

Abstract
Rationale
Arginine methylation by protein N-arginine methyltransferases (PRMTs) is an important post-translational modification in the regulation of protein signaling. PRMT2 contains a highly conserved catalytic Ado-Met binding domain, but the enzymatic function of PRMT2 with respect to methylation is unknown. The JAK-STAT pathway is proposed to be regulated through direct arginine methylation of STAT transcription factors, and STAT3 signaling is known to be required for leptin regulation of energy balance.
Objective
To identify the potential role of STAT3 arginine methylation by PRMT2 in the regulation of leptin signaling and energy homeostasis.
Methods and Results
We identified that PRMT2-/- mice are hypophagic, lean, and have significantly reduced serum leptin levels. This lean phenotype is accompanied by resistance to food-dependent obesity and an increased sensitivity to exogenous leptin administration. PRMT2 co-localizes with STAT3 in hypothalamic nuclei, where it binds and methylates STAT3 through its Ado-Met binding domain. In vitro studies further clarified that the Ado-Met binding domain of PRMT2 induces STAT3 methylation at the Arg31 residue. Absence of PRMT2 results in decreased methylation and prolonged tyrosine phosphorylation of hypothalamic STAT3, which was associated with increased expression of hypothalamic pro-opiomelanocortin following leptin stimulation.
Conclusions
These data elucidate a molecular pathway that directly links arginine methylation of STAT3 by PRMT2 to the regulation of leptin signaling, suggesting a potential role for PRMT2 antagonism in the treatment of obesity and obesity-related syndromes.
Keywords: PRMT2, leptin, methylation, STAT3
Introduction
Protein methylation regulates gene expression by post-translational modification of chromatin, structural proteins and transcription factors. Methylation of arginine residues is catalyzed by protein N-arginine methyltransferase (PRMT) enzymes utilizing an S-adenosylmethionine (Ado-Met) binding domain as the methyl donor. Most mammalian PRMTs (PRMT1 - 8) are classified as type I or type II according to the formation of ω-NG-monomethylarginine and either ω-NG, NG-asymmetric or ω-NG, N’G-symmetric dimethylarginine residues respectively.
PRMT2, a type I enzyme, contains a highly conserved catalytic Ado-Met binding domain and unique Src homology (SH) 3 domain that binds proteins with proline-rich motifs.1,2 While not initially described to have methyltransferase activity,2 subsequent research indicated PRMT2 binds estrogen receptor-α and enhances estrogen-related transcription through its Ado-Met domain, indirectly suggesting methyltransferase activity.3 However, despite further studies indicating PRMT2 exerts biologic activity,4-6 in vivo methyltransferase activity has not been convincingly demonstrated, and its designation as a type I enzyme has been challenged.7
Concurrently, mounting evidence suggests JAK-STAT signaling is regulated by arginine methylation.8-11 This includes the finding that PRMT1 associates with and methylates STAT1 on Arg31.9 This is of relevance, as Arg31 is conserved in other STAT members, with both STAT3 and STAT6 also able to undergo arginine methylation.10,11 Thus, methylation of STAT3 by PRMT enzymes may regulate its signaling pathways, possibly including the leptin-induced STAT3-dependent pathway.
Leptin, transcribed from the ob gene, is produced mainly in white adipose tissue and regulates energy status by activating Ob-Rb, the leptin long receptor isoform.12 Binding of leptin to Ob-Rb induces receptor conformational changes and JAK2 activation, with subsequent phosphorylation of tyrosine residues in Ob-Rb.13 Tyrosine phosphorylation of Ob-Rb Tyr1138 recruits STAT3 to an Ob-Rb-JAK2 complex, allowing phosphorylation of a Tyr705 residue in STAT3.14,15 Ob-Rb is highly expressed in nuclei of basomedial hypothalamus, including arcuate (ARC), dorsomedial hypothalamic and ventromedial hypothalamic (VMH) nuclei, collectively known as the “satiety center”.16-18 Via a STAT3-dependent pathway, leptin binding to Ob-Rb increases pro-opiomelanocortin (POMC) production in neurons projecting from the ARC nucleus.14,15,17-22 In turn, POMC generates an anorectic signal through α melanocyte-stimulating hormone binding to melanocortin 3 and 4 receptors.23-26 Multiple animal models have now verified the importance of STAT3-dependent signaling in regulating feeding, energy expenditure and body weight. These models have included rodents with mutant Ob-Rb (Tyr1138→Ser);15 loss of POMC;23 and the inactivation of STAT3 in central neural tissues,27 POMC-expressing neurons22 or Ob-Rb-expressing neurons.28
Given these reports we hypothesized that STAT3 methylation may regulate leptin signaling. In particular, based on preliminary observations we speculated that PRMT2 may fulfill this function.4,5 We identified that PRMT2-/- mice exhibit reduced body weight, resistance to obesity and increased leptin sensitivity. PRMT2 co-localized with STAT3 in hypothalamic nuclei, where it bound and methylated STAT3 through its catalytic domain. Absence of PRMT2 resulted in decreased methylation and prolonged tyrosine phosphorylation of STAT3, and increased expression of hypothalamic POMC following leptin stimulation. These findings delineate a novel mechanism of regulation of the leptin-STAT3-melanocortin pathway and confirm the methyltransferase activity of PRMT2 in vivo.
Materials and Methods
Complete Materials and Methods are available in the Online Expanded Materials and Methods supplement.
Generation of PRMT2+/- and PRMT2-/- mice
As previously described, PRMT2-/- mice were generated by deleting a 3 helix segment involved in the Ado-Met domain.4,5 All Animal care and experimental protocols were reviewed and approved by Animal Care Use Committee of National Heart, Lung, and Blood Institute in accordance with National Institutes of Health guidelines.
Statistical analyses
Data are presented as mean ± standard error of the mean (SEM). Comparisons between experimental groups were performed using unpaired Student’s t test or ANOVA when appropriate. Differences were considered significant if P < 0.05. As a minimum, all experimental groups were of at least n = 3.
