within 1 s or less. Further study revealed that the initial β clamp was left behind on the completed DNA, and that polymerase dissociation from β occurred within 1 s on finishing the template (13). Hence, the lagging strand polymerase hops from one β clamp to the next as illustrated in Fig. 1 A–C. The kinetics of polymerase transfer to a new clamp fit nicely within the time frame of the Okazaki fragment cycle and in vitro, and studies of replication forks demonstrated that the single γ complex within the holoenzyme repeatedly loads β onto the lagging strand during replication (11). However, the mechanism by which the polymerase achieved the knowledge that replication was complete, and how it coupled this intelligence to dissociation from β and DNA, remained unknown.
The ability of a highly processive polymerase to become suddenly distributive and dissociate from DNA is referred to as a “processivity switch.” A recent study (12) of this switch within Pol III holoenzyme revealed that the τ subunit, in addition to the polymerase and β, is needed for the switch to occur. That study also demonstrated that the τc section of τ is sufficient to elicit the processivity switch. τc can bind DNA, and it was suggested that τ may sense the difference in structure between primed DNA and completed DNA and may couple this DNA sensing to separation of the polymerase from the clamp. However, core polymerase and β also bind DNA, and, therefore, the DNA sensing role could reside in any of these proteins.
The present study demonstrates that the DNA sensor of the processivity switch is contained entirely within τ and reveals the detailed mechanism by which it parts the polymerase from the clamp in response to DNA structure. We demonstrate here that τ binds the extreme C terminus of the polymerase and that this same C-terminal tail of the polymerase is an essential attachment site to the β clamp. The τ subunit completes with β for this polymerase C-terminal tail. The winner of the competition is decided by the structure of DNA bound to the τ subunit. With primed DNA bound, τ loses affinity for the polymerase C-tail, thereby allowing the polymerase to function with β for processive DNA synthesis. The completed duplex DNA no longer holds τ back, and it binds the polymerase C-tail, displacing it from the β clamp.
Experimental Procedures
Materials. A C-terminal 20-residue deletion of α was constructed by recombinant methods, purified, and combined with ε and θ to reconstitute core Δ20, followed by purification from unbound subunits as described (15). The τC domain was purified as described (12). [32P]β contains a six-residue C-terminal tag that can be phosphorylated by using protein kinase as described (16). Peptides (HPLC-purified) were purchased from Biosynthesis (Lewisville, TX). Labeled nucleotides were from Dupont-NEB, and unlabeled nucleotides were from Pharmacia-LKB. Buffer A contains 20 mM Tris·HCl (pH 7.5), 0.5 mM EDTA, 2 mM DTT, and 10% glycerol. Superdex-12 gel filtration buffer contains 20 mM Tris·HCl (pH 7.5), 0.5 μM EDTA, 2 mM DTT, 4% glycerol, and 100 mM NaCl.
β-Dependent Replication Assays Using Primed M13mp18 ssDNA. Replication assays were assembled on ice and contained 1 nM singly primed M13mp18 circular ssDNA, 0.42 μM ssDNA-binding protein (as tetramer), 2 nM γ-complex (γ3δδ′χψ), and 30 nM β in 25 μl of 20 mM Tris·Cl (pH 7.5), 0.1 mM EDTA, 4% glycerol, 40 μg/ml BSA, 5 mM DTT, 8 mM MgCl2, 0.5 mM ATP, 60 μM each of dGTP, dATP, dTTP and dCTP, and 20 μM [α-32P]dTTP. Reactions were brought to 37°C, and replication was initiated on addition of core or core Δ20. After 30 s at 37°C, reactions were quenched on adding 25 μl of 1% SDS and 50 mM EDTA. A 15-μl sample was analyzed for nucleotide incorporation by spotting onto DE81 paper filter paper as described (17). Peptide inhibition assays were performed in the same fashion except the α C-tail 20-mer peptide was present before addition of the polymerase. Peptide was at concentrations of either 34 μM, 68 μM, 272 μM, or 544 μM.
β-Independent DNA Synthesis Assays. Assays contained 2.5 μg of activated calf thymus DNA and 0.2 μg of either core or core Δ20 in a final volume of 25 μl of 20 mM Tris·HCl (pH 7.5), 8 mM MgCl2, 5 mM DTT, 0.5 mM EDTA, 40 μg/ml BSA, 4% glycerol, 1 mM ATP, 60 μM each dCTP, dGTP, and dATP, and 20 μM [α-32P]dTTP. Assay mixtures were assembled on ice followed by incubation at 37°C for the indicated time. DNA synthesis was quantitated by using DE81 paper as described above.
