

Abstract
Background
Preconditioning using lipopolysaccharide (LPS), a toll-like receptor 4 (TLR4) ligand, has been demonstrated to reduce ischaemia/reperfusion injury (IRI) in some organs, but its effect in the liver has not been elucidated. We examined the liver protective mechanism and correlated signalling pathway of LPS preconditioning in mice.
Methods
BALB/c and TLR4 mutant mice underwent 90 min of 70% hepatic ischaemia. Lipopolysaccharide (100 µg/kg) was injected intraperitoneally 20 h or 30 min before ischaemia. Liver damage after reperfusion was examined using serum samples and liver specimens. To analyse the mechanism of preconditioning in detail, phosphorylation of representative signalling mediators to nuclear factor-κB (NF-κB) activation, Akt and interleukin-1 receptor-associated kinase-1 (IRAK-1), and expression of a negative feedback inhibitor, suppressor of cytokine signalling-1 (SOCS-1), were evaluated by Western blotting.
Results
Pretreatment with LPS only 20 h before ischaemia elicited a preconditioning effect; however, preconditioning was absent in TLR4 mutant mice. Lipopolysaccharide significantly decreased serum alanine aminotransferase, tumour necrosis factor-α, hepatocyte necrosis and NF-κB activity after reperfusion. Phosphorylated IRAK-1 was suppressed by LPS, whereas no difference was observed in phosphorylated Akt. Pre-ischaemic LPS provided early induction of SOCS-1.
Discussion
Late-phase LPS preconditioning provided liver protection against IRI through the downregulation of the TLR4 cascade derived from early induction of SOCS-1 during ischaemia/reperfusion.
Keywords: liver, preconditioning, lipopolysaccharide, ischaemia/reperfusion, SOCS-1
Introduction
Systemic circulation of lipopolysaccharide (LPS), a component of the Gram-negative bacteria cell wall, can result in serious consequences for patients, such as shock and disseminated intravascular coagulation. These undesirable complications are caused by stimulation of the toll-like receptor 4 (TLR4) signalling pathway, which regulates the innate immune system against invasion by pathogens and multiple other endogenous ligands.1 As a result of TLR4 signalling, the transcription factor nuclear factor-κB (NF-κB), which modulates induction of cytokines and expression of proinflammatory genes, is activated.2 By contrast, LPS has also been demonstrated to exert a tissue-protective effect in the heart, brain, kidney and other organs.3 Pretreatment with a non-toxic amount of LPS increases resistance to tissue damage after ischaemia/reperfusion (I/R), an effect designated as LPS preconditioning.4–6
Preconditioning that attenuates ischaemia/reperfusion injury (IRI) can be classified based on the protective timeframe and mechanism.7–9 Early-phase preconditioning exhibits a large degree of tissue protection for a short period compared with late-phase preconditioning. Each class of preconditioning exerts specific protective mechanisms with corresponding signalling pathways, such as through phosphatidylinositol 3 (PI3) kinase/Akt or TLR4.10–12 The PI3 kinase/Akt pathway has been demonstrated to be involved in hepatic ischaemic preconditioning, an early-phase preconditioning, by suppressing hepatocyte apoptosis.10 The PI3 kinase/Akt pathway has also been associated with cardiac LPS preconditioning, a late-phase event, which reduces myocardial infarct size.3 By contrast, the TLR4 pathway has been reported to participate in early-phase hepatic protection using high mobility group box-1 (HMGB-1), a TLR4 ligand similar to LPS.12 It is interesting that preconditioning with the same TLR4 ligand produces tissue protection in different phases and through the modulation of different signalling pathways in the heart and liver.
Hepatic preconditioning with LPS has not been adequately described in previous studies. Therefore, the present study aimed to investigate the protective effect of LPS pretreatment against liver IRI and the mechanism of LPS preconditioning.
Materials and methods
Animals and hepatic ischaemia
Male wild-type mice (BALB/c, C3H/HeN) and TLR4-mutant mice (C3H/HeJ) (weight 20–30 g) were prepared for all experiments.
All mice underwent laparotomy under general anaesthesia. Warm hepatic ischaemia of 70% was induced using a microvascular clip. Blood flow was recovered after 90 min of ischaemia. All experimental procedures with animals were carried out according to guidelines for the care and use of laboratory animals in place at Kagawa University.
Experimental design
All BALB/c mice received intraperitoneal pretreatment with 100 µg/kg of either LPS or normal saline. The mice were divided into an LPS-non-treated group (LPC[−]), which received normal saline injection; an early-phase LPS preconditioning group (Early-LPC), which underwent pretreatment with 100 µg/kg LPS 30 min before ischaemia, and a late-phase LPS preconditioning group (Late-LPC), which was subjected to 100 µg/kg LPS pretreatment 20 h before ischaemia, sustained ischaemia as previously described and reperfusion. After pretreatment, mice were either subjected to ischaemia for 90 min before reperfusion or killed at the scheduled time-point. TLR4-mutant mice (C3H/HeJ) and the corresponding wild-type mice (C3H/HeN) received pretreatment with or without LPS before I/R induction.