Results
PRMT2-/- mice are lean and exhibit reduced food intake
We observed that PRMT2-/- mice were born at the expected Mendelian frequency, lived for > 1.5 years without gross abnormalities, and were fertile. However, we noted that PRMT2-/- male mice gained less weight than age-matched control wild-type mice when weaned onto a chow diet. This weight difference was apparent at 6 weeks and was sustained throughout the period of chow diet administration until 30 weeks of age (Figure 1a and Online Table I). The weight of PRMT2+/- males was intermediate between that of wild-type and PRMT2-/- mice (Online Table I), suggesting that PRMT2 exerts a dose-responsive effect over body weight. While there were no differences between the weights of female wild-type and PRMT2-/- mice at 12 weeks, by 30 weeks of age, PRMT2-/- females were also lean compared with wild-type females (Online Table I). Measurement of snout-anus length at 12 weeks of age revealed that male and female PRMT2-/- mice were 3 - 4 % shorter than wild-type controls (Online Table I), indicating that PRMT2 may also contribute to linear growth regulation.
Figure 1.
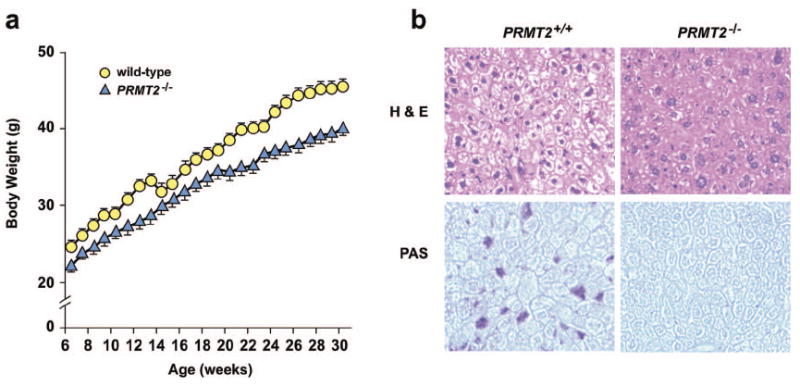
PRMT2-/- mice are lean, have reduced hepatic glycogen stores, and are resistant to diet-induced obesity.
a) Growth curves of age-matched male wild-type and PRMT2-/- mice fed a standard chow diet for 30 weeks after weaning (n = 12 in each group). Selected statistical comparisons are provided in Online Table I.
b) Hematoxylin & Eosin (H & E) (upper) and Periodic acid-Schiff (PAS) (lower) staining of liver sections from 8 week old wild-type and PRMT2-/- male mice. Original magnification ×40.
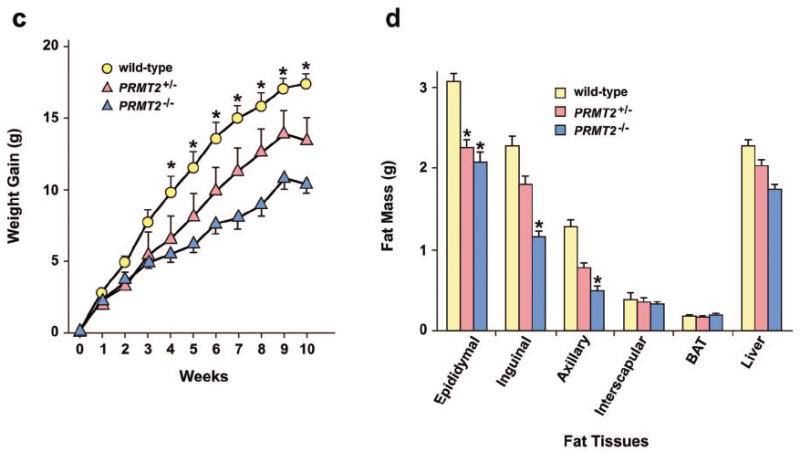
c) Weight gain of age-matched male wild-type (n = 7), PRMT2+/- (n = 5), and PRMT2-/- (n = 8) mice fed a high-fat diet for 10 weeks. *P < 0.001, PRMT2-/- vs. wild-type.
d) Liver, brown adipose tissue (BAT) and fat pad mass (epididymal, inguinal, axillary, interscapular) of age-matched male wild-type (n = 7), PRMT2+/- (n = 5), and PRMT2-/- (n = 8) mice fed a high-fat diet. *P < 0.001, PRMT2-/- and PRMT2+/- vs. wild-type.
Leanness in PRMT2-/- mice could result from decreased food intake, increased energy expenditure, and/or malabsorption. To address whether decreased food consumption was responsible for reduced weight gain, the food intake of wild-type and PRMT2-/- mice was monitored for 14 days. Average food intake was significantly decreased in both male and female PRMT2-/- mice compared with age-matched wild-type mice (Online Table I), indicating that reduced food intake by PRMT2-/- mice may play a role in their diminished weight gain. The stools of PRMT2-/- mice did not show any gross changes compared with those from wild-type mice (data not shown), suggesting that malabsorption, in particular lipid malabsorption, was not contributory.
Reduced liver glycogen content in PRMT2-/- mice
Blood pressure recording and complete necropsies were performed to assess for organ abnormalities. We identified that hepatic cytoplasmic vacuoles were less numerous in PRMT2-/- mice, and that hepatic cords and sinusoids were relatively more distinct than those of wild-type mice (Figure 1b, upper panels), suggesting reduced liver glycogen content in PRMT2-/- mice. Indeed, upon further analysis, hepatic glycogen content was markedly decreased in PRMT2-/- mice (Figure 1b, lower panels). No gross or microscopic abnormalities were observed in other PRMT2-/- organs or tissues, including heart, thymus, spleen, pancreas, kidney, skeletal muscle, and brown adipose tissue (data not shown). Normalized heart weight and blood pressure did not differ between wild-type and PRMT2-/- animals (Online Table II).