Protein–Protein Interaction Analysis by Gel Filtration. Gel filtration analysis of protein mixtures was performed by using a Fast Protein Liquid Chromatography Superose 12 column equilibrated in buffer A containing 100 mM NaCl. Analysis of β interaction with core was performed by mixing 6.5 μM core or core Δ20 with 6.5 μM β in 100 μl buffer A containing 100 mM NaCl. Analysis of τ interaction with core used 6.5 μM core or core Δ20 with 26 μM τ (as monomer) in 100 μl of buffer A containing 100 mM NaCl. Protein mixtures were incubated at 15°C for 30 min, and then the sample was injected onto the column. After the first 6.6 ml, fractions of 170 μl were collected. Fractions were analyzed in 10% SDS/polyacrylamide gels stained with Coomassie Blue.
Protein–Protein Interaction Analysis by PAGE Mobility Shift. Protein mobility shift assays contained 90 nM [32P]β and 506 nM Pol III core in 15 μl of buffer A containing 100 mM NaCl and 50 μg/ml BSA for 4 min at 37°C. When present, the α C-tail 20-mer peptide was added to a concentration of either 2.5, 4.8, 9.5, 19, 38, 76, 153, 306, or 613 μM. Five microliter aliquots were analyzed in a 4% native polyacrylamide gel developed in TBE buffer (90 mM Tris/90 mM boric acid/2.5 mM EDTA, pH 8.3) at 17 mA and 22°C. Gels were dried and then analyzed by using a PhosphorImager (Molecular Dynamics).
Microtiter Protein-Binding Assays. N-terminal biotinylated 20-mer peptides were diluted in PBS (30 μl) and incubated in 96-well microtiter plates for 1 h at 23°C. After washing three times with PBS, 44 nM [32P]β (and τ at either 0, 0.8, 1.75 or 3.5 μM) added in 30 μl Buffer A plus 4% glycerol and 40 μg/ml BSA. When present either 50 μM gel purified primed template (62-mer:31-mer, (12)) or double strand 62-mer was added along with [32P]β. Plates were incubated for 1h, then washed, dried and analyzed by using a PhosphorImager (Molecular Dynamics).
Fluorescence Measurements. The β subunit can be uniquely labeled at Cys-333 by using maleimide derivatives (18). β (3 mg) was labeled by using Oregon Green 488 maleimide (Molecular Probes) in 1 ml of 50 mM potassium phosphate (pH 7.5) and 100 mM NaCl. The Oregon Green maleimide (90 nmol) was dissolved in 100 μl of DMF; then, 80 μl was added to β with gentle stirring at 4°C, followed by overnight incubation at 4°C in the dark. βog was separated from unreacted reagent on a 50-ml column of BioGel 6 and contained ≈0.91 molecules of Oregon Green per β monomer as determined from protein absorbance at 280 nM (ε280 = 14,890 M–1·cm–1) and Oregon Green at 490 nm (ε491 = 76,000 M–1·cm–1).
Titration of wild-type core or core Δ20 into βog was performed as follows. Reactions contained 50 nM β2og and the indicated amount of core or core Δ20 in 60 μl of 20 mM Tris·Cl (pH 7.5), 0.5 mM EDTA, 1 mM DTT, and 50 mM NaCl, on ice. Reactions were shifted to 22°C for 15 min; then, 51 μl was placed in a 3 × 3-mm cuvette. Excitation was at 490 nm, and emission was monitored from 500–600 nm in a PTI (South Brunswick, NJ) Quantamaster spectrofluorimeter. Fluorescence emission at 517 nm was used for analysis. Data points were fit according to the model, A + B ↔ AB by using origin software (Microcal Software, Northampton, MA). Titrations using the N-terminal rhodamine (TAMRA)-labeled α C-tail 20-mer peptide contained 1 μM peptide and unlabeled β subunit, or 0.5 μM peptide and τ subunit, as indicated. When present, the synthetic gel-purified-primed template (62-mer:31-mer) or 62-mer homoduplex was added to a concentration of 2.5 μM. Excitation was at 545 nm, and emission was monitored from 550–630 nm. Fluorescence emission at 577 nm was used for analysis as described above.