Cytokine measurement
Serum concentrations of tumour necrosis factor-α (TNF-α) were measured using enzyme-linked immunosorbent assay (ELISA) kits (Pierce Biotechnology, Inc., Rockford, IL, USA).
Assessment of liver damage
Hepatic damage after I/R was evaluated by measurement of serum alanine aminotransferase (ALT) levels and histological findings. Histological assessment was based on the modified Suzuki's score,13 which grades necrotic areas on a scale of 0–4. A score of 0 indicates no evidence of necrosis, whereas a score of 4 represents > 60% necrosis.
Sample preparation
All nuclear and cytoplasmic protein extracts were prepared from homogenized liver specimens using a nuclear extraction kit (Pierce Biotechnology, Inc.) according to the manufacturer's instructions. Protein concentrations of extracts were determined using a BCA (bicinchoninic acid) protein assay kit (Pierce Biotechnology, Inc.) and standardized.
SDS-PAGE and Western blotting
Cytoplasmic proteins were electrophoresed and transferred to polyvinylidene difluoride membranes (Immobilon-P; Millipore Corp., Bedford, MA, USA). After appropriate blocking, membranes were incubated with primary antibodies: phosphorylated Akt (1:1000; Cell Signalling Technology, Inc., Beverly, MA, USA); phosphorylated interleukin-1 receptor-associated kinase-1 (IRAK-1) (1:1000; Cell Signalling Technology, Inc.), and suppressor of cytokine signalling-1 (SOCS-1) (1:500; Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA). The membranes were then incubated with horseradish peroxidase (HRP) conjugated secondary antibodies (Santa Cruz Biotechnology, Inc.) at the recommended dilution. Target proteins were visualized using an enhanced chemiluminescence system (Amersham Pharmacia, Amersham, UK).
Electrophoretic mobility shift assay
Nuclear factor-κB DNA binding activity was evaluated by electrophoretic mobility shift assay (EMSA) using standardized nuclear extracts as described above.
Quantitative reverse-transcription polymerase chain reaction
Each specimen was snap-frozen in liquid nitrogen and stored at −80 °C after sampling. Total RNA was isolated from murine tissue by guanidinium isothiocyanate-acid phenol extraction and quantified by measuring absorbance at 260 nm. One µg of total RNA was used for reverse transcription, and specific mRNAs were determined by real-time polymerase chain reaction (PCR) using Power SYBR Green PCR Master Mix (Applied Biosystems, Inc., Foster City, CA, USA) on ABI Prism 7000. Oligonucleotide primers and probes were: TNF-α, forward primer: 5′-AGC CTG TAG CCC ACG TCG TA-3′ and TNF-α, reverse primer: 5′-TGG CAC CAC TAG TTG GTT GTC T-3′, and SOCS-1, forward primer: 5′-GCA TCC CTC TTA ACC CGG TAC T-3′ and SOCS-1, reverse primer: 5′-ATA AGG CGC CCC CAC TTA AT-3′. 18S rRNA was amplified as an endogenous control. Primers and probes for 18S rRNA were obtained from a Pre-Developed TaqMan Assay Reagent Kit (Applied Biosystems AB, Stockholm, Sweden).
Statistical analysis
Results are expressed as mean ± standard error of the mean (SEM). Group comparisons were performed using Student's t-test or anova. Differences were considered significant at P < 0.05.
Results
Effect of LPS preconditioning on serum ALT
To examine the effect of LPS pretreatment against liver IRI, 100 µg/kg LPS was administered 20 h or 30 min before ischaemia. Late-phase LPS pretreatment significantly reduced serum levels of ALT compared with no pretreatment, whereas early-phase LPS pretreatment did not (Fig. 1A). To analyse underlying factors, expression of TNF-α, a pleiotropic cytokine which mediates multiple immune responses secondary to LPS stimulation, was measured by RT-PCR before induction of ischaemia. Expression of TNF-α was 40-fold higher in the Early-LPC group than in the Late-LPC group before ischaemia (Fig. 1B). This suggests that LPS pretreatment 30 min before ischaemia leads to an inflammatory state associated with the intense induction of cytokines in the pre-ischaemic period.
Figure 1.
