

Abstract
Intermittent hypoxia (IH) associated with sleep apnea leads to cardio-respiratory morbidities. Previous studies have shown that IH alters the synthesis of neurotransmitters including catecholamines and neuropeptides in brainstem regions associated with regulation of cardio-respiratory functions. GABA, a major inhibitory neurotransmitter in the central nervous system, has been implicated in cardio-respiratory control. GABA synthesis is primarily catalyzed by glutamic acid decarboxylase (GAD). Here, we tested the hypothesis that IH like its effect on other transmitters also alters GABA synthesis. The impact of IH on GABA synthesis was investigated in pheochromocytoma 12 (PC12) cells, a neuronal cell line which is known to express active form of GAD67 in the cytosolic fraction and also assessed the underlying mechanisms contributing to IH-evoked response. Exposure of cell cultures to IH decreased GAD67 activity and GABA level. IH-evoked decrease in GAD67 activity was due to increased cAMP - protein kinase A (PKA) - dependent phosphorylation of GAD67, but not as a result of changes in either GAD67 mRNA or protein expression. PKA inhibitor restored GAD67 activity and GABA levels in IH treated cells. PC12 cells express dopamine 1 receptor (D1R), a G-protein coupled receptor whose activation increased adenylyl cyclase (AC) activity. Treatment with either D1R antagonist or AC inhibitor reversed IH-evoked GAD67 inhibition. Silencing D1R expression with siRNA reversed cAMP elevation and GAD67 inhibition by IH. These results provide evidence for the role of D1R-cAMP-PKA signaling in IH mediated inhibition of GAD67 via protein phosphorylation resulting in down regulation of GABA synthesis.
INTRODUCTION
Humans with recurrent apneas are prone to develop cardio-respiratory abnormalities including hypertension, sympathetic activation, breathing irregularities, myocardial infarction and stroke (Nieto et al., 2000). Intermittent hypoxia (IH) is one of the major contributing factors for cardio-respiratory morbidities associated with sleep apneas (Foster et al., 2007; Prabhakar et al., 2007). Studies on rodents showed that IH elevated the levels of neurotransmitters including dopamine (DA) (Raghuraman et al., 2009) and C-terminally amidated neuropeptides such as substance P and neuropeptide Y (Sharma et al., 2009) in the brainstem regions and norepinephrine in the adrenal medulla (Kumar et al., 2006) that are known to involve in the regulation of cardiovascular function and sympathetic activity. The augmentation of catecholamines and bioactive peptide levels by IH is, in part, due to increased synthesis via activation of their respective rate-limiting synthesizing enzymes, tyrosine hydroxylase (TH) and peptidylglycine-α-amidating monooxygenase involving post-translational protein phosphorylation (Raghuraman et al., 2009) and proteolytic processing (Sharma et al., 2009), respectively. It remains to be determined whether the effects of IH also extend to other transmitter systems including amino acid transmitters.
GABA, a major inhibitory amino acid neurotransmitter in the central nervous system (Watanabe et al., 2002), has been implicated in the regulation of blood pressure and sympathetic activity (Schreihofer and Guyenet, 2002). In addition to its role as a neurotransmitter, GABA also functions as metabolite and as neurotrophic and neurodifferentiating signal molecule during early ontogenesis (Waagepetersen et al., 1999; Owens and Kriegstein, 2002). GABA is synthesized by enzymatic decarboxylation of L-glutamate involving pyridoxal-L-phosphate (PLP) requiring glutamic acid decarboxylase (GAD; EC 4.1.1.15). Following its pre-synaptic release, GABA is taken up by either neurons or glia by high affinity GABA transporters and subsequently metabolized by GABA-transaminase (GABA-T) to succinic semialdehyde, and then to succinate via oxidation. Two distinct molecular forms of GAD, viz., cytosolic 67-kDa (GAD67) and vesicular 65-kDa (GAD65) forms are known (Kaufman et al., 1991). Although both isoforms generate GABA, GAD67 exhibits a greater affinity for the co-factor PLP than GAD65 and exists in an active PLP-bound holoGAD form (Martin and Rimvall, 1993). On the other hand, GAD65 exists in an inactive PLP-unbound apoGAD form and requires binding of PLP for activation (Martin et al., 2000). The activities of GAD67 and GAD65 are known to be regulated in vivo by a variety of post-translational mechanisms that include protein phosphorylation and dephosphorylation, cysteine oxidation, palmitoylation and activity-dependent proteolytic processing (Wei and Wu, 2008). The effects of reversible protein phosphorylation on the activity of GAD isoforms have been well documented. In vitro studies show that GAD67 is inhibited by phosphorylation involving protein kinase A (PKA) whereas GAD65 is activated by phosphorylation mediated by protein kinase Cε (Wei et al., 2004). Threonine 91 has been identified as the major phosphorylation site of GAD67; however, the site of phosphorylation for GAD65 has not yet been identified. Multiple protein phosphatases (PP) including PP1, PP2A and PP2B have been shown to dephosphorylate GAD in vitro (Wei et al., 2004; Wei and Wu, 2008). Both GAD isoforms contain redox sensitive cysteine residues and oxidation of these residues results in enzyme inactivation (Wei and Wu, 2005). Furthermore, GAD undergoes proteolytic cleavage in vivo generating truncated forms. While truncated GAD65 was more active than the full length form the reverse is true for GAD67 (Sha et al., 2008).
Pheochromocytoma 12 (PC12) cells are derived from rat adrenal medullary tumors and they express multiple transmitters including DA and acetylcholine (Greene and Rein, 1977). PC12 cells are oxygen sensing cells wherein they release DA in response to acute hypoxia (Kumar et al., 1998) and they respond to continuous hypoxia (CH) by up-regulating hypoxia-inducible transcription factors 1 and 2 (HIF-1 and HIF-2) (Nanduri et al., 2009) and HIF-regulated genes such as TH and vascular endothelial growth factor (Seta et al., 2004). Previously, using this cell culture model, we showed that IH not only increases DA level (Kumar et al., 2003) but also facilitates acute hypoxia-evoked DA release (Kim et al., 2004). More over, IH-induced increase in cellular DA level is, in part, due to activation of TH, the rate-limiting enzyme in catecholamine biosynthesis via increased phosphorylation of serine-40 (Kumar et al., 2003). In addition to DA, PC12 cells also express GABA and its synthesizing enzymes, GAD65 and GAD67 and exposure of the cell cultures to CH has been shown to increase GAD65 and GAD67 protein and mRNA expressions (Kobayashi and Millhorn, 2001). Studies in bovine and rat adrenal medulla showed that GABA is stored in chromaffin granules and plays a paracrine or autocrine role for catecholamine secretion (Peters et al., 1989; Matsuoko et al., 2008). Although the role of GABA in PC12 cells is not known, based on the above studies in adrenal medullary chromaffin cells from which PC12 cells were originally derived, it is likely that GABA may function as a modulator of stress-evoked neurotransmitter secretion.
In the present study, our objectives are to determine the effects of IH on GAD activity and GABA and to elucidate the associated signaling mechanism(s) in PC12 cells. Previous studies have shown that DA via activation of either dopamine 1 or 2 receptors (D1R or D2R) differentially regulates mRNA expression of GAD65 and GAD67 in the rat striatum (Lindefors, 1993; Laprade and Soghomonian, 1995). D1R activation facilitates adenylyl cyclase (AC) - PKA signaling (Fimia and Sassone-Corsi, 2001) and PKA-dependent phosphorylation inhibited GAD67 activity (Wei et al., 2004). These findings led us to examine the involvement of D1R-AC-PKA signaling in IH-induced alterations in GAD activity and GABA in PC12 cells.
EXPERIMENTAL PROCEDURES
Materials
All the chemicals and reagents used in this study were of analytical grade and obtained from Sigma-Aldrich, unless otherwise stated. Gentamycin and other cell culture components were obtained from Invitrogen.