Results
The C-Terminal Tail of the Polymerase Interacts with β. To determine the precise location of the contact site between β and the α polymerase subunit of the heterotrimeric Pol III core (αεθ), we synthesized an overlapping set of peptides (20-mers) that spanned the region of α (812–991) previously reported to bind β (19, 20). Peptides were immobilized via N-terminal biotin to streptavidin-coated microtiter plates and then assayed for the ability to retain [32P]β in the well. The results, in Fig. 2A, showed no interaction strong enough to retain [32P]β. Next, we tested N- and C-terminal peptides and found a strong positive signal with the peptide corresponding to the C-terminal 20 residues of the α protein (Fig. 2 A). This peptide (see Fig. 2B) contains a sequence (QVELEFD) reminiscent of a recently proposed β-binding motif (QLxLF) found in other proteins that interact with β (19). It should be noted that the possibility exists that secondary structures required for protein–protein interactions are missing in the peptides spanning the internal regions of the α protein predicted by the earlier studies, and therefore other contact sites between α and β besides the C terminus of α could have evaded detection in our assay.
Fig. 2.

The α C terminus of Pol III core binds to the β clamp. (A) N-terminal biotinylated 20-mer peptides corresponding to an internal region and the N and C termini of α (as indicated by shading) were attached to streptavidin-coated microtiter plates and assayed for binding to [32P]β.(B) Sequence corresponding to the C-terminal 20 residues of α. (C) Use of rhodamine-labeled α C-tail 20-mer to measure the KD of interaction with the β clamp by fluorescence. (D) Native PAGE mobility shift assay of [32P]β interaction with core. Lanes 4–12 contain α C-tail peptide at either 2.5, 4.8, 9.5, 19, 38, 76, 153, 306, or 613 μM. (E) The α C-tail peptide inhibits β-dependent replication of singly primed M13mp18 ssDNA catalyzed by core polymerase, β, and γ complex.
Next, we quantitated the interaction by fluorescence using the α C-tail peptide labeled with rhodamine at the N terminus. In Fig. 2C, β was titrated into the rhodamine-labeled α C-tail peptide, and the resulting fluorescent enhancement indicated an interaction with a Kd value of ≈3 μM. To further examine the functional relevance of the α C-tail interaction with β, we asked whether the α C-tail peptide could displace wild-type core from β. Pol III core shifts the position of [32P]β in a native polyacrylamide gel, as demonstrated in Fig. 2D (compare lanes 1 and 2). Titration of the α C-tail peptide into the reaction results in dissociation of [32P]β from core (lanes 4–12). This result indicates that the C-terminal sequence of α is required for attachment of core to β because occupation of the peptide-binding site on β by the α C-tail 20-mer peptide prevents core-β complex formation. Further, the α C-tail peptide inhibits β dependent replication by core in the singly primed M13mp18 ssDNA replication assay (Fig. 2E).
In the experiments of Fig. 3, we removed the C-terminal 20 residues of the α polymerase subunit (αΔ20) of Pol III core by using recombinant methods and compared it with wild-type core in ability to function with β. The αΔ20 mutant was combined with the ε 3′-5-exonuclease and the θ subunit to reconstitute core, and the resulting “core Δ20” heterotrimer was purified from excess subunits. The core Δ20 was as active as wild-type core in a simple gap-filling assay on activated calf thymus DNA (Fig. 3A). Additional characterization of wild-type core and core Δ20 showed that they have a similar KM value for dNTP, and Kd value for primed DNA template (data not shown). Next, the activity of core and core Δ20 were compared on an ssDNA-binding protein-coated, singly primed M13mp18 circular ssDNA template. Pol III core requires the β clamp and γ complex clamp loader for activity on this substrate. The result shows that core Δ20 is substantially inactive in this assay (Fig. 3B), suggesting that core Δ20 has lost the ability to functionally interact with β.
Fig. 3.

The C-terminal polymerase tail is essential to function with β. Comparison of core and core Δ20 activity on activated calf thymus DNA (A) and singly primed and ssDNA-binding protein-coated M13mp18 ssDNA (B) in the presence of β and the γ complex clamp loader. (C) Analysis of β interaction with core and core Δ20 by gel filtration on an FPLC Superose 12 column. Shown are reaction mixtures contained β plus either (Top to Bottom) core, core Δ20, or no core. Column fractions were analyzed in an SDS/10% polyacrylamide gel. (D) Fluorescence determination of the KD value for β interaction with either core or core Δ20.