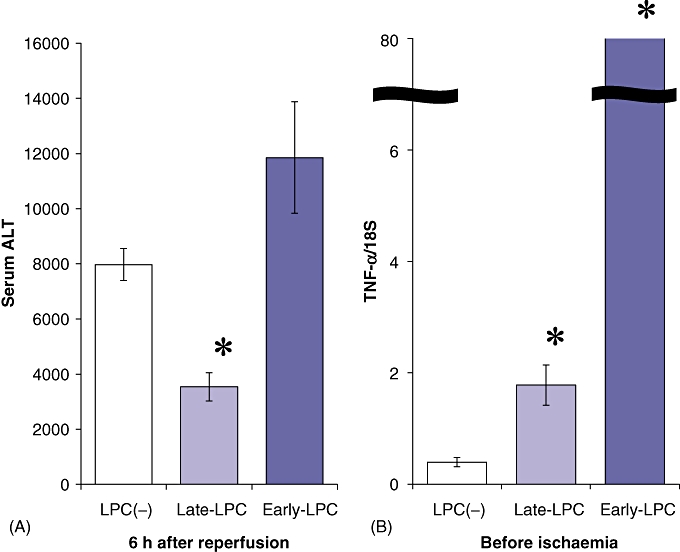
Lipopolysaccharide (LPS) pretreatment reduced serum levels of alanine aminotransferase (ALT) after ischaemia/reperfusion, but in late-phase treatment only. (A) LPS (100 µg/kg) was administered 20 h or 30 min before ischaemia (n = 6). Mice underwent 90 min of ischaemia and subsequent reperfusion. Serum levels of ALT in each sample were measured 6 h after reperfusion. (B) Expression of tumour necrosis factor-α (TNF-α) was examined for evaluation of the pre-ischaemic inflammatory response by reverse-transcription polymerase chain reaction (n = 6). As an endogenous control, the amplification of 18S rRNA was used. Results are expressed as mean ± standard error of the mean. *P < 0.05, compared with the LPC(−) group and the Early-LPC group. LPC(−), LPS-non-treated group; Late-LPC, late-phase LPS preconditioning group; Early-LPC, early-phase LPS preconditioning group
Late-phase LPS pretreatment confers hepatic protection
The Late-LPC group showed significantly less hepatic necrosis than the LPC(−) group (Fig. 2A). However, early-phase LPS pretreatment resulted in the same level of liver injury after I/R as did pretreatment without LPS. Liver specimens obtained from the LPC(−) group and the Early-LPC group showed > 60% necrosis of hepatocytes and severe sinusoidal congestion, whereas parenchymal damage was minimal in the Late-LPC group (Fig. 2B).
Figure 2.
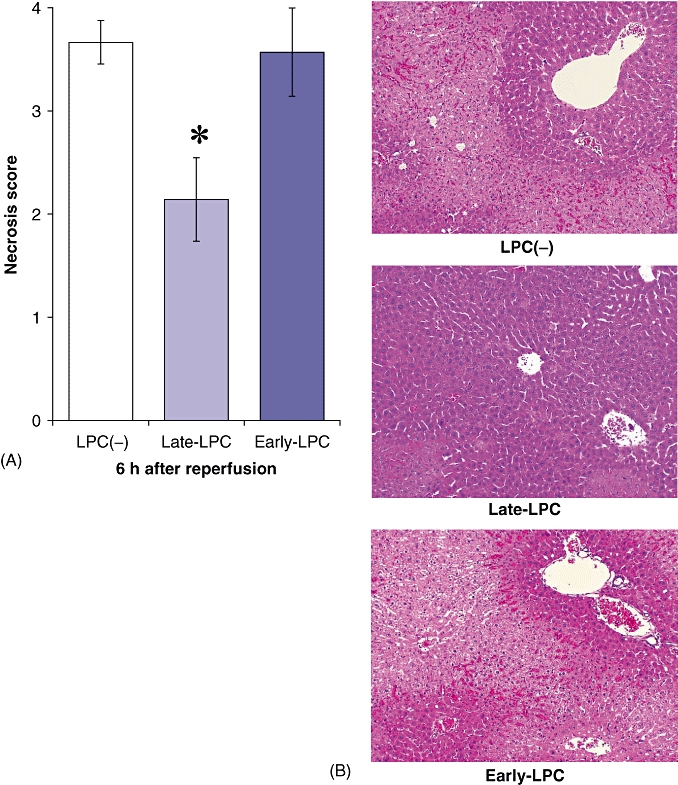
Late-phase lipopolysaccharide (LPS) pretreatment attenuated liver parenchymal necrosis after ischaemia/reperfusion, whereas early-phase pretreatment was unable to prevent liver damage. Mice were subjected to LPS pretreatment, ischaemia and reperfusion as described above. Liver specimens were sampled 6 h after perfusion (n = 6). (A) Effect of LPS pretreatment on necrosis of hepatocytes following ischaemia/reperfusion was evaluated according to the pathological damage score. (B) Liver sections of each group (haematoxylin and eosin stain; original magnification × 100). Results are expressed as mean ± standard error of the mean. *P < 0.05, compared with the LPC(−) group and the Early-LPC group. LPC(−), LPS-non-treated group; Late-LPC, late-phase LPS preconditioning group; Early-LPC, early-phase LPS preconditioning group
Late LPS preconditioning attenuates NF-κB activity independent of the PI3K/Akt pathway
We examined whether the protective effect of late-phase LPS preconditioning was associated with the PI3K/Akt signalling pathway by Western blotting. No difference in Akt phosphorylation was observed between the LPC(−) and Late-LPC groups (Fig. 3A). In addition, binding activity of NF-κB was evaluated using EMSA to confirm that liver protection was associated with NF-κB regulation. Less NF-κB activity was observed in the LPS pretreated mice compared with controls 1 h after reperfusion (Fig. 3B). Hence, LPS preconditioning downregulated NF-κB, but did not involve the PI3K/Akt signalling pathway.