Cell culture
PC12 cells were grown in Dulbecco’s modified Eagle’s medium (DMEM, Invitrogen) supplemented with 5% fetal bovine serum, 10% horse serum and 50 μg/L gentamycin in a humidified atmosphere of 10% CO2 at 37°C. Experiments were performed on cells grown to ~80% confluence between passages 2-6. Prior to either gas challenges described below or to drug treatments (for 30 min), the cells were grown in serum- and antibiotic-free medium for 16 h.
Exposure to IH
PC12 cells were exposed to IH as described earlier (Yuan et al., 2008) with few modifications. Briefly, petri-dishes containing serum starved cells were placed in a plexi-glass chamber and exposed to alternating cycles of hypoxia (1.5% O2 for 30 sec) and normoxia (21% O2 for 5 min) at 37°C for 10, 30, 60 and 120 cycles. Cells exposed to normoxia (21% O2) for similar duration served as controls.
Isolation of soluble and membrane-enriched fractions
PC12 cells exposed to either normoxia or IH were homogenized in 25 mM Tris-HCl buffer, pH 7.5, containing 250 mM sucrose, 20 mM MgCl2, 0.1 mM EGTA, 0.2 mM phenyl methyl sulfonyl fluoride, 1 mM 2-aminoethylisothiouronium bromide, protease (Roche) and phosphatase (Calbiochem) inhibitor cocktail. The extract was centrifuged at 5,500 x g for 15 min to remove the nuclear and mitochondrial fractions and the resulting supernatant was further centrifuged at 40,000 x g for 30 min. Both the supernatant and the pellet containing the soluble and the membrane-enriched fraction, respectively were stored at −80°C till further analysis of GAD activity.
Assay of GAD activity
GAD activity was determined using a fluorimetric assay described previously (Lowe et al., 1958). Briefly, one milliliter of the reaction mixture containing 100 mM phosphate buffer, pH 6.4, 12 mM L-glutamic acid and 10 μg protein equivalent of the cellular fraction was incubated at 37°C for 60 min. The amount of GABA formed during the reaction was determined by incubating 100 μl of reaction mixture first with 20 μl of 14 mM ninhydrin in 0.5M sodium carbonate buffer, pH 9.9 for 30 min at 60°C and then, after cooling on ice, with 20 μl copper-tartarate reagent for 15 min at room temperature. At the end of the incubation, relative fluorescence intensity was measured using excitation and emission wavelengths of 375 and 485 nm, respectively. GAD activity was expressed as nanomoles of GABA formed per hour per milligram protein. The protein concentration was determined using the DC protein assay (Bio-Rad Lab) following manufacturer’s instructions.
Analysis of GABA-T activity
GABA-T activity in PC12 cell extracts was determined using a coupled enzyme assay (Crooks et al., 2007). The reaction mixture containing 100 mM pyrophosphate buffer (pH 8.4), 3.5 mM β-mercaptoethanol, 5 mM α-ketoglutarate, 1 mM NAD+, and 18 mM GABA was incubated at 4 °C for 30 min and the change in absorbance at 340 nm was measured. GABA-T activity was expressed as nmoles of product formed per hour per milligram protein.
Determination of GABA turnover
GABA turnover was determined by measuring the rate of accumulation of GABA in cells following 2-aminooxy-acetic acid (AOAA, Sigma Chemical Company, St. Louis, MO) - induced inhibition of GABA-T activity as described previously (Grattan and Selmanoff, 1993). Cells were treated with AOAA (1 μM) for 30 min prior to their exposure to either normoxia or IH. Cells exposed to gas challenges in the absence of AOAA served as controls. The relative increase in GABA concentrations in AOAA-treated cells compared with the non-AOAA-treated cells was used as a measure of GABA turnover which was expressed as nanomoles of GABA per hour per milligram protein.
Analysis of GABA content
PC12 cells were extracted with 0.4N perchloric acid in a micro ultrasonic cell disrupter. The extract was centrifuged at 18,000 x g for 5 min and the supernatant was neutralized, derivatized with o-phthalaldehyde (OPA) as described previously (Donzanti and Yamamoto, 1988). OPA derivatives of amino acids were separated on a reverse-phase C18 column (Phenomenex, CA) by isocratic elution at 26°C with a mobile phase consisting of 28% (vol/vol) methanol, 0.1 M sodium phosphate, 0.13 mM EDTA adjusted to pH 6.5 with phosphoric acid. Under the experimental conditions, GABA eluted around ~ 45 min. GABA concentration was determined from a standard curve relating the concentration of GABA and the peak height and expressed as nanomoles of GABA per milligram protein.
Assay of PKA activity
Cells were homogenized in 25 mM Tris-HCl, pH 7.4, containing complete protease inhibitor cocktail (Roche) and phosphatase inhibitor cocktail set I (Calbiochem) at 4°C. The homogenate was centrifuged at 18,000 x g for 1 h at 4°C and the resulting supernatant was used for protein kinase assay. PKA activity was measured using the PKA activity assay kit (EKS-390A; Assay Designs, MI) and expressed as the change in absorbance at 450 nm per hour per milligram protein. The specificity of the assay was assessed using PKA specific inhibitory peptide I (PKAI; 1 μM).
Determination of cAMP content
Cellular level of cAMP was determined using a commercially available cAMP assay kit (cAMP-Screen Direct chemiluminescent system; Applied Biosystems) following manufacturers’ instructions. cAMP level was expressed as picomoles per milligram protein.
Immuno-precipitation of GAD67 and analysis of phospho-GAD67
Aliquots of the soluble fraction were incubated with 10 μg anti-GAD67 monoclonal antibody for 18 h at 4°C, followed by incubation with 30 μl Protein-G bead suspension. The immune complexes were separated by SDS-PAGE and immunoblotted with either anti-phospho threonine, or anti-GAD67 or anti-β-actin antibodies (loading control). Protein band intensities were quantitated using ImageJ software.
Reverse transcription-polymerase chain reaction
Total RNA was isolated from cells using Trizol reagent. The reverse transcription was carried out on 5 μg of total RNA using SuperScript II reverse transcriptase and random primers following manufacturers’ instructions (Invitrogen, Carlsbad, USA). Aliquots of first-strand cDNA were amplified by PCR using gene specific primers. Primers were constructed based on the reported cDNA sequence of GAD67 (Michelsen et al., 1991) in rats. The sequences and the predicted length of the amplified DNA products are as follows: GAD67: 5 ′-CACCCGTGTTTGTTCTTATG-3 ′ and 5 ′-GCTCCAGGCATTTGTTGATC-3 ′ (800 bp). Amplification of 28SrRNA was performed as control. The primers are: 5 ′-TGAACTATGCTTGGGCAGGG-3 ′ and 5 ′-AGCGCCATCCATTTTCAGGG-3 ′ (500 bp). Primers for D1R were : 5 ′-CAGGAGATACCGTTAGCAAGCC-3 ′ and 5 ′-TGTTATCATAGCCTGGACTGCAA-3 ′ (182 bp) (Zanassi et al., 1999). DNA was amplified using PCR conditions as reported earlier (Kobayashi and Millhorn, 2001). All PCR reactions were done in duplicate and the level of GAD67 and D1R expression was normalized to 28SrRNA.
Immunoblot analysis of GAD and D1R protein expression
Either total cell-free extract or cytosolic fraction of PC12 cells containing 40 μg of protein were separated on a 10% SDS–PAGE gel and the proteins were transferred to nitrocellulose membranes and immunolabeled overnight at 4°C with antibodies against either GAD 65/67 (rabbit polyclonal, 1:1000; Millipore Corp.,), or phospho threonine (rabbit polyclonal; 1:500; Millipore Corp.,) or β-actin (mouse monoclonal, 1:5000; Sigma-Aldrich), or D1R (mouse monoclonal, 1:2000; Sigma-Aldrich) followed by incubation with appropriate horseradish peroxidase conjugated secondary antibody for 1 h at 37°C. Immunolabeled proteins were detected by enhanced chemiluminescence detection system (Amersham). Image densities were quantified using ImageJ software.
