

Abstract
A patient with Budd-Chiari syndrome who underwent orthotopic liver transplantation and developed recurrent disease is described. The immediate postoperative period was complicated by multiple thrombotic episodes, followed by a period of apparent remission associated with the initiation of coumadin and persantine therapy. After discontinuation of such antithrombotic therapy in order to biopsy the liver, the patient experienced another series of clinically overt vascular thromboses and ultimately died of sepsis 15 mo posttransplantation after a prolonged and complicated terminal hospital course. At autopsy, recurrent Budd-Chiari syndrome as well as thromboses in numerous other organs was demonstrated.
The Budd-Chiari syndrome (1,2) is a rare condition characterized by occlusion of the major hepatic veins. It is characterized by hepatomegaly, progressive ascites, and distension of the veins of the abdominal wall. There are a variety of conditions that are known to predispose the individual to the Budd-Chiari syndrome. These include hepatic tumor, membranous obstruction of the inferior vena cava at the level of the hepatic vein ostia (3), estrogen therapy, oral contraceptives, and disease of hematopoietic stem cells such as polycythemia rubra vera, idiopathic myelofibrosis, and paroxysmal nocturnal hemoglobinuria. Often, however, no underlying cause is apparent. Thrombosis in one or more extrahepatic sites is not uncommon in such cases. Obstruction of the smaller hepatic veins, without large hepatic vein involvement is known to occur also and has been termed “venoocclusive disease.” This latter condition is thought to be a proliferative rather than a thrombotic process. Chemotherapeutic agents (4) and ingestion of plant alkaloids (5) are factors that are known to produce such vascular lesions. With conventional therapy, patients with either condition generally have a poor prognosis. Therefore, liver transplantation potentially is an attractive alternative treatment option which might be considered, at least in some, and actually has been performed in a few cases (5,6). Because of the idiopathic nature of most cases or the persistent presence of an underlying, predisposing disease being unaffected by hepatic transplantation, recurrent Budd-Chiari syndrome in the transplanted liver is a theoretical possibility. Herein, we report such a case.
Report of a Case
A 21-yr-old white female was admitted to Presbyterian-University Hospital in April 1981 for a follow-up visit and liver biopsy 1 yr after a liver transplant for Budd-Chiari syndrome. The original diagnosis of Budd-Chiari syndrome was made in 1975 when she presented with ascites. A diagnostic evaluation consisting of radiologic efforts to visualize the hepatic veins and inferior vena cava failed to identify the hepatic veins but demonstrated an otherwise normal inferior vena cava. Other studies included six separate platelet counts, all of which were normal (mean ± SEM: 247,000 ± 32,000/mm3), numerous determinations of hemoglobin (12.6 ± 0.3 g/dl), and multiple white blood cell count determinations (8.6 ± 0.2 × 103 cells/mm3), which were also normal. Similarly, standard coagulation studies were performed and were found to be normal. Initial limited therapeutic success was achieved with a side-to-side portocaval shunt and the placement of several LeVeen shunts. Ultimately she failed to respond, despite such therapies, and developed progressive ascites and very large bilateral pleural effusions. As a last resort, she received an orthotopic liver transplant at the University of Colorado Medical Center in March 1980.
The donor was a young, healthy adult who died of a gunshot wound and had no known history of clotting abnormalities.
Of interest is the fact that a 675-g congested spleen with scattered, small foci of erythroblasts and rare megakaryocytes in the red pulp was removed from the recipient at the time of transplantation.
Her postoperative course was prolonged and complicated with severe adult respiratory distress syndrome, small bowel obstruction, recurrent mesenteric venous thromboses with intestinal infarction, multiple pulmonary emboli, deep vein thrombosis of the legs, and thrombosis of the superior vena cava as well as the right and left subclavian veins. Because of these numerous thrombotic vascular complications, a repeat hematologic evaluation was initiated again and resulted only in the further documentation of normal values for hemoglobin, white blood cell and platelet counts, prothrombin, partial thromboplastin, and clotting times. She was placed on Coumadin and persantine. She had had no previous history of major illness or hospitalization. Medication at the time of her final admission included cyclosporin A, prednisone, coumadin, cimetidine, and persantine. Exercise tolerance was limited by fatigue and shortness of breath after ascent of one flight of stairs. On physical examination, she was noted to have decreased breath sounds at the right lung base, prominent venous collaterals over the anterior thorax, a 10-cm liver span in the midclavicular line, and multiple incisional hernias. There was no ascites. The hemoglobin was 14.9 gm/dl, hematocrit 46.2%, white blood cells 17,500/mm3, and platelets 1,190,000/mm3. Gamma glutamyl transpeptidase was 5.6 times the upper limit of normal, while the serum glutamic oxaloacetic transaminase (SGOT), serum glutamic pyruvic transaminase (SGPT), and bilirubin levels were all within normal limits. The prothrombin time (PT) was 14.6 s (control 11.0 s), and activated partial thromboplastin time was 35.6 s (control 25.1 s). A large right-sided pleural effusion was noted on chest x-ray.