Figure 3.
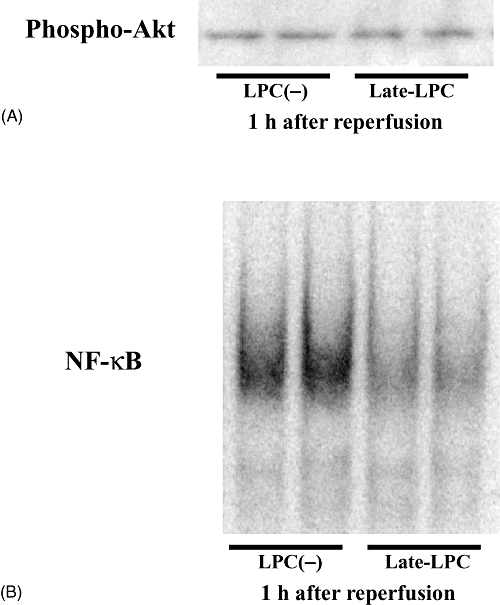
Late-phase lipopolysaccharide (LPS) preconditioning made no difference in the phosphorylation of Akt, but inhibited the activation of transcription factor nuclear factor (NF)-κB. Mice underwent 100 µg/kg LPS pretreatment 20 h before 90 min ischaemia and reperfusion. Samples were obtained 1 h after reperfusion (n = 6). (A) Effect of LPS preconditioning on the phosphorylation of Akt was examined by Western blotting. (B) NF-κB binding activity was investigated by electrophoretic mobility shift assay using nuclear extracts prepared from liver samples. LPC(−), LPS-non-treated group; Late-LPC, late-phase LPS preconditioning group; Phospho-, phosphorylated
Involvement of TLR4 in LPS preconditioning
The role of TLR4 signalling, which can also activate NF-κB, was examined in LPS preconditioning using TLR4-mutant mice and the associated wild type. Similar levels of hepatic injury occurred in mutant mice after I/R both with and without LPS preconditioning, including localized parenchymal necrosis and mild sinusoidal congestion (Fig. 4A). By contrast, LPS preconditioning ameliorated the severe damage of IRI in wild-type mice (Fig. 4B). These findings indicate that TLR4 signalling is involved in hepatic protection after I/R.
Figure 4.
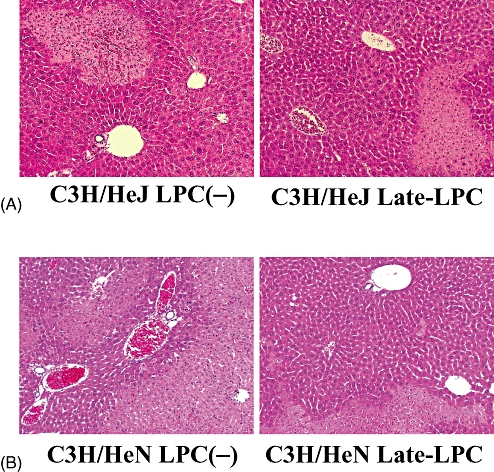
Toll-like receptor 4 (TLR4) signalling pathway was associated with hepatic protection provided by lipopolysaccharide (LPS) preconditioning. TLR4-mutant mice (C3H/HeJ) and wild-type mice for TLR4-mutant (C3H/HeN) were subjected to ischaemic procedures for 90 min with or without LPS pretreatment. After 6 h of reperfusion, both strains of mice were killed and liver specimens were obtained. Liver sections of (A) mutant mice and (B) wild-type mice (haematoxylin and eosin stain; original magnification × 100). LPC(−), LPS-non-treated group; Late-LPC, late-phase LPS preconditioning group
Inhibition of inflammatory cascade via the TLR4 pathway
Western blot analysis was performed to investigate whether LPS preconditioning had an actual effect on the TLR4 pathway in hepatic I/R. The phosphorylation of IRAK-1, a key molecule in the TLR4 cascade that leads to NF-κB activation, was examined. The Late-LPC group exhibited lower expression of phosphorylated IRAK-1 6 h after reperfusion in comparison with the LPC(−) group (Fig. 5A). As an indicator of inflammatory progression, serum levels of TNF-α were measured using ELISA (Fig. 5B). Lipopolysaccharide preconditioning significantly reduced serum TNF-α compared with negative controls. Overall, pre-ischaemic LPS treatment appears to inhibit post-I/R inflammation via the TLR4 cascade.