Altered metabolic homeostasis in PRMT2-/- mice
To further explore the possible metabolic derangements caused by PRMT2 deficiency, blood glucose and serum insulin levels were assessed in 8 - 12 week old mice. Blood glucose levels in fasting and fed PRMT2-/- male mice were significantly lower than wild-type male mice, while fasting glucose levels were also significantly reduced in female PRMT2-/- mice compared with wild-type females (Online Table I). In addition, serum insulin and triglyceride concentrations were reduced in male and female PRMT2-/- mice compared with wild-type mice (insulin: P = 0.07 and P < 0.05, respectively; triglycerides: P = 0.06 and P < 0.05, respectively) (Online Table I).
We also investigated potential changes in pro-inflammatory cytokine production with high-fat feeding. Wild-type and PRMT2-/- mice were fed a high-fat or normal chow diet for 10 weeks and the following cytokines were measured: interleukin (IL)-1α, IL-2, IL-4, IL-5, IL-6, IL-17, tumor necrosis factor-α (TNFα), interferon (IFN)-γ (Online Table III). We identified that following high-fat feeding, IL-2 was increased in PRMT2-/- versus wild-type mice, while IL-17 levels in PRMT2-/- mice were decreased with high-fat feeding as compared to normal chow diet. However, the magnitude of these changes were small, and the majority of the examined cytokines were unchanged (Online Table III). Taken together, these results demonstrate that the absence of PRMT2 results in minimal alterations to inflammatory regulation, but significant changes in glucose, insulin, and lipid metabolism.
Resistance to food-dependent obesity in PRMT2-/- mice
High-fat feeding induces weight gain and obesity, which is associated with increased visceral fat mass, glucose intolerance and insulin resistance in non-obese rodents.29 To determine whether PRMT2-/- mice are resistant to diet-induced obesity, wild-type and PRMT2-/- mice were fed a high-fat diet for 10 weeks. PRMT2-/- mice gained significantly less weight than heterozygote or wild-type mice (Figure 1c). This relative leanness was associated with reduced fat mass, with epididymal, inguinal, and axillary fat pads weighing significantly less in PRMT2-/- than wild-type mice (Figure 1d). No differences were observed in liver, interscapular fat pad or brown adipose tissue mass. These data indicate that PRMT2-/- mice are relatively protected against food-dependent obesity and increased adiposity induced by high-fat feeding.
Regulation of leptin signaling by PRMT2 in vivo
The leanness and reduced adiposity of PRMT2-/- mice suggested that PRMT2 may modulate leptin signaling. Therefore, we evaluated leptin regulation in PRMT2-/- mice, and identified that serum leptin concentrations in male and female PRMT2-/- mice were significantly decreased compared with wild-type mice (Online Table I). Next, to directly assess leptin sensitivity, we measured the responses of wild-type and PRMT2-/- mice to exogenous leptin. As the response to peripheral leptin injection can vary by rodent body weight,30 we studied wild-type and PRMT2-/- mice with similar body weights at 4 days prior to leptin injection (32.1 ± 1.3 g vs. 32.4 ± 2.2 g, P = not significant [n.s.]), with similar weights confirmed again at day 0 immediately prior to the first leptin dose (32.1 ± 1.3 g vs. 32.6 ± 2.1 g, P = n.s.). Consistent with previous reports,31 we observed no major changes in body weight following 6 days of intraperitoneal (i.p.) leptin treatment (0.1 μg/g body weight, twice daily) in wild-type mice (Figure 2a). In contrast, PRMT2-/- mice continuously lost weight during this period of leptin administration (Figure 2a). In addition, by comparison to wild-type mice, PRMT2-/- mice exhibited reduced food intake during leptin treatment (Figure 2b). Thus, PRMT2-/- mice, which are lean and have lower circulating levels of leptin, are more sensitive to exogenous leptin administration.
Figure 2.
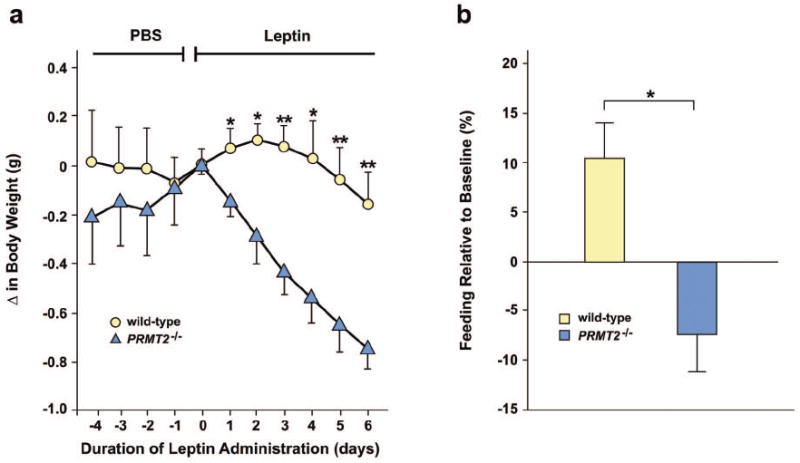
PRMT2-/- mice exhibit enhanced sensitivity to exogenous leptin. Mice at 12 -16 weeks of age (n = 8 males in both groups) were injected i.p. with phosphate buffered saline (PBS) for 4 days then for 6 days with recombinant mouse leptin (0.1 μg/g twice daily).
a) Change in body weight in response to leptin in wild-type and PRMT2-/- mice. *P < 0.05, **P < 0.005, PRMT2-/- vs. wild-type.
b) Change in total 6 day food intake (%) for wild-type vs. PRMT2-/- mice during leptin treatment, *P < 0.05.