In vitro dephosphorylation of GAD67 by PP1, PP2A and PP2B
Cell-free extracts were incubated with the catalytic subunit of either PP1 (2.5 units) or PP2A (0.2 units) or PP2B (0. 2 units) in 25 mM Tris-HCl buffer, pH 7.0 for 15 min at 25°C. For control samples, the extracts were incubated with buffer. Following dephosphorylation, the reaction mixtures were assayed for GAD67 activity as described above.
siRNA transfection
Transfection of PC12 cells with siRNAs specific for either D1R or scrambled sequence (control) was performed at a concentration of 60 picomoles per millilitre by following the procedure recommended by the manufacturer (Santa Cruz Biotechnology, Santa Cruz, CA). A mixture of 3 siRNAs, each of 20-25 nucleotides in size, designed to specifically knockdown target gene expression was used. For control, siRNA consisting of a scrambled sequence was used. One day prior to transfection, cells were plated in growth medium without antibiotics and were transiently transfected with individual siRNA separately. After 8 h, the medium was replaced with a complete culture medium and the transfection was continued for an additional 36 h. Following the completion of transfection, cells were exposed to either normoxia or 60 cycles of IH as described above. The efficiency of siRNA knockdown of D1R was confirmed by western blot analysis.
Experimental protocols
Series 1
GAD activity was determined in triplicate in the cytosolic and membrane-enriched fractions of PC12 cells (~3 × 106) exposed to either IH (for 10, 30, 60 and 120 cycles) or normoxia (n = 4 experiments in each group). GABA level was determined in triplicate in acid extracts of PC12 cells (~3×106) exposed to either IH (60 cycles) or normoxia (n = 4 experiments in each group).
Series 2
The mRNA and protein expression of GAD67 and GABA level in either total cell-free extract or soluble fraction of PC12 cells (~3 × 106) exposed to either normoxia or 60 cycles of IH were analyzed in triplicate (n = 4 experiments in each group).
Series 3
GAD67 phosphorylation was assessed in the soluble fraction isolated from IH treated (60 cycles) and normoxic cells (~10×106; n = 4 experiments in each group). PKA activity was measured in triplicate in PC12 cells (~3 × 106) exposed to either normoxia or 60 cycles of IH (n = 4 experiments in each group).
Series 4
The effects of PP1, PP2A and PP2B treatments on GAD67 activity was determined in duplicate in extracts of PC12 cells (~3 × 106) exposed to either normoxia or 60 cycles of IH (n = 4 experiments in each group). GAD67 activity was measured in duplicate in PC12 cells exposed to either normoxia or 60 cycles of IH in the presence of inhibitors specific to either PKA [i.e., PKA inhibitor-α (14–22) amide, Myr-N-Gly-Arg-Thr-Gly-Arg-Arg-Asn-Ala-Ile-NH2, 0.2 and 1 μM], or protein kinase C (PKC) (bisindolylmaleimide, BIS; 0.1 and 1 mM) or AC (dideoxyadenosine, DiDA; 5 and 50 μM; n = 4 experiments in each group). Inhibitors were added to the cell culture medium 30 min prior to either normoxic or IH exposure and remained in the medium during the entire duration of gas challenges.
Series 5
Protein and mRNA expression of D1R in PC12 cells (~3 × 106) exposed to either normoxia or 60 cycles of IH were analyzed in duplicate (n = 4 experiments in each group). GAD67 activity was measured in triplicate in PC12 cells exposed to either normoxia or 60 cycles of IH in the presence of D1R antagonist (SCH 23390, 10 and 100 nM n = 4 experiments in each group; Tocris Bioscience, MO). The antagonist was added to the cell culture medium 30 min prior to either normoxic or IH exposure and remained in the medium during the entire duration of gas challenges.
Series 6
Protein expression of D1R was assessed in duplicate in PC12 cells (~3 × 106) transfected with siRNA specific for either D1R or scrambled sequence (n = 4 experiments in each group). GAD67 activity and cAMP level in mock as well as D1R siRNA transfected PC12 cells exposed to either normoxia or 60 cycles of IH were determined in triplicate (n = 4 experiments in each group).
Data analysis
All data derived from the 6 series of experiments as outlined above were expressed as mean ± SEM. Statistical significance was evaluated by unpaired t-test or one-way ANOVA for repeated measures. p value <0.05 was considered significant.
RESULTS
Expression of GAD isoforms in PC12 cells
To determine the relative abundance of GAD isoforms in PC12 cells, GAD enzyme activity and isoform expression in the cytosolic and membrane-enriched fractions were analyzed. We found that more than 90% of GAD activity occurs in the cytosolic fraction (cytosol, 4.2 ± 0.4 nmol per h per milligram versus membrane, 0.2 ± 0.03 nmol per h per milligram). Western blot analysis revealed that GAD67 is exclusively expressed in the cytosolic fraction whereas GAD65 is localized to the membrane fraction (see Supplemental Fig. S1). Therefore, for subsequent analysis of regulation of GAD activity by IH only the cytosolic fraction of PC12 cells expressing GAD67 was used. In the following sections, the activity due to GAD67 is referred as “GAD67 activity”.
Effects of IH on GAD67 and GABA-T activities and GABA turnover
Exposure of cells to IH progressively decreased GAD67 activity in a cycle-dependent manner. A significant decrease in GAD67 activity compared to normoxia treated cells was seen with 30 cycles of IH which reached a plateau with 60 cycles of IH (Fig. 1A). IH-evoked down regulation of GAD67 activity was associated with a marked reduction in the steady-state level of GABA (Fig. 1B). To assess the potential contribution of GABA turnover to the observed decrease in cellular GABA level, cells were treated with AOAA (1 μM), an inhibitor of GABA-T, prior to exposure to either normoxia or IH. The increase in GABA level in the presence of AOAA was used as a measure of GABA turnover. IH caused a modest but significant decrease in GABA turnover (Fig. 1C). Further, GABA-T activity in IH treated cells was higher than that of normoxic cells (Fig. 1D). The decrease in GAD67 activity seen in IH treated cells was not due to decrease in either GAD67 mRNA or protein level (Fig. 2A&B).
Figure 1. Effects of IH on GAD67 and GABA-T activities, GABA level and GABA turnover.
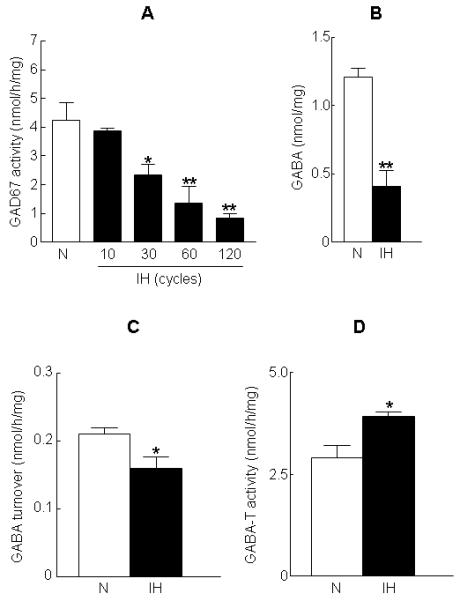
PC12 cells were exposed to 10, 30, 60 and 120 cycles of IH or to normoxia (N). (A) GAD67 activity was determined using fluorimetric assay as described in “Methods”. (B) GABA level in normoxia and IH treated cells. GABA level was determined using HPLC coupled with electrochemical detection as described in “Methods”. (C) GABA turnover and (D) GABA-T activity in normoxia and IH treated cells. GABA turnover and GABA-T activity were determined as described in “Methods”. Data from 4 independent experiments are expressed as mean ± SEM. * & ** denote p< 0.05 and p < 0.01 respectively.