Figure 5.
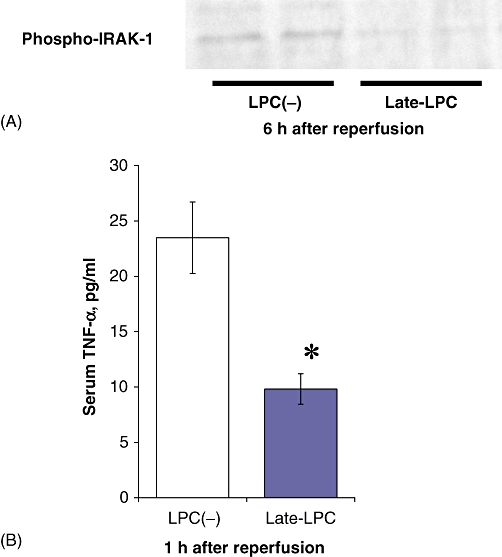
Lipopolysaccharide (LPS) preconditioning inhibited inflammatory response after ischaemia/reperfusion via the toll-like receptor 4 (TLR4) signalling pathway. Mice underwent LPS pretreatment and ischaemic procedures as described above. Serum and liver samples were obtained 1 h and 6 h after reperfusion, respectively (n = 6). (A) Western blotting showed the difference between the LPC(−) group and the Late-LPC group in phosphorylation of interleukin-1 receptor-associated kinase-1 (IRAK-1). (B) Serum levels of tumour necrosis factor-α (TNF-α) were measured for evaluation of the inflammation after reperfusion (n = 6). Results are expressed as mean ± standard error of the mean. *P < 0.05, compared with the LPC(−) group. LPC(−), LPS-non-treated group; Late-LPC, late-phase LPS preconditioning group; Phospho-, phosphorylated
Modulation of the TLR4 pathway by induction of SOCS-1
Post-ischaemic expression of SOCS-1, a feedback inhibitor of TLR4 signalling, was examined to analyse its potential protective effect against hepatic IRI. RT-PCR revealed a significant difference in SOCS-1 expression 1 h after reperfusion between the LPC(−) and Late-LPC groups (Fig. 6A). Lipopolysaccharide preconditioning led to earlier induction of SOCS-1 in the reperfusion period (Fig. 6B). These data suggest that modulation of TLR4 signalling through induction of SOCS-1 in the early phases of I/R contributes to the liver protection derived from LPS preconditioning.
Figure 6.
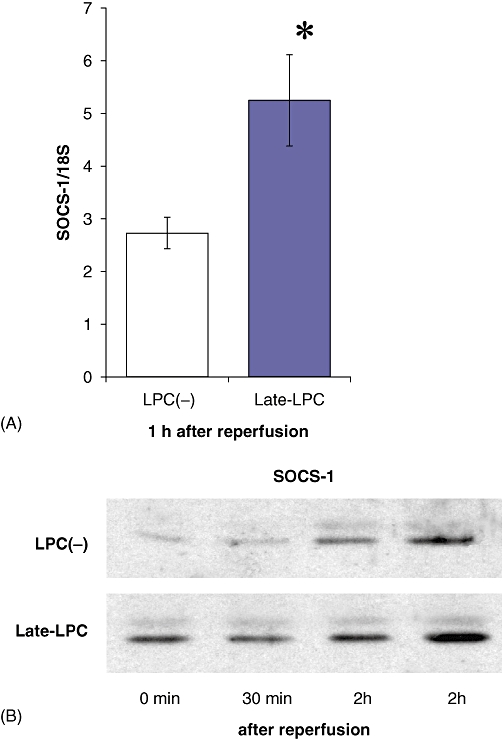
Lipopolysaccharide (LPS) preconditioning induced suppressor of cytokine signalling-1 (SOCS-1) in the earlier post-ischaemic period. Mice were subjected to pretreatment, ischaemia and reperfusion as described above. Protein extracts were prepared from liver parenchyma sampled after reperfusion (n = 5). Expression of SOCS-1 was investigated using (A) reverse transcription polymerase chain reaction and (B) Western blotting. As an endogenous control, the amplification of 18S rRNA was used. Results are expressed as mean ± standard error of the mean. *P < 0.05, compared with the LPC(−) group. LPC(−), LPS-non-treated group; Late-LPC, late-phase LPS preconditioning group
Discussion
Since the initial report of hepatic injury after I/R by Toledo-Pereyra and colleagues,14 many studies have been conducted to clarify the mechanisms underlying this injury. The process of hepatic injury during I/R has been elucidated in detail.15–17 Diverse types of intrahepatic cells participate in the inflammatory response triggered by circulatory blockade, including hepatocytes, Kupffer cells, sinusoidal endothelial cells and neutrophils. Moreover, these cells produce reactive oxygen species and other proinflammatory mediators such as cytokines, chemokines and adhesion molecules. The interaction among these cells and mediators leads to the development of a complicated inflammatory response induced by I/R. A series of inflammatory responses activates transcription factor NF-κB.18–20 NF-κB in turn regulates the gene expression of many proinflammatory mediators, in particular TNF-α, which is induced in the relatively early ischaemic phase and augments the subsequent inflammatory signal indirectly such as by recruiting neutrophils.21 Therefore, NF-κB is considered to represent a promising therapeutic target for suppressing IRI.