PRMT2 and STAT3 co-localize in the hypothalamus
It is known that anorexigenic POMC neurons in the ARC nucleus express both Ob-Rb and STAT3, and that the paraventricular hypothalamic (PVH) nucleus is also a prominent site of Ob-Rb and STAT3 expression that detects and integrates anorexigenic signals arising from the ARC nucleus.14,15,17-22,24 Therefore, given our finding that leptin sensitivity is increased in PRMT2-/- mice, we examined whether PRMT2 localizes to the hypothalamus and if it might interact with STAT3 at these sites. Using in-situ-hybridization we observed abundant expression of PRMT2 mRNA in the anterior and medial hypothalamus of wild-type mice, including the ARC, PVH, VMH and supraoptic hypothalamic nuclei, while PRMT2 mRNA was not detected in PRMT2-/- mice (Figure 3a). In a similar anatomical distribution to PRMT2, STAT3 mRNA was also detected in the ARC, PVH and VMH nuclei (Figure 3b). Quantification of these results revealed a modest reduction in STAT3 mRNA levels in the PVH nucleus for PRMT2-/- compared to WT mice, with similar STAT3 mRNA levels in the ARC nucleus for WT and PRMT2-/- animals (Figure 3b, Online Figure I).
Figure 3.
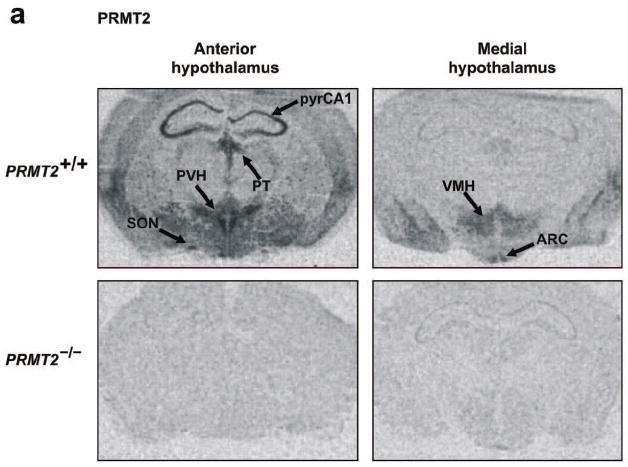
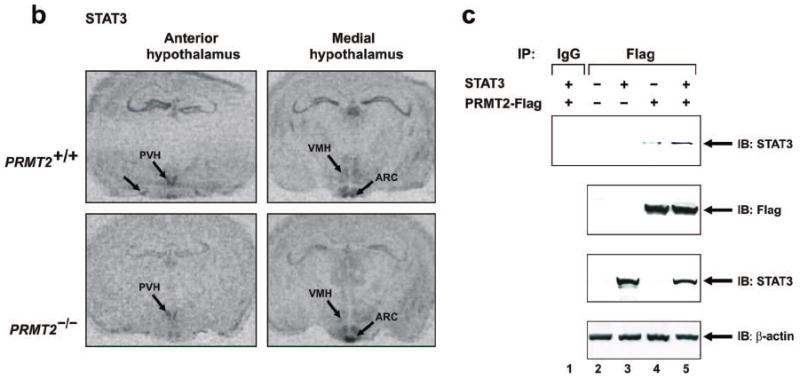
PRMT2 and STAT3 co-localize in the hypothalamus.
a) Anterior (left) and medial (right) hypothalamic expression of PRMT2 mRNA by in-situ hybridization in PRMT2+/+ (upper) but not PRMT2-/- (lower) male mice. Extrahypothalamic areas expressing PRMT2 include the CA1 hippocampal pyramidal cell layer (pyrCA1) and paraventricular thalamic nucleus (PT). SON = supraoptic nuclei.
b) Anterior (left) and medial (right) hypothalamic expression of STAT3 mRNA by in-situ hybridization in PRMT2+/+ (upper) and PRMT2-/- (lower) male mice.
c) 293 cells were transiently transfected with expression vectors encoding mouse PRMT2-Flag and/or STAT3. After incubation for 24 hours, cells were lysed and samples were immunoprecipitated with anti-Flag antibody or with preimmune rabbit IgG, followed by immunoblotting with anti-STAT3 antibody (upper panel). Control immunoblots (Westerns) with anti-Flag antibody (second panel), anti-STAT3 antibody (third panel) and anti-β-actin antibody (lower panel) were performed on the same samples. IB = immunoblot.
To confirm this possible association between PRMT2 and STAT3 in vitro co-precipitation experiments were performed. We transfected 293 cells with Flag-tagged full-length PRMT2 cDNA and/or STAT3 cDNA, immunoprecipitated with anti-Flag antibody, and immunoblotted with anti-STAT3 antibody. As suggested by our in vivo in-situ-hybridization data, endogenous STAT3 was found to co-precipitate with transfected PRMT2 (Figure 3c, lane 4), with the interaction between PRMT2 and STAT3 increasing when both substrates were co-transfected (Figure 3c, lane 5). These data, particularly when taken in conjunction with our prior finding of enhanced leptin sensitivity in PRMT2-/- mice and the expression pattern of PRMT2 RNA, suggest that PRMT2 may influence appetite and body weight via the hypothalamic leptin-STAT3 signaling pathway.
The Ado-Met binding domain of PRMT2 methylates STAT3 at Arg31
The Arg31 STAT residue is known to undergo methylation in STAT1 and is conserved across other STAT family members.9-11 To investigate the possibility that STAT3 may also undergo methylation at Arg31 we first conducted methylation assays, transfecting wild-type or mutated PRMT2 cDNAs lacking a functional Ado-Met domain into 293 or mouse embryonic fibroblast (MEF) cells and using a glutathione S-transferase-STAT3 (GST-STAT3) protein as substrate and Flag fusion proteins as an enzyme source. We identified that in 293 cells, wild-type PRMT2 methylated GST-STAT3 (Figure 4a, lane 3), while mutant PRMT2 or vector alone did not (Figure 4a, lanes 4, 5). Cell extracts from wild-type MEFs also methylated GST-STAT3, while methylation of STAT3 was abolished in PRMT2-/- MEFs (Figure 4b, lane 2 vs. 4). Using wild-type MEFs transfected with a Arg31→Ala GST-STAT3 mutant protein we observed that methylation was abolished (Figure 4b, lane 3), suggesting that the Arg31 residue of STAT3 is a likely target for PRMT2 methylation.