Figure 2. Effects of IH on GAD67 mRNA and protein expression and GAD67 phosphorylation.
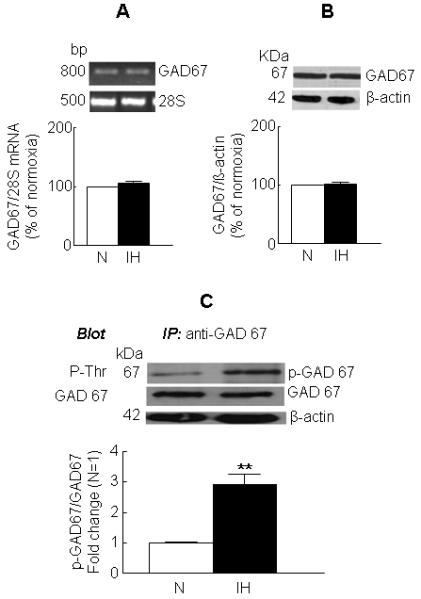
Cells were exposed to 60 cycles of IH or to normoxia (N). GAD67 mRNA (A) and protein (B) were determined by RT-PCR and western blot analysis, respectively. Representative example (top panel) and average data (bottom panel) are shown. 28SrRNA and β-actin served as normalization controls. (C) Effect of IH on GAD67 phosphorylation. GAD67 from the soluble fraction was immuno-precipitated with a monoclonal anti-GAD67 antibody and the immune complex was resolved on SDS-PAGE and probed with either anti-phospho threonine or polyclonal anti-GAD67 or anti- β-actin (loading control) antibodies. Representative example of immunoblot (top panel) and average data (bottom panel) are shown. Data from 4 independent experiments are expressed as mean ± SEM. ** denote p < 0.01.
In the subsequent experiments, we focused on the mechanisms by which IH down regulates GAD67 activity in PC12 cells. For these studies, cells exposed to 60 cycles of IH which showed a maximum inhibition of GAD67 activity were used.
IH increases GAD67 phosphorylation
Because threonine phosphorylation results in inhibition of GAD67 activity (Bao et al., 1995), the possible role of phosphorylation in IH-evoked down regulation of GAD67 activity was investigated. GAD67 was immuno-precipitated from the cytosolic fraction of control and IH-exposed cells and the resulting immuno-precipitates were analyzed by western blot using anti-phospho threonine antibody. Low levels of phosphorylated form of GAD67 were detected in cells exposed to normoxia. However, exposure to IH resulted in a ~2.8-fold increase in GAD67 phosphorylation as compared to the level seen in the normoxic cells (Fig. 2C).
PKA mediated phosphorylation contributes to IH-induced GAD67 inhibition
Previous in vitro studies showed that PKA, which requires cAMP for activation, mediates the phosphorylation of GAD67 at Thr-91 (Wei et al., 2004) and the subsequent inhibition of GAD67 activity. Therefore, we examined whether regulation of PKA contributes to IH-evoked phosphorylation of GAD67 and inhibition of GAD67 activity. IH induced a ~4-fold increase in PKA activity (Fig. 3A). Pretreatment with PKAI, a membrane permeable PKA-specific inhibitor, dose-dependently attenuated IH-induced GAD67 inhibition (Fig. 3B), whereas it had no significant effect on GAD67 activity in normoxic control cells (data not shown). A similar reversal of GAD67 inhibition by IH was not seen in cells pretreated with BIS, a PKC-specific inhibitor (Fig. 3B). More over, PKAI reversed IH-evoked decrease in GABA level (Table 1). Furthermore, dephosphorylation via treatment of cell extracts with the catalytic subunit of either PP1 or PP2A or PP2B restored GAD activity in IH treated cells (Table 2) but not in normoxic cells (data not shown).
Figure 3. IH up regulates protein kinase A (PKA) activity and PKA inhibitor prevents IH-induced GAD67 inhibition.
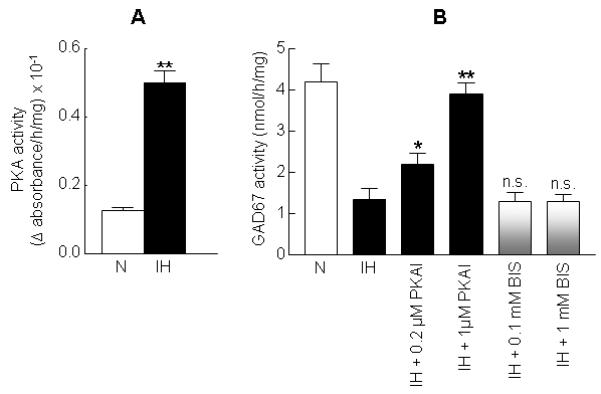
(A) Effect of 60 cycles of IH on PKA activity. (B) Effects of PKA and PKC inhibitors on GAD67 activity in IH treated cells. Cells were treated with either PKA inhibitor-α (14-22) amide (PKAI; 0.2 and 1 μM), an inhibitor of PKA or bisindolylmaleimide (BIS; 0.1 and 1 mM), a PKC-specific inhibitor prior to IH treatment. Data from 4 independent experiments are expressed as mean ± SEM. * & ** denote p< 0.05 and p < 0.01 respectively, n.s. – not significant and N – normoxia.
Table 1.
Effects of pharmacological agents on GABA level in PC12 cells exposed to intermittent hypoxia (IH)$
| DRUG TREATMENT | GABA (NMOLE/MG) |
|---|---|
| Normoxia | 1.11 ± 0.23 |
| IH | 0.39 ± 0.15 |
| IH + PKAI (1 μM) | 0.99 ± 0.08** |
| IH + BIS (1 mM) | 0.40 ± 0.22n.s. |
| IH + DiDA (50 μM) | 1.21 ± 0.18** |
| IH + SCH 23390 (100 nM) | 1.02 ± 0.19** |
GABA level was determined in acid extracts of PC12 cells exposed to either normoxia or IH (60 cycles) in the absence or presence of indicated drugs using HPLC coupled with electrochemical detection.
denotes p < 0.01 compared to IH treatment
not significant compared to IH.
Table 2.
Effect of protein phosphatase (PP)-mediated dephosphorylation on GAD67 activity in IH treated PC12 cells$
| TREATMENT | GAD67 ACTIVITY (NMOL/H/MG) |
|---|---|
| Normoxia | 4.19±0.56 |
| IH | 1.35±0.59 |
| IH + PP1 (2.5 U) | 2.79±0.26** |
| IH + PP2A (0.2 U) | 3.29±0.28** |
| IH + PP2B (0.2 U) | 3.59±0.11** |
denotes p < 0.01 compared IH treatment.
denotes p < 0.01 compared IH treatment.
D1R mediates cAMP/PKA-dependent GAD67 inhibition by IH
Thus far, our results suggest that activation of PKA is required for IH-evoked inhibition of GAD67 activity. PKA activity in vivo is regulated by the second messenger cAMP, whose level is determined by the activity of AC which requires G-protein coupled receptor activation (Hanoune and Defer, 2001). PC12 cells not only express but also release DA in response to a variety of stimuli including acute hypoxia (Kumar et al., 1998). Exposure of PC12 cells to IH results in augmented DA release in response to acute hypoxia (Kim et al., 2004). More over, D1R, a G-protein coupled receptor which is positively linked to AC activation, is expressed in PC12 cells (Zachor et al., 2000; Jin et al., 2008). Therefore, in the following series of experiments, we assessed whether D1R mediated AC activation plays a role in PKA-dependent GAD67 inhibition by IH.