Several treatments have been investigated to reduce hepatic IRI through direct or indirect inhibition of NF-κB activation.22,23 In the liver, pretreatment that modulates PI3K/Akt signalling and TLR4 signalling has been demonstrated to suppress I/R-induced NF-κB activation.10–12 The PI3K/Akt pathway is crucial to cell survival as it regulates apoptosis in response to various stimuli.24,25 Tissue protection through the inhibition of apoptosis in reperfused organs has been reported in hepatic ischaemic preconditioning10 and cardiac LPS preconditioning.4 However, the TLR4 pathway protects the innate immune system from a variety of endogenous or exogenous stimuli, including hepatic I/R.26–29 Accordingly, the negative regulation of this pathway may drive preconditioning. Preconditioning by HMGB-1 in the liver has been demonstrated to inhibit inflammatory signalling via the TLR4 cascade.12 We found that control groups expressed similar levels of phosphorylated Akt and higher levels of phosphorylated IRAK-1 compared with LPS pretreatment groups. Hepatic LPS preconditioning has been demonstrated to involve TLR4 signalling, but not PI3K/Akt. As mentioned above, Ha et al. showed that the PI3K/Akt pathway is associated with cardiac LPS preconditioning.4 The current study has not elucidated the differences between heart and liver in implicated signalling pathways of LPS preconditioning.
Preconditioning, a tissue-protective procedure, has been developed in diverse organs because injury after I/R inflicts severe damage under clinical conditions. Recently, many preconditioning techniques, using non-lethal invasive procedures and drugs, have been reported.9,30 Early-phase preconditioning provides protection immediately after the procedure, which is maintained for 2–3 h. Early preconditioning primarily acts through direct influence upon parenchymal and non-parenchymal cellular function. By contrast, late preconditioning produces protection after 12–24 h and protects tissues for 3–4 days. Late-phase preconditioning influences protein synthesis through the activation of several stress-responsive genes. Although early preconditioning may exert a larger protective effect, late preconditioning may be more appealing, given its longer effective duration.7,8 Some studies indicate that LPS produces late preconditioning, whereas HMGB-1 produces early preconditioning.4–6 In the present study, early LPS pretreatment failed to provide hepatic protection. Histological assessment exhibited the same level of hepatic necrosis in mice with and without early LPS pretreatment. However, early LPS pretreatment did produce extremely high TNF-α expression before ischaemia, which suggests that a hyperinflammatory state may accompany the upregulation of signalling mediators. These differences in results may be caused by inadequate recovery from the septic state in the early pretreatment group. Lipopolysaccharide pretreatment requires sufficient time to produce a preconditioning effect.
Low-dose LPS has been shown to induce tolerance to subsequent toxic doses of LPS and upregulate SOCS-1 and interleukin-1 receptor-associated kinase M (IRAK-M), negative feedback inhibitors of TLR4.31,32 We also demonstrated that LPS preconditioning induced immediate upregulation of SOCS-1 in the early reperfused period. In addition, HMGB-1 preconditioning has been reported to upregulate IRAK-M.12 Taken together, pretreatment using a TLR4 ligand, such as LPS or HMGB-1, appears to provide tissue protection against stressors by inducing negative feedback inhibitors of this cascade.
In summary, we have demonstrated that pretreatment with LPS attenuated liver injury following I/R. This liver-protective effect was induced by pretreatment 20 h before ischaemia, but not 30 min before ischaemia. Lipopolysaccharide preconditioning against liver IRI involves TLR4 signalling and its negative feedback inhibitor, SOCS-1. Therefore, negative feedback inhibitors of the TLR4 cascade may serve as therapeutic targets to reduce hepatic IRI and other inflammatory diseases.
Conflicts of interest
None declared.