Figure 4.
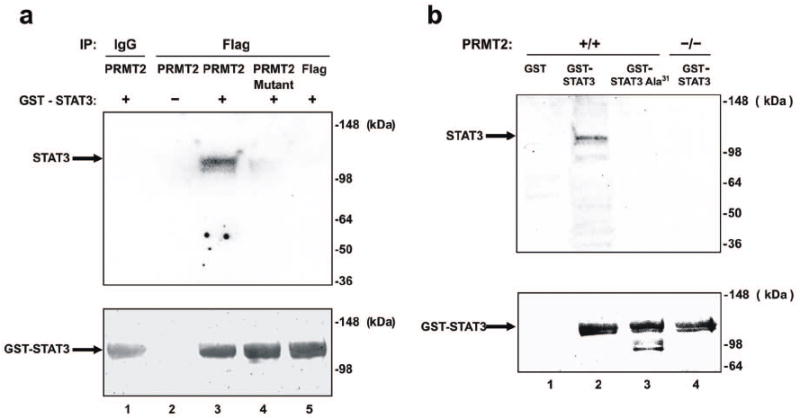
Methylation of STAT3 at Arg31 by PRMT2.
a) 293 cells were transiently transfected with expression vectors encoding Flag-tagged wild-type or mutant mouse PRMT2 lacking the Ado-Met binding domain. After transfection, cell lysates were incubated with Anti-Flag M2 Affinity Gel, and immune complexes were used as the enzyme source for the methylation reaction. IP products were subjected to an in vitro methylation assay using GST-STAT3 and the methyl donor [3H]-Ado-Met. Samples were analysed by SDS-PAGE followed by autoradiography. Molecular weight markers are indicated on the right of the panels. The amount of GST protein for each lane is shown by Coomassie staining (lower panel).
b) Wild-type and PRMT2-/- MEF extracts were subjected to an in vitro methylation assay using GST, GST-STAT3 or GST-STAT3 Arg31→Ala31, under the same conditions, analyzed and presented as for Figure 4a.
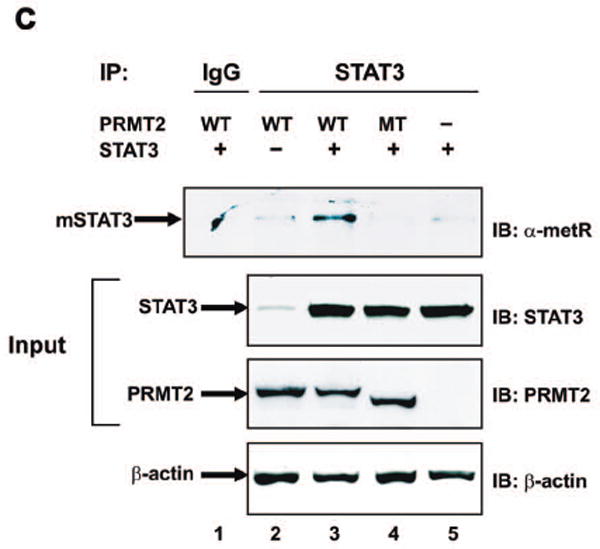
c) 293 cells were transiently transfected with expression vectors encoding mouse PRMT2, mutant PRMT2 lacking an Ado-Met binding domain and/or STAT3. After transfection, cells were lysed and samples were immunoprecipitated with anti-STAT3 antibody or with preimmune rabbit IgG, followed by immunoblotting with anti-arginine (mono- and di-methyl) antibody (α-metR) (upper panel). Protein loading conditions were verified by immunoblotting (Westerns) on the same samples with anti-STAT3 (second panel), anti-PRMT2 (third panel) and anti-β-actin (lower panel) antibodies. WT = wild-type; MT = mutant; mSTAT3 = methylated STAT3.
To further confirm that STAT3 is arginine-methylated by PRMT2 via its Ado-Met domain, we performed transient transfection into 293 cells using wild-type and mutated PRMT2 cDNA lacking a functional Ado-Met domain, followed by immunoprecipitation (IP) with anti-STAT3 antibody and then immunoblotting with an anti-α-metR antibody that recognizes free and bound NG, NG-dimethyl or monomethyl arginine. Endogenous methylated STAT3 was observed, at the extreme limits of detection, in cells transfected with either wild-type PRMT2 (Figure 4c, lane 2, upper panel) or STAT3 (Figure 4c, lane 5, upper panel). However, methylated STAT3 was markedly increased when STAT3 was co-transfected with PRMT2 (Figure 4c, lane 3, upper panel). Importantly, methylated STAT3 was not observed following transfection with a catalytic defective PRMT2 mutant (Figure 4c, lane 4, upper panel). As a whole, these three separate experiments provide strong evidence to suggest that the Ado-Met binding domain of PRMT2 induces STAT3 methylation at Arg31.
PRMT2-/- mice exhibit reduced STAT3 methylation
To confirm that PRMT2 is implicated in the methylation of STAT3 in vivo, the STAT3 methylation status of wild-type and PRMT2-/- mice was examined in the hypothalamus and other organs with and without leptin stimulation. These experiments identified a significant reduction in baseline STAT3 methylation in the hypothalamus of PRMT2-/- versus wild-type mice (Figure 5a, 1st row: lane 2 vs. lane 6; Figure 5b). Upon leptin stimulation, hypothalamic STAT3 methylation was decreased in wild-type animals, but remained unchanged in PRMT2-/- mice (Figure 5a, 1st row: lane 4 vs. lane 2 and lane 8 vs. lane 6; Figure 5b).
Figure 5.