Next, we examined core Δ20 for its ability to bind β. Core Δ20 was mixed with β, and interaction between them was analyzed by gel filtration on a Superose 12 sizing column (Fig. 3C). The Middle panel shows that β does not comigrate with core Δ20 but instead elutes in the position of free β (Bottom). The control, using wild-type core (Top), shows that β comigrates with core. Inability of core Δ20 to bind β indicates that the C-terminal residues of α are indeed important to the interaction of core with β.
The above results demonstrate that the polymerase requires the C-terminal residues to bind and function with β. The Kd value of peptide binding to β accounts for ≈74% (Kd ≈ 4 μM, 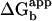 ≈ 7.36 kcal/mol) of the energy of interaction between core and β (Kd ≈ 50 nM,
≈ 7.36 kcal/mol) of the energy of interaction between core and β (Kd ≈ 50 nM, 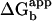 ≈ 9.95 kcal/mol) (12). In Fig. 3D, we compared the affinity of core Δ20 and core to bind β tagged with a fluorescent reporter. As expected, the affinity of core Δ20 for β is significantly reduced relative to wild-type core.
≈ 9.95 kcal/mol) (12). In Fig. 3D, we compared the affinity of core Δ20 and core to bind β tagged with a fluorescent reporter. As expected, the affinity of core Δ20 for β is significantly reduced relative to wild-type core.
The C Terminus of the Polymerase also Interacts with the τ Subunit.
We next asked whether the α C-terminal tail is a target of the τ subunit processivity switch protein. For example, τ may somehow block the α C terminus from binding β in response to polymerase finishing a DNA template, thus separating the polymerase from its clamp. τ could do so either by steric occlusion (e.g., driving a wedge between β and α), or perhaps by directly binding the C-terminal tail of α. To examine these issues, we determined whether τ directly binds the α C-tail peptide, and whether τ has a lower affinity for core Δ20 compared with wild-type core. First, we analyzed the interaction of τ with core and core Δ20 by gel filtration on a Superose 12 column. The result, in Fig. 4, demonstrates that wild-type core forms a stable complex with τ, but core Δ20 does not. This finding is consistent with an earlier study (20) showing that a mutant α subunit lacking 48 C-terminal residues could not form a stable complex with τ. To measure the affinity of τ to the α C-tail peptide, we used the 24-kDa C-terminal section of τ (τc), which contains the domain responsible for binding α. In Fig. 4B, τc is titrated into the N-terminal-labeled rhodamine α C-tail peptide. The result demonstrates that τc binds this peptide with a Kd value of 350 nM, ≈10-fold tighter than β. Hence, it seems likely that τ could out-compete β for attachment to the polymerase tail, an action at the center of the processivity switch.
Fig. 4.

The τ subunit binds the polymerase C-tail, and the strength is modulated by DNA. (A) Interaction of τ with core (Upper gel) and lack thereof by using core Δ20 (Lower gel) as analyzed by gel filtration using a Superose 12 column. The affinity of τc for the N-terminal rhodamine labeled α C-tail 20-mer peptide was determined by fluorescence by using no DNA (B), primed DNA (C), or duplex DNA (D). τ was 0, 0.2, 0.4, 0.6, 0.8, 1.0, 1.2, 1.6, 2.0, or 3.0 μM. E and F demonstrate that DNA has little effect on the affinity of β for the α C-tail peptide. Reactions were performed as in C and D, respectively, except that β (at 0, 0.3, 0.7, 1.1, 1.5, 2.6, 3.5, 4.5, or 8 μM) replaced τc. (G) To determine whether τ and β compete for the α C-tail, biotinylated α C-tail 20-mer was immobilized in wells of a strepavidin-coated 96-well plate and then incubated with [32P]β (44 nM) and τ (0, 0.8, 1.75, or 3.5 μM) before washing the wells and examining them by PhosphorImager. The synthetic primed template abrogates the ability of τ to displace [32P]β from the α C-tail (Middle). Double-stranded DNA has little effect on the competition (Bottom).
If this τ-α C-tail interaction is a centerpiece of the processivity switch, two predictions should be upheld. One is that τ should compete with β for this α C-tail peptide, and the other is that the affinity of τ for the α C-tail should be regulated by DNA structure in the same manner as the processivity switch. Specifically, primed DNA should decrease the interaction between τ and the α C-tail in order that the α C-tail of the polymerase can bind to β for processive Okazaki fragment extension. On finishing replication to produce duplex DNA, τ should regain affinity for the α C-tail, thereby disrupting the α-β interaction and causing core polymerase to dissociate from β and DNA.