Examination of her peripheral blood smear demonstrated a striking thrombocytosis with numerous visible large and occasionally gigantic platelets. However, no megakaryocyte fragments were seen. The red blood cells appeared normal; specifically no target, oval, polychromatic, macrocytes, or teardrop cells were seen. The neutrophils were moderately hypersegmented and basophils were increased in number.
Complete coagulation study on the second hospital day revealed a blocking or “lupus-like” inhibitor of the PT and APTT systems, normal to elevated levels of antithrombin III, and no coumadin effect (i.e., normal levels of vitamin K-dependent factors) despite the use of coumadin 5 mg/day. As a result of these findings and in anticipation of doing a liver biopsy, the coumadin and persantine were discontinued. Bone marrow aspirate and biopsy revealed a normocellular (~50:50 ratio of fat/cells) marrow with increased number of megakaryocytes but with a completely normal appearing morphology of all the cellular elements. There was no increase in fibroblasts, and reticulum stains revealed normal amounts of reticulum material. The bone marrow aspirate and biopsy specimen were interpreted as being nondiagnostic but not incompatible with a very early phase of idiopathic myelofibrosis. The leukocyte alkaline phosphatase was elevated at 190 (nl 40–100). The bone marrow was cultured and found to be Philadelphia chromosome negative. The marrow aspirate also was cultured and studied for the production of colonies of cells grown in methylcellulose. Colonies of granulocyte-macrophages (CFU-GM) and of erythroid cells (BFU-E) were moderately reduced in number: 5 CFU-GM/1003 marrow cells plated were seen (normal 37–84) and 10 BFU-E/2 × 105 marrow cells plated were seen (normal 30–90). These results allowed no specific diagnostic interpretation. It was thought that her primary hematologic diagnosis was “primary thrombocythemia.” She was started on treatment with hydroxyurea. One day after starting the hydroxyurea, an episode of acute left flank pain occurred, which was diagnosed clinically as renal vein thrombosis. The patient subsequently developed fever, respiratory distress, and hypotension. She was treated with antibiotics, corticosteroids, and anticoagulation with heparin and Coumadin. On the 35th hospital day, a laparotomy was performed for small bowel obstruction that was thought to be secondary to adhesions. Acute tubular necrosis, neutropenia, and Enterobacter sepsis ensued. On the 52nd day, an intraabdominal abscess was drained, and a Tenckhoff peritoneal dialysis catheter was placed because of azotemia. Peritonitis with various combinations of Enterobacter cloacae, Serratia marscesens, Herella, Streptococus faecalis, and Candida albicans developed. The platelet count fell to 30,000/mm3. On the 65th day the patient was reexplored because of feculent drainage from the Tenckhoff catheter. The expected bowel perforation was not found, but a constrictive pericarditis was identified and relieved. The platelet count fell further, and a bone marrow aspirate revealed decreased megakaryocytes, an increased M solidus E ratio, and a normal morphology of all cell lines. The patient developed bacteremia with antibiotic-resistant Mima polymorpha. She died on the 75th hospital day.
Pathology
The original liver, removed at the time of transplantation, weighed 1700 g and had near total occlusion of the main hepatic veins by old thrombus. Microscopically, there were organized thrombi in the terminal hepatic veins, centrilobular congestion, and disappearance of liver cells in this zonal area (zone 3 of Rappaport). Large regenerating nodules were also present (Figure 1), Needle liver biopsy of the orthotopic transplant obtained on days 35 and 52 of the terminal hospitalization showed thrombosis of the terminal hepatic veins with organization.
Figure 1.
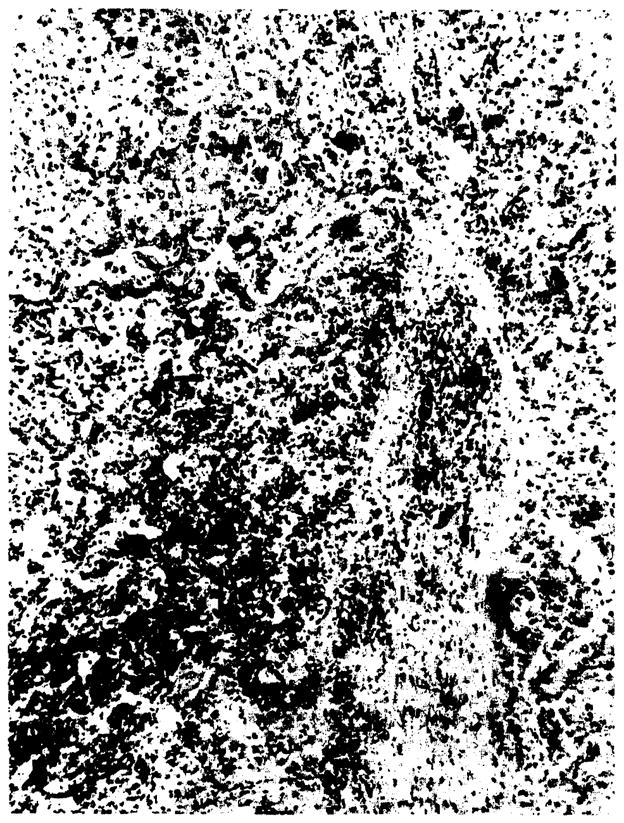
Original liver with dilated and congested sinusoids. The two-cell-thick plates indicate regeneration. Note partial “occlusion of radicle of terminal hepatic vein by organizing thrombosis. (H & E; ×93.)