References
- 1.Takeda K, Akira S. TLR signalling pathways. Semin Immunol. 2004;16:3–9. doi: 10.1016/j.smim.2003.10.003. [DOI] [PubMed] [Google Scholar]
- 2.Kawai T, Akira S. Signalling to NF-kappaB by toll-like receptors. Trends Mol Med. 2007;13:460–469. doi: 10.1016/j.molmed.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 3.Biswas SK, Lopez-Collazo E. Endotoxin tolerance: new mechanisms, molecules and clinical significance. Trends Immunol. 2009;30:475–487. doi: 10.1016/j.it.2009.07.009. [DOI] [PubMed] [Google Scholar]
- 4.Ha T, Hua F, Liu X, Ma J, McMullen JR, Shioi T, et al. Lipopolysaccharide-induced myocardial protection against ischaemia/reperfusion injury is mediated through a PI3K/Akt-dependent mechanism. Cardiovasc Res. 2008;78:546–553. doi: 10.1093/cvr/cvn037. [DOI] [PubMed] [Google Scholar]
- 5.Rosenzweig HL, Lessov NS, Henshall DC, Minami M, Simon RP, Stenzel-Poore MP. Endotoxin preconditioning prevents cellular inflammatory response during ischaemic neuroprotection in mice. Stroke. 2004;35:2576–2581. doi: 10.1161/01.STR.0000143450.04438.ae. [DOI] [PubMed] [Google Scholar]
- 6.Heemann U, Szabo A, Hamar P, Müller V, Witzke O, Lutz J, et al. Lipopolysaccharide pretreatment protects from renal ischaemia/reperfusion injury: possible connection to an interleukin-6-dependent pathway. Am J Pathol. 2000;156:287–293. doi: 10.1016/S0002-9440(10)64729-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stein AB, Tang XL, Guo Y, Xuan YT, Dawn B, Bolli R. Delayed adaptation of the heart to stress: late preconditioning. Stroke. 2004;35:2676–2679. doi: 10.1161/01.STR.0000143220.21382.fd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bolli R. The late phase of preconditioning. Circ Res. 2000;87:972–983. doi: 10.1161/01.res.87.11.972. [DOI] [PubMed] [Google Scholar]
- 9.Carini R, Albano E. Recent insights on the mechanisms of liver preconditioning. Gastroenterology. 2003;125:1480–1491. doi: 10.1016/j.gastro.2003.05.005. [DOI] [PubMed] [Google Scholar]
- 10.Izuishi K, Tsung A, Hossain MA, Fujiwara M, Wakabayashi H, Masaki T, et al. Ischaemic preconditioning of the murine liver protects through the Akt kinase pathway. Hepatology. 2006;44:573–580. doi: 10.1002/hep.21298. [DOI] [PubMed] [Google Scholar]
- 11.Harada N, Hatano E, Koizumi N, Nitta T, Yoshida M, Yamamoto N, et al. Akt activation protects rat liver from ischaemia/reperfusion injury. J Surg Res. 2004;121:159–170. doi: 10.1016/j.jss.2004.04.016. [DOI] [PubMed] [Google Scholar]
- 12.Izuishi K, Tsung A, Jeyabalan G, Critchlow ND, Li J, Tracey KJ, et al. Cutting edge: high-mobility group box 1 preconditioning protects against liver ischaemia-reperfusion injury. J Immunol. 2006;176:7154–7158. doi: 10.4049/jimmunol.176.12.7154. [DOI] [PubMed] [Google Scholar]
- 13.Suzuki S, Toledo-Pereyra LH, Rodriguez FJ, Cejalvo D. Neutrophil infiltration as an important factor in liver ischaemia and reperfusion injury: modulating effects of FK506 and cyclosporine. Transplantation. 1993;55:1265. doi: 10.1097/00007890-199306000-00011. [DOI] [PubMed] [Google Scholar]
- 14.Toledo-Pereyra LH, Simmons RL, Najarian JS. Protection of the ischaemic liver by donor pretreatment before transplantation. Am J Surg. 1975;129:513–517. doi: 10.1016/0002-9610(75)90308-6. [DOI] [PubMed] [Google Scholar]
- 15.Lentsch AB, Kato A, Yoshidome H, McMasters KM, Edwards MJ. Inflammatory mechanisms and therapeutic strategies for warm hepatic ischaemia/reperfusion injury. Hepatology. 2000;32:169–173. doi: 10.1053/jhep.2000.9323. [DOI] [PubMed] [Google Scholar]
- 16.Teoh NC, Farrell GC. Hepatic ischaemia reperfusion injury: pathogenic mechanisms and basis for hepatoprotection. J Gastroenterol Hepatol. 2003;18:891–902. doi: 10.1046/j.1440-1746.2003.03056.x. [DOI] [PubMed] [Google Scholar]
- 17.Jaeschke H. Molecular mechanisms of hepatic ischaemia-reperfusion injury and preconditioning. Am J Physiol Gastrointest Liver Physiol. 2003;284:15–26. doi: 10.1152/ajpgi.00342.2002. [DOI] [PubMed] [Google Scholar]