Since stimulus-evoked DA release is a prerequisite for activation of DA signaling, we first determined whether IH-induced facilitation of DA release by hypoxia contributes to GAD67 inhibition by IH. Cells were treated with either 2-aminoethoxydiphenyl borate (2-APB), an inhibitor of intracellular Ca2+-mobilization or CdCl2, an inhibitor of calcium entry via voltage-activated Ca2+ channel prior to IH exposure. Previously, we found that these inhibitors of calcium trafficking inhibited IH-evoked augmentation of DA release by hypoxia (Kim et al., 2004). Both 2-APB and CdCl2 reversed IH-evoked GAD67 inhibition (see Supplemental Fig. S2) without affecting GAD67 activity in normoxic control cells (data not shown).
We then determined the effect of IH on AC activity by measuring cellular cAMP levels. In normoxic cells, cAMP level was 6.8 ± 0.5 pmol per milligram protein and IH increased cAMP level by nearly 3-fold (Fig. 4A). DiDA, a potent AC inhibitor, dose-dependently reversed IH-induced GAD67 inhibition (Fig. 4B), whereas it had no effect on GAD67 activity in normoxic control cells (data not shown). Furthermore, DiDA prevented IH-induced decrease in GABA level (Table 1).
Figure 4. Elevation of cAMP level by IH and reversal of IH-evoked GAD67 inhibition by adenylyl cyclase inhibitor.
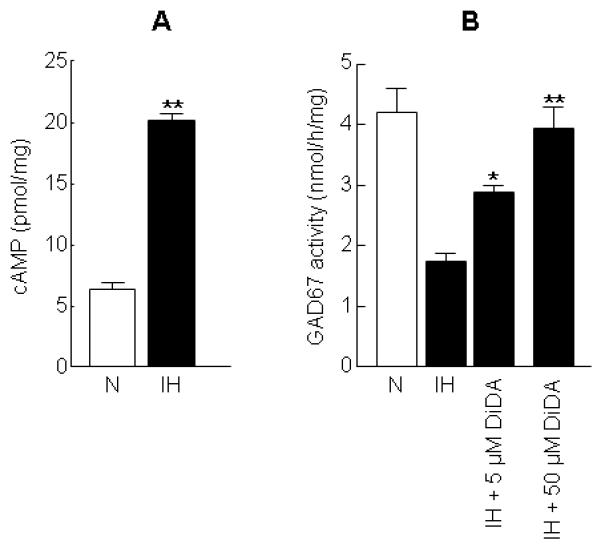
(A) Augmentation of cAMP level by 60 cycles of IH. (B) Effect of dideoxyadenosine (DiDA), an adenylyl cyclase inhibitor on GAD67 activity in IH treated cells. Data from 4 independent experiments are expressed as mean ± SEM. * & ** denote p< 0.05 and p < 0.01 respectively and N -normoxia.
To determine whether D1R contributes to IH-induced activation of both AC and PKA and the ensuing inhibition of GAD67 and decrease in GABA level, cells were treated with SCH 23390 (10 and 100 nM), a D1R-specific antagonist, prior to exposure to either normoxia or IH. D1R antagonist (100 nM) not only attenuated IH-evoked increases in cAMP (Fig. 5A) and PKA activity (Fig. 5B) but also prevented GAD67 phosphorylation (Fig. 5C), GAD67 inhibition (Fig. 5D) and GABA down regulation (Table 1) evoked by IH. By contrast, in normoxic cells, SCH 23390 treatment was without any effect on either PKA (normoxia alone, 0.2 ± 0.02 versus normoxia + SCH 23390, 0.22 ± 0.01 ng of active PKA per milligram protein) or GAD67 activity (-SCH23390, 4.2±0.3 versus +SCH23390, 4.3±0.1 nmol per h per milligram protein). Notably, IH also increased D1R mRNA and protein expression by 3- and 2.8-fold, respectively (Fig. 6A).
Figure 5. Dopamine D1 receptor (D1R) antagonist prevents IH-evoked alterations in cAMP, PKA, GAD67 phosphorylation and GAD activity in PC12 cells.
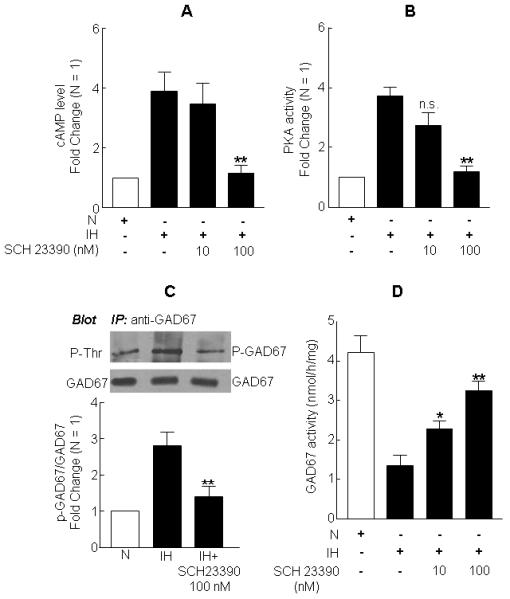
Cells were treated with either 10 or 100 nM of SCH 23390, a D1R-specific antagonist prior to exposure to either normoxia (N) or 60 cycles of IH. Analyses of cAMP level (A) and PKA activity (B) are shown. (C) Effect of SCH 23390 on GAD67 phosphorylation in IH treated cells. Representative example of immunoblot (top panel) and average data (bottom panel) are shown. (D) Effect of D1R specific antagonist on GAD67 activity in IH cells. Data from 4 independent experiments are expressed as mean ± SEM. * & ** denote p< 0.05 and p < 0.01 respectively and n.s. – not significant.
Figure 6. Up-regulation of D1R mRNA and protein by IH and reversal of IH responses of cAMP and GAD67 activity by siRNA silencing of D1R expression.
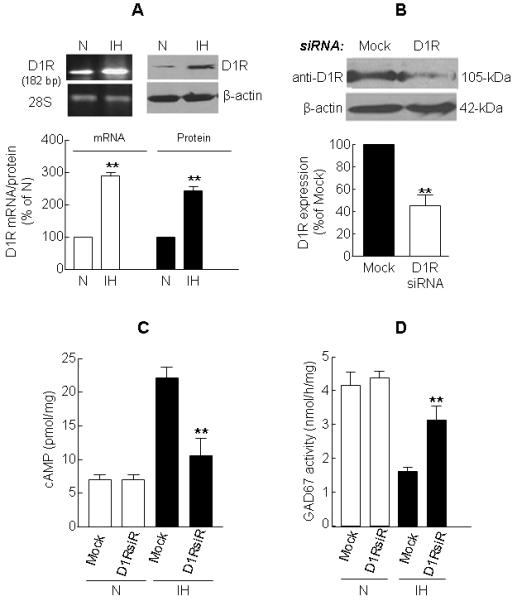
(A) IH increases D1R mRNA (left panel) and protein (right panel) expression. (B) Immunoblot analysis of D1R protein expression in mock (control) and D1R siRNA transfected cells. Representative example (top panel) and average data (bottom panel) are shown. (C) Effect of D1R specific siRNA on cAMP level in normoxic (N) and IH treated cells. (D) Reversal of IH-induced decrease in GAD67 activity by siRNA silencing of D1R expression. Data from 4 independent experiments are expressed as mean ± SEM. ** denotes p < 0.01 and N - normoxia.
A similar attenuation of IH-induced GAD67 inhibition was also seen with SKF 83566 (at 0.5 and 5 nM), which is a more potent D1R antagonist than SCH 23390 (see Supplemental Fig. S3). However, blockade of β-adrenergic receptor with ICI 118551 (at 100 and 1000 nM) had no significant effect on GAD67 activity in IH-treated cells (see Supplemental Fig. S3).