At autopsy, the transplanted liver weighed 3700 g. All of the surgical anastomoses of the transplanted liver were patent. In the inferior vena cava, at the hepatic vein orifice (part of the graft), a thrombus containing early granulation tissue and occluding ~5% of the orifice was noted. The portal vein distant from the vascular anastomosis had a 50% occlusion by old, recanalized thrombus. In scattered foci, centrilobular areas were noted to be red and sunken below the cut surface. Sinusoidal congestion was prominent throughout. Histologically there was moderate hepatocyte proliferation such that many liver plates were two or more cells thick. Some areas demonstrated dropout of centrilobular liver cells with associated lipofuchsin-containing macrophages. Occluded central (terminal hepatic) veins could be readily identified in these areas (Figures 2 and 3). Fibrin stain confirmed the thrombotic origin of these occlusions while standard stains clearly ruled out the proliferative lesion characteristic of venoocclusive disease. No histologic evidence of liver rejection was seen.
Figure 2.

Transplant liver with occlusion of terminal hepatic vein due to organizing thrombosis with partial recanalization. Area of zonal necrosis is indicated by arrows. (H & E; ×40; inset, ×93.)
Figure 3.
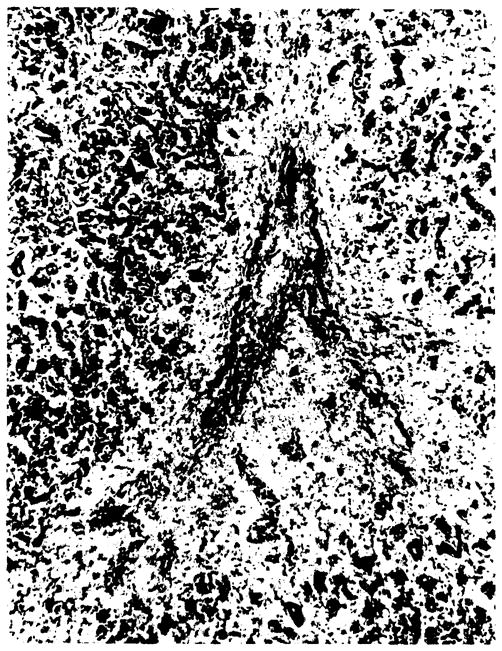
Transplant liver with area of zonal necrosis in centrilobular area (Rappaport zone 3) with organizing thrombosis of vein in its center. (Verhoff-VanGieson; ×93.)
Additional important findings at autopsy included pneumonia, bronchiectasis, recent and old pulmonary thromboemboli, a recent left renal vein thrombosis, an old thyroid vein thrombus, old cerebral cortical vein thromboses, and positive postmortem cultures for Serratia in the lung and Herrelia, Enterobacter, and Candida in the blood. The cause of death was attributed to multiple organ failure and sepsis.
Discussion
To our knowledge, this is the first reported case of recurrent Budd-Chiari syndrome in a patient after orthotopic liver transplantation. This is particularly noteworthy in that, to our knowledge, orthotopic liver transplantation has been performed six times for treatment of the Budd-Chiari syndrome, and none of the other 5 cases have experienced recurrence despite survivals as long as 7.5 yr and two pregnancies after transplantation in one of our own cases.
No cause for hepatic vein thrombosis could be identified in the patient described in this report despite a thorough hematologic evaluation and search for recognized other causes (2). Her liver, examined at time of transplantation, had extensive large vessel thrombosis typical of the Budd-Chiari syndrome. It eventually became abundantly clear that she had a generalized “hypercoagulable” state, as the list of thrombosed veins became progressively longer.