We used the rhodamine-labeled α C-tail peptide to determine whether primed DNA or duplex DNA affects the affinity of τc for the α C-tail peptide. The presence of primed DNA resulted in a large decrease (>20-fold) in affinity of τc for the α C-tail peptide (Fig. 4C). This result is in keeping with the behavior expected for the processivity switch. In the presence of duplex DNA, τc retained tight affinity for the α C-tail peptide, again consistent with the predicted behavior of a processivity switch (Fig. 4D). To determine whether this DNA effect is specific to τ, we examined the effect of DNA on β interaction with the α C-tail peptide. In Fig. 4 E and F, β was titrated into the rhodamine-labeled α C-tail peptide in the presence of the primed template or duplex DNA. The results show that neither DNA structure has a significant effect on the strength of interaction between β and the α C-tail peptide. Next, we turned our attention to the question of whether τ competes with β for the α C-tail peptide.
The τ Subunit Senses DNA Structure and Generates a Processivity Switch. To examine whether β and τ compete for the α C-tail peptide, the N-biotinylated α C-tail peptide was immobilized to wells of a streptavidin-coated 96-well plate. The wells were then treated with [32P]β, followed by addition of increasing concentrations of τ, and then washed. If τ competes with β for the α C-tail peptide, [32P]β should be displaced and washed out of the wells. The results of the experiment, in the Top row of Fig. 4G demonstrate that addition of τ indeed results in loss of [32P]β from the wells, consistent with direct competition between τ and β for the α C-tail.
A critical characteristic of the processivity switch is its regulation by DNA structure. A primed template suppresses the switch, whereas a completed duplex DNA structure does not. The results of Fig. 4 demonstrated that these DNA structures modulate the affinity of τ for the α C-tail peptide in a way that is consistent with the processivity switch. Hence, one may expect that repeating the competition experiment in the presence of a primed template should result in negating the ability of τ to displace [32P]β from the wells. The results show that the primed template indeed prevents τ from displacing [32P]β from the wells (Fig. 4G Middle). Finally, the experiment was repeated by using duplex DNA. Now, the results show that τ maintains the ability to compete [32P]β from the α C-tail peptide (Fig. 4G Bottom). Hence, different DNA structures modulate the effectiveness of τ as a competitor for the β-α C-tail interaction, precisely as expected for the workings of a processivity switch.
Discussion
This report investigates the mechanism by which the τ subunit regulates association of the polymerase with the β clamp. We find that τ binds the extreme C-terminal residues of the polymerase (α subunit), and that the affinity of τ for this polymerase C-tail peptide is modulated by DNA bound to τ. The same α C-tail peptide also forms a necessary contact to the β clamp. The τ subunit competes with β for the polymerase C-tail. τ senses a primed template structure and lowers its affinity to the polymerase C-tail, allowing the polymerase to remain attached to the β sliding clamp for high processivity (Fig. 1D Left). On completion of an Okazaki fragment, the τ subunit senses that the DNA is finished and binds the polymerase C-tail peptide with high affinity, thereby sequestering this critical polymerase contact away from the β clamp and displacing the polymerase from β and DNA (Fig. 1D Right). Release of the polymerase from DNA allows it to seek out a new primed site that will suppress the τ peptide switch and free the polymerase C-tail for interaction with a new β clamp to begin the next Okazaki fragment.
The competition between τ and β for the polymerase C-tail would, at first glance, suggest that τ releases from the polymerase whereas it functions with β, thereby uncoupling the polymerases from one another at the replication fork. However, previous studies (12, 21) have demonstrated that τ remains attached to the polymerase–β complex on primed DNA, consistent with studies (20, 22) indicating the presence of multiple contact points between τ and the polymerase (see Fig. 1D). Moreover, τ stimulates β-dependent synthesis by core on primed DNA, supporting the observation that τ attaches to the core–β complex on the DNA substrate (23). In the absence of DNA, τ does not bind tightly to the polymerase lacking the C-terminal residues, indicating that the other site(s) of attachment between α and τ require DNA. The exact nature and location of this DNA-induced interaction between τ and core is currently under investigation.