To further confirm the role of D1R signaling in IH-evoked GAD67 inhibition, we transiently transfected PC12 cells with siRNA specific for D1R. As shown in Fig. 6B, siRNA treatment effectively attenuated D1R protein expression. Silencing D1R expression with siRNA partially reversed elevation in cAMP levels (Fig. 6C) as well as GAD67 inhibition (Fig. 6D) by IH. However, in normoxic cells, D1R siRNA silencing had no significant effect on either cAMP level or GAD67 activity.
DISCUSSION
The objectives of the present study were to determine the effect of IH on GAD67 activity and to elucidate the underlying cellular mechanism(s). Our results demonstrated that IH inhibits GAD67 activity in PC12 cells via increased phosphorylation of GAD67 resulting in decreased GABA level. More importantly, IH-induced post-translational phosphorylation of GAD67 was mediated by mechanisms involving activation of D1R-cAMP-PKA signaling pathway.
In contrast to the inhibitory effect of IH on GAD67 activity, Kobayashi and Millhorn (2001) reported an increase in GAD activity in PC12 cell cultures following exposure to CH lasting several hours to days. In preliminary studies, we found that exposure of cells to CH for 90 min (which is equivalent to the cumulative duration of hypoxia seen in 60 cycles of IH) neither affects the activity nor alters phosphorylation of GAD67. It is, therefore, conceivable that the inhibitory effect of hypoxia on GAD67 activity is specific to IH. IH-evoked GAD67 inhibition was not associated with decreases in either mRNA or protein expression of GAD67, whereas CH-evoked up-regulation of GAD activity was associated with concomitant increases in GAD67 mRNA and protein expression (Kobayashi and Millhorn, 2001). Taken together, these findings suggest that IH and CH exert distinct and diametrically opposite modulatory effects on GAD67 activity.
In vitro studies have identified several protein modification reactions including phosphorylation of threonine residue, oxidative modification of sulfhydryls and truncation via proteolytic cleavage lead to inhibition of GAD67 activity (Wei and Wu, 2008). Our results showed that IH neither alters GAD67 protein expression nor facilitates the generation of truncated GAD67 whereas it markedly increases the level of phosphorylated form of GAD67 compared to untreated control. Furthermore, post-IH treatment with protein phosphatases reversed GAD67 inhibition by IH. Taken together, these findings support a critical role for post-translational phosphorylation in IH-induced GAD inhibition.
Cyclic AMP-dependent PKA plays a major role in GAD67 phosphorylation and the resultant GAD67 inhibition (Wei et al., 2004). The following lines of evidence support the role for PKA in IH-induced GAD67 inhibition. First, IH increased the level of cAMP and PKA activity. Second, incubation of cell cultures, prior to IH exposure, with a PKA-specific inhibitor prevented GAD67 inhibition by IH. Third, DiDA, an inhibitor of AC which generates cAMP required for PKA activation, prevented IH-induced inhibition of GAD67. Thus, facilitation of GAD67 phosphorylation by IH seems to be primarily mediated via mechanisms involving cAMP-dependent PKA activation. Consistent with this observation is the finding that in the rat medullary brainstem regions, IH up-regulates PKA-dependent serine phosphorylation of TH, the rate-limiting enzyme in catecholamine biosynthesis (Raghuraman et al., 2009). Results from this study further showed that chronic IH, in addition to PKA, also activated Ca2+-calmodulin-dependent kinase and extracellular signal-regulated kinase with a concomitant down regulation of PP2A resulting in marked sustained up-regulation of TH phosphorylation and activity (Raghuraman et al., 2009). However, it remains to be determined whether down regulation of PP also contributes to IH-induced GAD67 inhibition in PC12 cells.
A major finding of the present study is that IH-induced PKA activation and the ensuing GAD67 phosphorylation and inhibition of GAD67 activity is coupled to activation of D1R. PC12 cells express a number of DA receptor subtypes including D1R which is positively coupled to AC (Zachor et al., 2000; Neve et al., 2004). Several lines of evidence suggest that D1R activation is up-stream to IH-evoked phosphorylation and initiates a cascade of reactions that lead to inhibition of GAD67. First, IH has been shown to facilitate the release of DA by acute hypoxia (Kim et al., 2004) which may trigger D1R activation in an autocrine manner. Second, blockade of DA release by inhibiting either intracellular Ca2+ mobilization and/or calcium entry via voltage-activated Ca2+ channel prevented GAD67 inhibition by IH. Third, D1R mRNA and protein expression was augmented by IH and D1R antagonist, not only attenuated IH-induced increases in cAMP and PKA activity but also reduced GAD67 phosphorylation and the ensuing GAD67 inhibition by IH. Fourth, silencing D1R expression with siRNA prevented IH-induced GAD67 inhibition. Previously, several studies have examined the role of DA receptors in GAD modulation in the rat striatum (Lindefors, 1993; Laprade and Soghomonian, 1995). Results from these studies demonstrated that DA via activation of D1R and D2R mediates the differential regulation of GAD65 and GAD67 mRNA expression in a region-specific manner. Notably, results from the present study provide evidence for an additional role for D1R in that it mediates IH-induced inhibition of GAD67 activity in the absence of any associated decrease in its mRNA level. Taken together, the above observations provide a compelling evidence for a novel role for D1R-cAMP-PKA signaling in IH-induced down regulation of GAD67 activity (Fig. 7).
Figure 7.
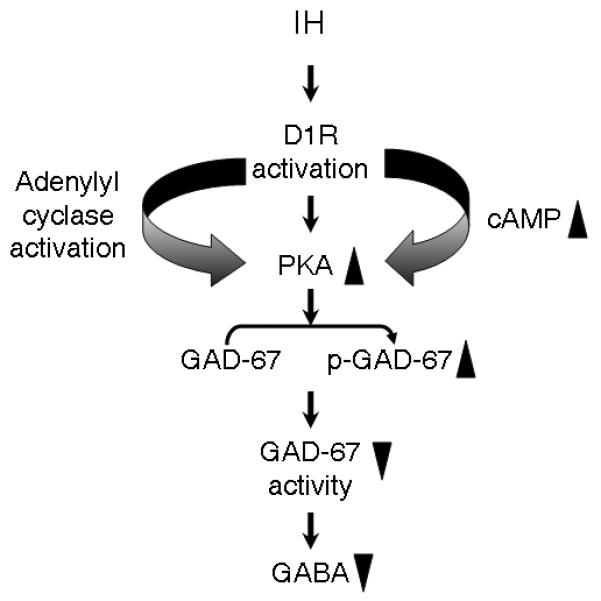
Schematic presentation of IH-induced activation of D1R-cAMP-PKA signaling pathway facilitating GAD67 phosphorylation mediated GAD67 inhibition and GABA down regulation. Key:▲– up-regulation; ▼– down-regulation; P-GAD67 – phospho-GAD67
Our results showed that IH attenuates steady-state GABA level in PC12 cells. Mechanism(s) involving either a decrease in GABA synthesis along with an increase in GABA catabolism via the concerted action of GABA-T and succinic semialdehyde dehydrogenase (Waagepetersen et al., 2003) or augmentation of GABA release and impairment in GABA re-uptake via alteration in GABA transporter expression may contribute to the decrease in GABA level in IH cells. Our results showed that despite an increase in GABA-T activity, GABA turnover tends to be lower in IH-treated cells than in normoxic cells. The findings that IH not only decreased GAD67 activity but also GABA turnover suggest that the reduction in the GABA level seen in IH treated PC12 cells may in part be due to attenuated GABA synthesis. Whether IH decreases the expression of GABA transporter, and if so, the attenuated GABA re-uptake is also responsible for IH-induced down regulation of GABA, however, remains to be investigated.