At the time of her final admission, she was found to have extreme thrombocytosis and a lupus-like inhibitor of the PT and APTT systems. Whether or not the lupus-like inhibitor might relate to the problems with thrombosis is unknown. Examination of her blood suggested that the thrombocytosis was due to a primary disease of the myeloid stem cell compartment rather than being a “reactive” thrombocytosis. Evidence for such a lesion process included the presence of numerous abnormally large and occasionally “giant” platelets, neutrophilia with hypersegmental neutrophils in the absence of a megaloblastic anemia, and increased blood basophils. Interestingly, the bone marrow was unremarkable except for the presence of increased megakaryocytes. The presence of minimal, but definite, myeloid hematopoiesis in the excised spleen at the time of liver transplantation indicates that extramedullar hematopoiesis was present then, although not detected. Potential hematologic diagnoses include idiopathic myelofibrosis in the cellular, nonfibrotic phase or essential thrombocythemia. It should be noted however, that neither diagnosis can be made with certainty, and the possibility that the hematopoietic abnormalities present terminally were due at least in part to liver transplantation cannot be excluded.
The transplanted liver, examined by needle biopsy on two occasions before death and at autopsy, showed evidence of recurrence of thrombosis in the hepatic veins. In addition, at autopsy a nonocclusive thrombosis of large hepatic veins was identified, although it probably was clinically unimportant. In contrast, the small intrahepatic terminal veins showed extensive occlusion by new and old thrombosis. Although the autopsy picture of small vein occlusion more closely resembles the so-called venoocclusive disease than classical Budd-Chiari syndrome, some of the vascular occlusions present at autopsy were shown to contain fibrin and therefore must have been due to hepatic venous thrombosis (the Budd-Chiari syndrome) rather than endothelial proliferation characteristic of venoocclusive disease.
Finally, within our own transplant experience, 3 other patients have had liver transplantation for the Budd-Chiari syndrome. Moreover, at least 2 other cases have been done by others. (7) All 5 patients are alive as of this report, 23–91 mo after transplantation. Clearly, Budd-Chiari syndrome cannot be an absolute contraindication to liver transplantation. It must be recognized, however, that based upon our experience with the present case, depending upon the cause of the coagulation abnormality, recurrent Budd-Chiari syndrome is at least possible and may affect the long-term course of such patients. Such poor risk patients would appear to be those who have an underlying disorder of hematopoietic stem cells such as those with polycythemia rubra vera, idiopathic myelofibrosis, and paroxysmal nocturnal hemoglobinuria. In contrast to these high risk patients and again based upon our own experience, albeit quite small, those patients with the Budd-Chiari syndrome, due presumably to oral contraceptive use, would appear to do quite well after transplantation as long as such potentially thrombogenic agents are avoided subsequently. Relevant to this latter issue is the fact that the patient in the present report appeared to do well after an initial stormy postoperative course until her thrombotic therapy was discontinued in anticipation of doing a follow-up liver biopsy. Whether or not her terminal course would not have occurred had such therapy not been discontinued has to be seriously considered.
In summary, we present a case of recurrent Budd-Chiari syndrome in a patient who underwent orthotopic liver transplantation for hepatic vein thrombosis that was not responsive to the usual measures used in the management of this unusual problem.
Acknowledgments
The authors acknowledge the help of Dr. R. H. Fennell in providing them with information concerning the patient’s hospital course while at the University of Colorado Medical Center.
References
- 1.Parker RGF. Occlusion of the hepatic veins in man. Medicine. 1959;38:369–402. doi: 10.1097/00005792-195912000-00002. [DOI] [PubMed] [Google Scholar]
- 2.Mitchel MC, Biotnott JK, Kaufman S, et al. Bucld Chiari syndrome: etiology, diagnosis and management. Medicine. 1982;61:199–218. doi: 10.1097/00005792-198207000-00001. [DOI] [PubMed] [Google Scholar]
- 3.Takeuchi J, Takada A, Hasumura Y, et al. Budd-Chiari syndrome associated with obstruction of the interior vena cava. Am J Med. 1971;51:11–20. doi: 10.1016/0002-9343(71)90319-6. [DOI] [PubMed] [Google Scholar]
- 4.Asbury RF, Rosenthal SN, Descalzi ME, et al. Hepatic venoocclusive disease due to DTIC. Cancer. 1980;45:2670–4. doi: 10.1002/1097-0142(19800515)45:10<2670::aid-cncr2820451031>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 5.Bras G, Jelliffe DB, Stuart KL. Venoocclusive disease of the liver with nonportal type of cirrhosis, occurring in Jamaica. Arch Pathol. 1954;57:285–300. [PubMed] [Google Scholar]
- 6.Putnam CW, Porter KA, Weil R, et al. Liver transplantation for Budd-Chiari syndrome. JAMA. 1976;236:1142–3. [PMC free article] [PubMed] [Google Scholar]
- 7.Calne RY, Williams R. Orthotopic liver transplantation: the first 60 patients. Br Med J. 1977;1:471. doi: 10.1136/bmj.1.6059.471. [DOI] [PMC free article] [PubMed] [Google Scholar]