Interaction between the C terminus of a DNA polymerase with its clamp has been observed in the T4 system (24). The cocrystal structure of the clamp of the T4 relative, RB69 phage (25) with a C-terminal peptide derived from the polymerase, reveals a striking structural similarity to the way the E. coli β clamp binds to the δ wrench subunit of γ complex (26), and to the structure of the human proliferating cell nuclear antigen clamp in complex with the p21 cell cycle inhibitor (27). The β, T4 gp45 and proliferating cell nuclear antigen sliding clamp structures are all similar to one another, and thus these observations suggest a common method by which diverse proteins bind to sliding clamps. It therefore seems quite likely that the α C-tail peptide of E. coli Pol III core binds to the same hydrophobic pocket of β as observed in the β·δ complex, as α and δ compete for β (28). Indeed, even the 20-mer α C-tail peptide used in this study competes with δ for β, further suggesting a same site location for these ligands on β (data not shown).
The critical nature of the interaction of the α C-tail with β, and of δ with β, combined with the fact that these interactions are relatively weak protein–protein interactions, suggests that this site on β may be a prime location for a small molecule inhibitor of replication. Indeed, as shown here, the small α C-tail peptide inhibits β-dependent DNA synthesis in vitro.
All cellular organisms use a sliding clamp–DNA polymerase complex as a processive unit for chromosome replication. Furthermore, replication in all cells is thought to occur in a semidiscontinuous fashion, where leading strand synthesis is continuous and the lagging strand is synthesized discontinuously. Processivity clamps and clamp loaders are conserved across all domains of life. Hence, the need for a processivity switch would seem to generalize to other organisms. It seems quite possible therefore that other chromosomal replicases may employ a peptide switch protein that acts like E. coli τ to disengage the polymerase from its clamp on completing lagging strand fragments.
Numerous proteins are known to bind to the β and proliferating cell nuclear antigen clamps (29, 30). Several of these clamp interactive proteins are involved in DNA repair, DNA modification, or cell cycle control. Others include different types of DNA polymerases, such as those that bypass lesions (31, 32). It is thought that, in some cases, two or more clamp interactive proteins act with the same clamp, but at different times. A current topic of interest is how the action of different proteins with the same clamp is orchestrated. This study on the τ peptide switch has implications for directing protein traffic flow on clamps. For example, β is known to interact with Pol I and ligase (33). These proteins are needed sequentially after an Okazaki fragment is complete, at which time Pol I replaces the RNA with DNA, and ligase joins the fragments. The τ switch not only clears Pol III from β but does so precisely where it is needed for action with Pol I and ligase. Moreover, interaction of various polymerases and repair factors with clamp proteins will result in a change of DNA structure (e.g., the DNA will undergo repair, modification, or extension). It seems likely that these proteins will also need to disengage from the clamp when the job at hand is complete. In light of the studies reported herein, one may anticipate that some of these processes may also employ a switch that responds to DNA structure, like the peptide switch within DNA polymerase III holoenzyme described here.
Acknowledgments
This work was supported by the Howard Hughes Medical Institute and by National Institutes of Health Grant GM38839.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: Pol III core, polymerase III core; ssDNA, single-stranded DNA.
References
- 1.Kornberg, A. & Baker, T. (1992) DNA Replication (Freeman, New York), 2nd Ed.