What is the functional significance of GAD67 inhibition and the ensuing decrease in GABA level seen in cell cultures treated with IH? In rats, treated with chronic IH, there was a significant reduction in GAD67 activity in the ventral but not in the dorsal medullary brainstem region (unpublished observations). The ventral medullary brainstem region has been shown to contribute to the central regulation of blood pressure and sympathetic activity (Avanzino et al., 1994; Schreihofer and Guyenet, 2002) both of which were elevated in obstructive sleep apnea patients (Nieto et al., 2000) as well as in rodents exposed to chronic IH (Bao et al., 1997; Kumar et al., 2006). Since GAD catalyzes the decarboxylation of glutamate to generate GABA, it is conceivable that IH-induced inhibition of GAD67 may decrease GABA levels in the brainstem. This GAD67 inhibition by IH is of functional significance in that it may result in loss of GABA-driven homeostatic control of sympathetic activity and blood pressure. Studies in guinea pigs showed that approximately 8% of the total flux through the tricarboxylic acid (TCA) cycle in cerebral cortical slices is mediated via “GABA shunt” (Balazs et al., 1970). Therefore, it is likely the IH-induced decrease in GABA level may also affect energy metabolism mediated via ‘GABA shunt’. The finding that IH induces a modest but significant decrease in GABA turnover along with a parallel decline in steady state GABA level supports such a possibility. A potential reduction in GABA shunt would be beneficial for optimal energy production during IH via augmented TCA cycle activity. However whether IH alters energy metabolism via augmentation of TCA cycle activity remains to be determined.
Supplementary Material
Footnotes
This work was supported by National Institutes of Health, Lung and Blood Institute Grants HL-90554 (to N. R. P) and HL-89616 (to G. K. K).
REFERENCES
- Avanzino GL, Ruggeri P, Blanchi D, Cogo CE, Ermirio R, Weaver LC. GABAB receptor-mediated mechanisms in the RVLM studied by microinjections of two GABAB receptor antagonists. Am. J. Physiol. 1994;266:H1722–H11728. doi: 10.1152/ajpheart.1994.266.5.H1722. [DOI] [PubMed] [Google Scholar]
- Balázs R, Machiyama Y, Hammond BJ, Julian T, Richter D. The operation of the gamma-aminobutyrate bypath of the tricarboxylic acid cycle in brain tissue in vitro. Biochem. J. 1970;116:445–461. doi: 10.1042/bj1160445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao G, Metreveli N, Li R, Taylor A, Fletcher EC. Blood pressure response to chronic episodic hypoxia: role of the sympathetic nervous system. J. Appl. Physiol. 1997;83:95–101. doi: 10.1152/jappl.1997.83.1.95. [DOI] [PubMed] [Google Scholar]
- Bao J, Cheung WY, Wu JY. Brain L-glutamate decarboxylase. Inhibition by phosphorylation and activation by dephosphorylation. J. Biol. Chem. 1995;270:6464–6467. doi: 10.1074/jbc.270.12.6464. [DOI] [PubMed] [Google Scholar]
- Crooks DR, Welch N, Smith DR. Low-Level Manganese Exposure Alters Glutamate Metabolism in GABAergic AF5 Cells. Neurotoxicology. 2007;28:548–554. doi: 10.1016/j.neuro.2007.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donzanti BA, Yamamoto BK. An improved and rapid HPLC-EC method for the isocratic separation of amino acid neurotransmitters from brain tissue and microdialysis perfusates. Life Sci. 1988;43:913–922. doi: 10.1016/0024-3205(88)90267-6. [DOI] [PubMed] [Google Scholar]
- Fimia GM, Sassone-Corsi P. Cyclic AMP signaling. J Cell Sci. 2001;114:1971–1972. doi: 10.1242/jcs.114.11.1971. [DOI] [PubMed] [Google Scholar]
- Foster GE, Poulin MJ, Hanly PJ. Intermittent hypoxia and vascular function: implications for obstructive sleep apnoea. Exp. Physiol. 2007;92:51–65. doi: 10.1113/expphysiol.2006.035204. [DOI] [PubMed] [Google Scholar]
- Grattan DR, Selmanoff M. Regional variation in γ-aminobutyric acid turnover: effect of castration on γ-aminobutyric acid turnover in microdissected brain regions of the male rat. J. Neurochem. 1993;60:2254–2264. doi: 10.1111/j.1471-4159.1993.tb03512.x. [DOI] [PubMed] [Google Scholar]
- Greene LA, Rein G. Release, storage and uptake of catecholamines by a clonal cell line of nerve growth factor (NGF) responsive pheo-chromocytoma cells. Brain Res. 1977;129:247–263. doi: 10.1016/0006-8993(77)90005-1. [DOI] [PubMed] [Google Scholar]
- Hanoune J, Defer N. Regulation and role of adenylyl cyclase isoforms. Ann. Rev. Pharmacol. Toxicol. 2001;41:145–174. doi: 10.1146/annurev.pharmtox.41.1.145. [DOI] [PubMed] [Google Scholar]
- Jin CM, Yang YJ, Huang HS, Lim SC, Kai M, Lee MK. Induction of dopamine biosynthesis by l-DOPA in PC12 cells: implications of L-DOPA influx and cyclic AMP. Eur. J. Pharmacol. 2008;591:88–95. doi: 10.1016/j.ejphar.2008.06.052. [DOI] [PubMed] [Google Scholar]
- Kaufman DL, Houser CR, Tobin AJ. Two forms of the gamma-aminobutyric acid synthetic enzyme glutamate decarboxylase have distinct intraneuronal distributions and cofactor interactions. J. Neurochem. 1991;56:720–723. doi: 10.1111/j.1471-4159.1991.tb08211.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DK, Natarajan N, Prabhakar NR, Kumar GK. Facilitation of dopamine and acetylcholine release by intermittent hypoxia in PC12 cells: involvement of calcium and reactive oxygen species. J. Appl. Physiol. 2004;96:1206–1215. doi: 10.1152/japplphysiol.00879.2003. [DOI] [PubMed] [Google Scholar]
- Kobayashi S, Millhorn DE. Hypoxia regulates glutamate metabolism and membrane transport in rat PC12 cells. J. Neurochem. 2001;76:1935–1948. doi: 10.1046/j.1471-4159.2001.00214.x. [DOI] [PubMed] [Google Scholar]
- Kumar GK, Kim DK, Lee MS, Ramachandran R, Prabhakar NR. Activation of tyrosine hydroxylase by intermittent hypoxia: involvement of serine phosphorylation. J. Appl. Physiol. 2003;95:536–544. doi: 10.1152/japplphysiol.00186.2003. [DOI] [PubMed] [Google Scholar]
- Kumar GK, Overholt JL, Bright GR, Hui KY, Lu H, Gratzl M, Prabhakar NR. Release of dopamine and norepinephrine by hypoxia from PC-12 cells. Am. J. Physiol. Cell Physiol. 1998;274:C1592–C1600. doi: 10.1152/ajpcell.1998.274.6.C1592. [DOI] [PubMed] [Google Scholar]
- Kumar GK, Rai V, Sharma SD, Ramakrishnan DP, Peng YJ, Souvannakitti D, Prabhakar NR. Chronic intermittent hypoxia induces hypoxia-evoked catecholamine efflux in adult rat adrenal medulla via oxidative stress. J. Physiol. 2006;575:229–239. doi: 10.1113/jphysiol.2006.112524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laprade N, Soghomonian JJ. Differential regulation of mRNA levels encoding for the two isoforms of glutamate decarboxylase (GAD65 and GAD67) by dopamine receptors in the rat striatum. Brain Res. Mol. Brain Res. 1995;34:65–74. doi: 10.1016/0169-328x(95)00139-j. [DOI] [PubMed] [Google Scholar]