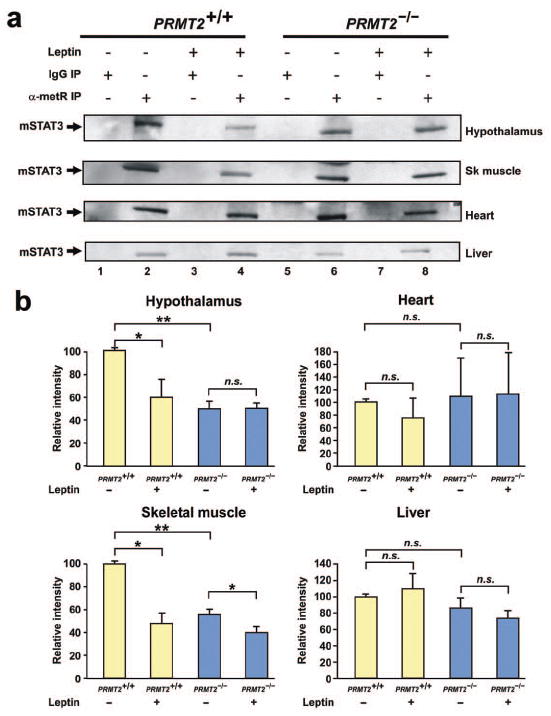
STAT3 methylation is reduced in PRMT2-/- tissues in vivo.
a) Methylated STAT3 levels were assessed in the hypothalamus, skeletal muscle, heart and liver of wild-type and PRMT2-/- male mice 90 min after peripheral leptin treatment (1.67 μg/g body weight). Total tissue lysates (3 mice per experimental group) were immunoprecipitated with α-metR antibody or with preimmune mouse IgG, followed by immunoblotting with anti-STAT3. Sk muscle = skeletal muscle. Experiments were performed in triplicate (36 mice in total).
b) Quantification of methylated STAT3 levels in hypothalamic, skeletal muscle, heart and liver extracts. The relative intensity of methylated STAT3 (as shown in Figure 5a) was determined by densitometry and normalized to the results from untreated wild-type mice. Data represent the mean ± SEM of 3 mice per group, *P < 0.05, **P < 0.01.
We also investigated the methylation status of STAT3 in other tissues that express PRMT2, including skeletal muscle, heart and liver. Baseline STAT3 methylation was decreased in skeletal muscle in PRMT2-/- versus wild-type mice. Following leptin stimulation, in a similar fashion to the hypothalamus, STAT3 methylation levels were reduced in the skeletal muscle of wild-type animals but were unchanged in PRMT2-/- mice (Figure 5a, 2nd row, Figure 5b). The levels of methylated heart and liver STAT3 were not influenced by PRMT2 deletion or leptin stimulation (Figure 5a, 3rd and 4th rows, Figure 5b). Collectively, these data confirm that the absence of PRMT2 induces a decrease in STAT3 methylation in vivo, including in the hypothalamus and skeletal muscle, but not in the heart or liver.
Prolonged STAT3 phosphorylation in PRMT2-/- cells and tissues
Arginine methylation of STAT1 plays a critical role in its tyrosine dephosphorylation, with inhibition of STAT1 methylation resulting in prolonged STAT1 tyrosine phosphorylation.32 Therefore, we examined whether absence of PRMT2 would modulate STAT3 tyrosine phosphorylation. We observed tyrosine phosphorylation of STAT3 in nuclear extracts from wild-type and PRMT2-/- vascular smooth muscle cells (VSMCs) following 10 min of stimulation with mouse leptin (Figure 6a), with significantly greater tyrosine phosphorylation present in PRMT2-/- compared with wild-type VSMCs after 30 min of leptin treatment (Figures 6a and b). These findings were confirmed by immune-staining with an antibody that recognizes a phosphorylated Tyr705 residue of STAT3 (Figure 6c).
Figure 6.
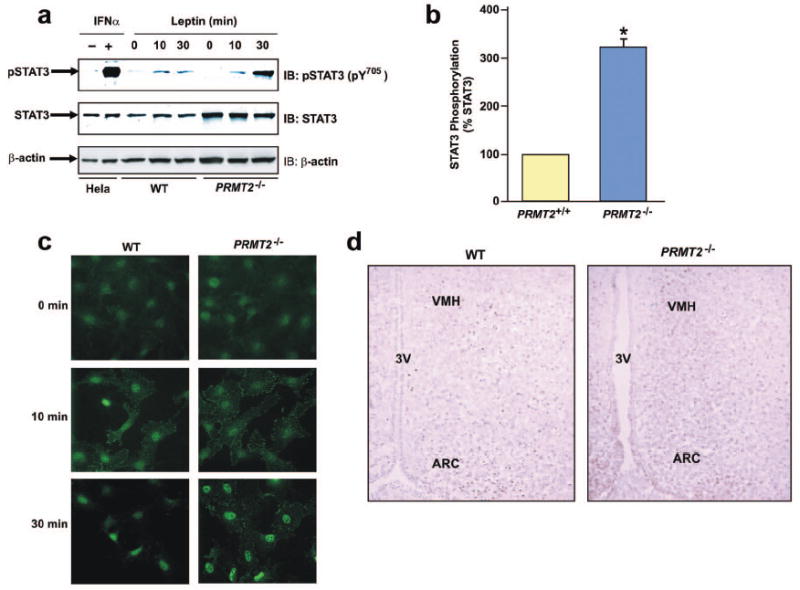
PRMT2 modulates leptin-induced STAT3 tyrosine phosphorylation.
a) Quiescent wild-type and PRMT2-/- VSMCs were treated with or without mouse leptin (100 nM) for the indicated times. Nuclear extracts were immunoblotted using antibodies against phospho-STAT3 [pSTAT3 (pY705)] (upper panel), STAT3 (middle panel) and β-actin (lower panel). Total cell extracts from HeLa cells prepared with or without IFN-α treatment are shown as positive and negative controls, respectively.