- 2.Kelman, Z. & O'Donnell, M. (1995) Annu. Rev. Biochem. 64, 171–200. [DOI] [PubMed] [Google Scholar]
- 3.Kong, X. P., Onrust, R., O'Donnell, M. & Kuriyan, J. (1992) Cell 69, 425–437. [DOI] [PubMed] [Google Scholar]
- 4.Baker, T. A. & Bell, S. P. (1998) Cell 92, 295–305. [DOI] [PubMed] [Google Scholar]
- 5.O'Donnell, M., Jeruzalmi, D. & Kuriyan, J. (2001) Curr. Biol. 11, R935–R946. [DOI] [PubMed] [Google Scholar]
- 6.Jeruzalmi, D., O'Donnell, M. & Kuriyan, J. (2001) Cell 106, 429–441. [DOI] [PubMed] [Google Scholar]
- 7.Blinkova, A., Hervas, C., Stukenberg, P. T., Onrust, R., O'Donnell, M. E. & Walker, J. R. (1993) J. Bacteriol. 175, 6018–6027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim, S., Dallmann, H. G., McHenry, C. S. & Marians, K. J. (1996) J. Biol. Chem. 271, 21406–21412. [DOI] [PubMed] [Google Scholar]
- 9.Onrust, R., Finkelstein, J., Naktinis, V., Turner, J., Fang, L. & O'Donnell, M. (1995) J. Biol. Chem. 270, 13348–13357. [DOI] [PubMed] [Google Scholar]
- 10.Gao, D. & McHenry, C. S. (2001) J. Biol. Chem. 276, 4441–4446. [DOI] [PubMed] [Google Scholar]
- 11.Yuzhakov, A., Turner, J. & O'Donnell, M. (1996) Cell 86, 877–886. [DOI] [PubMed] [Google Scholar]
- 12.Leu, F. P., Georgescu, R. & O'Donnell, M. (2003) Mol. Cell 11, 315–327. [DOI] [PubMed] [Google Scholar]
- 13.Stukenberg, P. T., Turner, J. & O'Donnell, M. (1994) Cell 78, 877–887. [DOI] [PubMed] [Google Scholar]
- 14.O'Donnell, M. E. (1987) J. Biol. Chem. 262, 16558–16565. [PubMed] [Google Scholar]
- 15.Studwell-Vaughan, P. S. & O'Donnell, M. (1993) J. Biol. Chem. 268, 11785–11791. [PubMed] [Google Scholar]
- 16.Kelman, Z., Naktinis, V. & O'Donnell, M. (1995) Methods Enzymol. 262, 430–442. [DOI] [PubMed] [Google Scholar]
- 17.Studwell-Vaughan, P. S. & O'Donnell, M. (1991) J. Biol. Chem. 266, 19833–19841. [PubMed] [Google Scholar]
- 18.Griep, M. A. & McHenry, C. S. (1988) Biochemistry 27, 5210–5215. [DOI] [PubMed] [Google Scholar]
- 19.Dalrymple, B. P., Kongsuwan, K., Wijffels, G., Dixon, N. E. & Jennings, P. A. (2001) Proc. Natl. Acad. Sci. USA 98, 11627–11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim, D. R. & McHenry, C. S. (1996) J. Biol. Chem. 271, 20690–20698. [DOI] [PubMed] [Google Scholar]
- 21.Stukenberg, P. T. & O'Donnell, M. (1995) J. Biol. Chem. 270, 13384–13391. [DOI] [PubMed] [Google Scholar]
- 22.Kim, D. R. & McHenry, C. S. (1996) J. Biol. Chem. 271, 20699–20704. [DOI] [PubMed] [Google Scholar]
- 23.Maki, S. & Kornberg, A. (1988) J. Biol. Chem. 263, 6561–6569. [PubMed] [Google Scholar]
- 24.Berdis, A. J., Soumillion, P. & Benkovic, S. J. (1996) Proc. Natl. Acad. Sci. USA 93, 12822–12827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shamoo, Y. & Steitz, T. (1999) Cell 99, 155–166. [DOI] [PubMed] [Google Scholar]
- 26.Jeruzalmi, D., Yurieva, O., Zhao, Y., Young, M., Stewart, J., Hingorani, M., O'Donnell, M. & Kuriyan, J. (2001) Cell 106, 417–428. [PubMed] [Google Scholar]
- 27.Gulbis, J. M., Kelman, Z., Hurwitz, J., O'Donnell, M. & Kuriyan, J. (1996) Cell 87, 297–306. [DOI] [PubMed] [Google Scholar]
- 28.Naktinis, V., Turner, J. & O'Donnell, M. (1996) Cell 84, 137–145. [DOI] [PubMed] [Google Scholar]
- 29.Warbrick, E. (2000) BioEssays 22, 997–1006. [DOI] [PubMed] [Google Scholar]
- 30.Lenne-Samuel, N., Wagner, J., Etienne, H. & Fuchs, R. P. (2002) EMBO Rep. 3, 45–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tang, M., Shen, X., Frank, E. G., O'Donnell, M., Woodgate, R. & Goodman, M. F. (1999) Proc. Natl. Acad. Sci. USA 96, 8919–8924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tang, M., Pham, P., Shen, X., Taylor, J. S., O'Donnell, M., Woodgate, R. & Goodman, M. F. (2000) Nature 404, 1014–1018. [DOI] [PubMed] [Google Scholar]
- 33.López de Saro, F. J. & O'Donnell, M. (2001) Proc. Natl. Acad. Sci. USA 98, 8376–8380. [DOI] [PMC free article] [PubMed] [Google Scholar]