- Lindefors N. Dopaminergic regulation of glutamic acid decarboxylase mRNA expression and GABA release in the striatum: a review. Prog. Neuropsychopharmacol. Biol. Psychiatry. 1993;17:887–903. doi: 10.1016/0278-5846(93)90018-n. [DOI] [PubMed] [Google Scholar]
- Lowe IP, Robins E, Eyerman GS. The fluorimetric measurement of glutamic decarboxylase and its distribution in brain. J. Neurochem. 1958;3:8–18. doi: 10.1111/j.1471-4159.1958.tb12604.x. [DOI] [PubMed] [Google Scholar]
- Martin DL, Rimvall K. Regulation of gamma-aminobutyric acid synthesis in the brain. J. Neurochem. 1993;60:395–407. doi: 10.1111/j.1471-4159.1993.tb03165.x. [DOI] [PubMed] [Google Scholar]
- Martin DL, Liu H, Martin SB, Wu SJ. Structural features and regulatory properties of the brain glutamate decarboxylases. Neurochem. Int. 2000;37:111–119. doi: 10.1016/s0197-0186(00)00014-0. [DOI] [PubMed] [Google Scholar]
- Matsuoko H, Harada K, Endo Y, Warashina A, Doi Y, Nakamura J, Inoue M. Molecular mechanisms supporting a paracrine role of GABA in rat adrenal medullary cells. J. Physiol. 2008;20:4825–4842. doi: 10.1113/jphysiol.2008.158709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michelsen BK, Peterson JS, Boel E, Moeldrup A, Dyrberg T, Madsen D. Cloning, characterization, and autoimmune recognition of rat islet glutamic acid decarboxylase in insulin-dependent diabetes mellitus. Proc. Natl Acad. Sci. U.S.A. 1991;88:8754–8758. doi: 10.1073/pnas.88.19.8754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanduri J, Wang N, Yuan G, Khan SA, Souvannakitti D, Peng YJ, Kumar GK, Garcia JA, Prabhakar NR. Intermittent hypoxia degrades HIF-2alpha via calpains resulting in oxidative stress: implications for recurrent apnea-induced morbidities. Proc. Natl. Acad. Sci. U. S. A. 2009;106:1199–1204. doi: 10.1073/pnas.0811018106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neve KA, Seamans JK, Trantham-Davidson H. Dopamine receptor signaling. J. Recept. Signal Transduct. Res. 2004;24:165–205. doi: 10.1081/rrs-200029981. [DOI] [PubMed] [Google Scholar]
- Nieto FJ, Young TB, Lind BK, Shahar E, Samet JM, Redline S, D’Agostino RB, Newman AB, Lebowitz MD, Pickering TG. Association of sleep-disordered breathing, sleep apnea, and hypertension in a large community-based study. Sleep Heart Health Study. JAMA. 2000;283:1829–1836. doi: 10.1001/jama.283.14.1829. [DOI] [PubMed] [Google Scholar]
- Owens DF, Kriegstein AR. Is there more to GABA than synaptic inhibition? Nat. Rev. Neurosci. 2002;3:715–727. doi: 10.1038/nrn919. [DOI] [PubMed] [Google Scholar]
- Peters JA, Lambert JJ, Cottrell GA. An electrophysiological investigation of the characteristics and function of GABAA receptors on bovine adrenomedullary chromaflfin cells. Pflugers Arch. 1989;415:95–103. doi: 10.1007/BF00373146. [DOI] [PubMed] [Google Scholar]
- Prabhakar NR, Dick TE, Nanduri J, Kumar GK. Systemic, cellular and molecular analysis of chemoreflex-mediated sympathoexcitation by chronic intermittent hypoxia. Exp. Physiol. 2007;92:39–44. doi: 10.1113/expphysiol.2006.036434. [DOI] [PubMed] [Google Scholar]
- Raghuraman G, Rai V, Peng YJ, Prabhakar NR, Kumar GK. Pattern-specific sustained activation of tyrosine hydroxylase by intermittent hypoxia: role of reactive oxygen species-dependent downregulation of protein phosphatase 2A and upregulation of protein kinases. Antioxid. Redox Signal. 2009;11:1777–1789. doi: 10.1089/ars.2008.2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreihofer AM, Guyenet PG. The baroreflex and beyond: control of sympathetic vasomotor tone by GABAergic neurons in the ventrolateral medulla. Clin. Exp. Pharmacol. Physiol. 2002;29:514–521. doi: 10.1046/j.1440-1681.2002.03665.x. [DOI] [PubMed] [Google Scholar]
- Seta KA, Ferguson TK, Millhorn DE. Discovery of oxygen-responsive genes in pheochromocytoma cells. Methods Enzymol. 2004;381:449–464. doi: 10.1016/S0076-6879(04)81030-9. [DOI] [PubMed] [Google Scholar]
- Sha D, Jin Y, Wu H, Wei J, Lin CH, Lee YH, Buddhala C, Kuchay S, Chishti AH, Wu JY. Role of mu-calpain in proteolytic cleavage of brain L-glutamic acid decarboxylase. Brain Res. 2008;1207:9–18. doi: 10.1016/j.brainres.2008.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma SD, Raghuraman G, Lee MS, Prabhakar NR, Kumar GK. Intermittent hypoxia activates peptidylglycine alpha-amidating monooxygenase in rat brain stem via reactive oxygen species-mediated proteolytic processing. J. Appl. Physiol. 2009;106:12–19. doi: 10.1152/japplphysiol.90702.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waagepetersen HS, Sonnewald U, Schousboe A. The GABA paradox: multiple roles as metabolite, neurotransmitter, and neurodifferentiative agent. J. Neurochem. 1999;73:1335–1342. doi: 10.1046/j.1471-4159.1999.0731335.x. [DOI] [PubMed] [Google Scholar]
- Waagepetersen HS, Sonnewald U, Schousboe A. Compartmentation of glutamine, glutamate, and GABA metabolism in neurons and astrocytes: functional implications. Neuroscientist. 2003;9:398–403. doi: 10.1177/1073858403254006. [DOI] [PubMed] [Google Scholar]
- Watanabe M, Maemura K, Kanbara K, Tamayama T, Hayasaki H. GABA and GABA receptors in the central nervous system and other organs. Int. Rev. Cytol. 2002;213:1–47. doi: 10.1016/s0074-7696(02)13011-7. [DOI] [PubMed] [Google Scholar]
- Wei J, Wu JY. Structural and functional analysis of cysteine residues in human glutamate decarboxylase 65 (GAD65) and GAD67. J. Neurochem. 2005;93:624–633. doi: 10.1111/j.1471-4159.2005.03046.x. [DOI] [PubMed] [Google Scholar]
- Wei J, Wu JY. Post-translational regulation of L-glutamic acid decarboxylase in the brain. Neurochem. Res. 2008;33:1459–1465. doi: 10.1007/s11064-008-9600-5. [DOI] [PubMed] [Google Scholar]
- Wei J, Davis KM, Wu H, Wu JY. Protein phosphorylation of human brain glutamic acid decarboxylase (GAD)65 and GAD67 and its physiological implications. Biochemistry. 2004;43:6182–6189. doi: 10.1021/bi0496992. [DOI] [PubMed] [Google Scholar]
- Yuan G, Nanduri J, Khan S, Semenza GL, Prabhakar NR. Induction of HIF-1alpha expression by intermittent hypoxia: involvement of NADPH oxidase, Ca2+ signaling, prolyl hydroxylases, and mTOR. J. Cell Physiol. 2008;217:674–685. doi: 10.1002/jcp.21537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zachor DA, Moore JF, Brezausek C, Therbert A, Percy AK. Cocaine inhibits NGF-induced PC12 cells differentiation through D(1)-type dopamine receptors. Brain Res. 2000;869:85–97. doi: 10.1016/s0006-8993(00)02355-6. [DOI] [PubMed] [Google Scholar]
- Zanassi P, Paolillo M, Montecucco A, Avvedimento EV, Schinelli S. Pharmacological and molecular evidence for dopamine D(1) receptor expression by striatal astrocytes in culture. J Neurosci Res. 1999;58:544–552. doi: 10.1002/(sici)1097-4547(19991115)58:4<544::aid-jnr7>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.