- 18.Zwacka RM, Zhang Y, Zhou W, Halldorson J, Engelhardt JF. Ischaemia/reperfusion injury in the liver of BALB/c mice activates AP-1 and nuclear factor kappaB independently of IkappaB degradation. Hepatology. 1998;28:1022–1030. doi: 10.1002/hep.510280417. [DOI] [PubMed] [Google Scholar]
- 19.Yoshidome H, Kato A, Edwards MJ, Lentsch AB. Interleukin-10 suppresses hepatic ischaemia/reperfusion injury in mice: implications of a central role for nuclear factor kappaB. Hepatology. 1999;30:203–208. doi: 10.1002/hep.510300120. [DOI] [PubMed] [Google Scholar]
- 20.Funaki H, Shimizu K, Harada S, Tsuyama H, Fushida S, Tani T. Essential role for nuclear factor kappaB in ischaemic preconditioning for ischaemia-reperfusion injury of the mouse liver. Transplantation. 2002;27:551–556. doi: 10.1097/00007890-200208270-00021. [DOI] [PubMed] [Google Scholar]
- 21.Teoh N, Field J, Sutton J, Farrell G. Dual role of tumour necrosis factor-alpha in hepatic ischaemia-reperfusion injury: studies in tumour necrosis factor-alpha gene knockout mice. Hepatology. 2004;39:412–421. doi: 10.1002/hep.20035. [DOI] [PubMed] [Google Scholar]
- 22.Suetsugu H, Iimuro Y, Uehara T, Nishio T, Harada N, Hatano E, et al. Nuclear factor κB inactivation in the rat liver ameliorates short-term total warm ischaemia/reperfusion injury. Gut. 2005;54:835–842. doi: 10.1136/gut.2004.043034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee CH, Kim SH, Lee SM. Effect of pyrrolidine dithiocarbamate on hepatic vascular stress gene expression during ischaemia and reperfusion. Eur J Pharmacol. 2008;595:100–107. doi: 10.1016/j.ejphar.2008.07.064. [DOI] [PubMed] [Google Scholar]
- 24.Williams DL, Ozment-Skelton T, Li C. Modulation of the phosphoinositide 3-kinase signalling pathway alters host response to sepsis, inflammation, and ischaemia/reperfusion injury. Shock. 2006;25:432–439. doi: 10.1097/01.shk.0000209542.76305.55. [DOI] [PubMed] [Google Scholar]
- 25.Fruman DA, Cantley LC. Phosphoinositide 3-kinase in immunological systems. Semin Immunol. 2002;14:7–18. doi: 10.1006/smim.2001.0337. [DOI] [PubMed] [Google Scholar]
- 26.Kawai T, Akira S. TLR signalling. Semin Immunol. 2007;19:24–32. doi: 10.1016/j.smim.2006.12.004. [DOI] [PubMed] [Google Scholar]
- 27.Zhai Y, Shen XD, O’Connell R, Gao F, Lassman C, Busuttil RW, et al. Cutting edge: TLR4 activation mediates liver ischaemia/reperfusion inflammatory response via IFN regulatory factor 3-dependent MyD88-independent pathway. J Immunol. 2004;173:7115–7119. doi: 10.4049/jimmunol.173.12.7115. [DOI] [PubMed] [Google Scholar]
- 28.Tsung A, Hoffman RA, Izuishi K, Critchlow ND, Nakao A, Chan MH, et al. Hepatic ischaemia/reperfusion injury involves functional TLR4 signalling in non-parenchymal cells. J Immunol. 2005;175:7661–7668. doi: 10.4049/jimmunol.175.11.7661. [DOI] [PubMed] [Google Scholar]
- 29.Zhai Y, Qiao B, Shen XD, Gao F, Busuttil RW, Cheng G, et al. Evidence for the pivotal role of endogenous toll-like receptor 4 ligands in liver ischaemia and reperfusion injury. Transplantation. 2008;85:1016–1022. doi: 10.1097/TP.0b013e3181684248. [DOI] [PubMed] [Google Scholar]
- 30.Selzner N, Rudiger H, Graf R, Clavien PA. Protective strategies against ischaemic injury of the liver. Gastroenterology. 2003;125:917–936. doi: 10.1016/s0016-5085(03)01048-5. [DOI] [PubMed] [Google Scholar]
- 31.Liu ZJ, Liu XL, Zhao J, Shi YJ, Yan LN, Chen XF, et al. The effects of SOCS-1 on liver endotoxin tolerance development induced by a low dose of lipopolysaccharide are related to dampen NF-kappaB-mediated pathway. Dig Liver Dis. 2008;40:568–577. doi: 10.1016/j.dld.2007.12.019. [DOI] [PubMed] [Google Scholar]
- 32.Liu ZJ, Yan LN, Li XH, Xu FL, Chen XF, You HB, et al. Upregulation of IRAK-M is essential for endotoxin tolerance induced by a low dose of lipopolysaccharide in Kupffer cells. J Surg Res. 2008;150:34–39. doi: 10.1016/j.jss.2007.12.759. [DOI] [PubMed] [Google Scholar]