b) The pSTAT3 (pY705) and STAT3 signals from blots obtained following 30 min leptin treatment were quantified by densitometry, and amounts of pSTAT3 in wild-type and PRMT2-/- cells were normalized to the amount of STAT3 in each sample. Results are expressed as mean ± SEM of 3 independent experiments. *P < 0.05 for PRMT2-/- vs. wild-type.
c) Tyrosine phosphorylated STAT3 was imaged by immune-staining in quiescent wild-type and PRMT2-/- VSMCs untreated or treated with mouse leptin (100 nM) for 10 or 30 min, using anti-pSTAT3 (pY705) antibody. Original magnification ×40.
d) Tyrosine phosphorylated STAT3 activity in hypothalamic sections from wild-type and PRMT2-/- male mice 90 min after peripheral leptin treatment (1.67 μg/g body weight) was imaged by immunohistochemistry using anti-pSTAT3 (pY705) antibody. 3V = Third ventricle. Original magnification ×20.
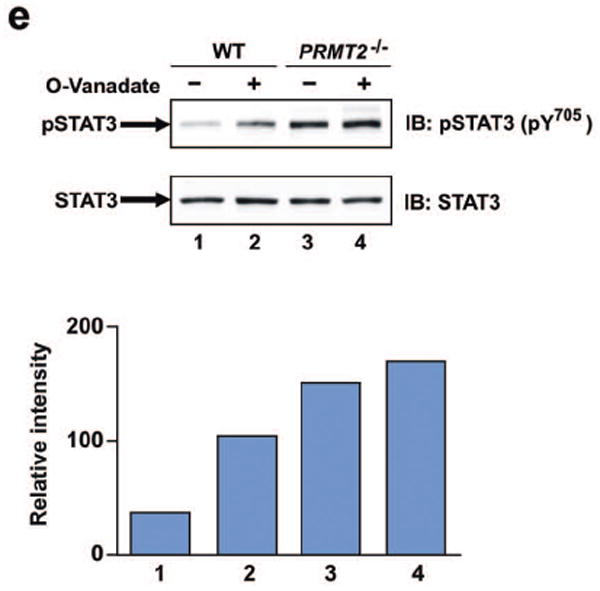
e) Quiescent wild-type and PRMT2-/- VSMCs were pretreated with or without o-vanadate (200 μM) for 15 min and then stimulated with recombinant mouse leptin (30min, 100 nM). Nuclear extracts were immunoblotted using anti-pSTAT3 (pY705) (upper panel) and anti-STAT3 antibodies (lower panel). The results of a representative experiment were quantitated by densitometry and are presented below with each column representing the corresponding lane. Data are from a representative experiment that was repeated in triplicate with similar results.
To further explore the central effects of leptin stimulation on STAT3 phosphorylation in vivo, cortical sections of wild-type and PRMT2-/- mice were analyzed by immunohistochemistry. In wild-type mice, tyrosine phosphorylation of hypothalamic STAT3 (pY705) peaked at 45 min after peripheral leptin administration (1.67 μg/g body weight), gradually declining by 180 min after injection (data not shown). At 90 min after leptin stimulation, tyrosine phosphorylated STAT3 remained detectable in the ARC and VMH of wild-type mice (Figure 6d, left panel). However, tyrosine phosphorylated STAT3 was significantly more abundant in the ARC and VMH of PRMT2-/- mice at the same time-point (Figure 6d, right panel), suggesting that PRMT2 regulates hypothalamic STAT3 phosphorylation in vivo.
We next assessed the tyrosine phosphorylation status of STAT3 in peripheral tissues. Phosphorylated STAT3 was not detected in sections of the heart and skeletal muscle in wild-type and PRMT2-/- mice (data not shown), but was readily detectable in the liver. However, unlike the hypothalamus, and consistent with the fact that leptin stimulation and PRMT2 deletion do not alter hepatic STAT3 methylation (Figures 5a and 5b), the levels of tyrosine phosphorylated STAT3 in wild-type and PRMT2-/- livers were similar and unaltered by systemic leptin administration (Online Figure II).
It has been reported that the single amino acid substitution of Arg31 for Ala in STAT1 modulates its ability to undergo tyrosine dephosphorylation.33 To determine the possible involvement of tyrosine phosphatase in the enhanced tyrosine phosphorylation of PRMT2-/- cells, we examined the effect of the phosphatase inhibitor o-vanadate on leptin-stimulated STAT3 tyrosine phosphorylation. These experiments indicated that in nuclear extracts from wild-type VSMCs, pretreatment with o-vanadate enhanced leptin-stimulated tyrosine phosphorylation of STAT3 (Figure 6e, lanes 1 and 2). In contrast, nuclear extracts from untreated PRMT2-/- VSMCs showed increased STAT3 tyrosine phosphorylation compared with untreated wild-type cells, while PRMT2-/- VSMCs did not exhibit any further enhancement of STAT3 tyrosine phosphorylation following o-vanadate treatment (Figure 6e lanes 3 and 4). Taken together, these data suggest that PRMT2 deletion results in augmented hypothalamic STAT3 tyrosine phosphorylation after leptin stimulation, and that augmented tyrosine phosphorylation in PRMT2-/- cells is likely due to altered tyrosine phosphatase activity.
Hypothalamic POMC expression in PRMT2-/- mice is increased in response to leptin stimulation
To determine whether PRMT2 deletion affects hypothalamic levels of anorexigenic POMC and orexigenic NPY following leptin treatment, mRNA expression was assessed by quantitative real time-PCR (qRT-PCR). Consistent with our finding that PRMT2-/- mice are more sensitive to exogenous leptin, administration of leptin at a higher dose than given previously (0.5 μg/g body weight, twice daily for 3 days) induced weight loss in PRMT2-/- (-0.03 ± 0.14 g vs. -1.0 ± 0.2 g; vehicle vs. leptin-treatment, P < 0.005) but not wild-type mice (-0.12 ± 0.16 g vs. -0.59 ± 0.29 g; vehicle vs. leptin-treatment, P = n.s.). There was no difference between the baseline expression levels of hypothalamic POMC mRNA in untreated wild-type versus PRMT2-/- mice. Expression of hypothalamic POMC mRNA in wild-type mice treated with leptin was 1.5 times greater than in non-treated wild-type mice (Figure 7a). However, leptin-treated PRMT2-/- mice exhibited significantly greater expression of hypothalamic POMC mRNA compared to leptin-treated wild-type mice (Figure 7a). In contrast and suggesting PRMT2-independence, baseline levels of hypothalamic NPY mRNA expression were slightly higher in PRMT2-/- than wild-type mice. Nevertheless, following leptin treatment hypothalamic NPY expression was significantly reduced, to a similar extent, in both PRMT2-/- and wild-type mice (Figure 7b). Taken in the context of the current literature (see above and introduction), these data suggest that PRMT2 modulates central melanocortin function by regulating hypothalamic Ob-Rb- and specific STAT3-dependent pathways, including POMC signaling.
Figure 7.
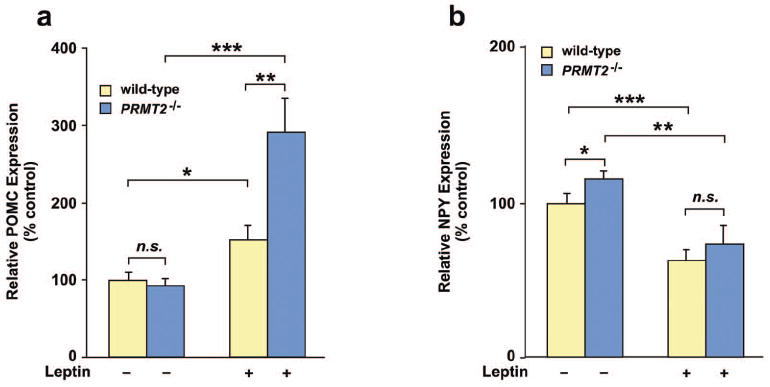
Hypothalamic POMC and NPY mRNA levels in wild-type and PRMT2-/- mice. Wild-type and PRMT2-/- male mice were injected with mouse leptin or PBS and hypothalamic POMC (a) and NPY (b) mRNA were quantitated by qRT-PCR. Data are expressed as a percent of the PBS control and represent the mean ± SEM of 5 mice per group, *P < 0.05, **P < 0.01, ***P < 0.005.
Finally, we sought to confirm that deletion of PRMT2 was not associated with compensatory changes in other PRMT isoforms that might have confounded these results. We surveyed the mRNA levels of PRMT isoforms 1 – 8 in the hypothalamus, liver, heart and skeletal muscle (Online Figure III). As expected, PRMT2 mRNA was not detectable in PRMT2-/- mice. Although isolated changes in the levels of certain isoforms were noted in specific tissues, the degree of change was generally modest and no consistent pattern was observed. Therefore, it appears unlikely that secondary alterations in the levels of these alternate PRMT isoforms are implicated in our results.
Discussion
Despite abundant evidence that other PRMT enzymes exhibit methyltransferase activity, proof that PRMT2 can function in this fashion in vivo has been elusive. While in vitro PRMT2 methyltransferase activity has been shown,34 in vivo evidence is limited7 and we believe our study to be the first to fully elucidate and characterize PRMT2 methyltransferase activity in vivo.
This work arose from our observation that PRMT2-/- mice are leaner than wild-type animals. This was associated with decreased food intake, perturbed energy metabolism and enhanced leptin sensitivity. These data led us to pursue the possibility that PRMT2 modulates leptin signaling. We identified that PRMT2 localizes with STAT3 within the hypothalamus including the ARC, VMH and PVH nuclei, which are recognized as critical sites for leptin functioning. Absence of PRMT2 was associated with reduced methylation and prolonged tyrosine phosphorylation of hypothalamic STAT3, likely contributing to enhanced leptin-induced POMC function. As part of the leptin signaling machinery we found that PRMT2 binds STAT3, methylating its Arg31 residue via the PRMT2 catalytic domain. Importantly, despite these data indicating the importance of hypothalamic leptin-STAT3-melanocortin signaling in PRMT2-/- mice, it cannot be ruled out that changes in STAT3 methylation in peripheral tissues, or other secondary changes such as reduced leptin levels, may have contributed to the observed phenotype. Nevertheless, taken as a whole, our findings ascribe clear methyltransferase activity to PRMT2, suggesting that this enzyme is a pivotal modulator of hypothalamic leptin-STAT3 signaling and energy homeostasis.
PRMT2-/- mice exhibited several physiologic and biochemical changes that, in humans, are associated with an improved cardiovascular risk profile. In addition to those mentioned above, PRMT2 deletion was associated with decreased triglyceride, glucose and insulin levels, suggesting that PRMT2 may facilitate the metabolic syndrome and diabetes. While not addressed by our studies, we speculate that this ‘anti-diabetogenic’ PRMT2-/- phenotype may be attributable to altered leptin-melanocortin signaling and decreased adiposity, resulting in favorable changes in glucose/energy metabolism. On the other hand, it remains possible that a more direct PRMT2 effect is operative. For example, STAT3 regulates hypothalamic insulin signaling,35 and it is possible that STAT3 methylation by PRMT2, or PRMT2-mediated methylation of other proteins, may directly regulate insulin activity and the progression to diabetes.
In addition to STAT3, other known PRMT2 targets include histone H4,34 IκB-α,4 estrogen receptor-α,3 the androgen receptor,6 the retinoblastoma gene product (RB),5 and hnRNP (heterogeneous nuclear ribonucleoprotein) E1B-AP5.36 Hypothetically, in addition to attenuated STAT3 methylation, the effects of PRMT2 deletion on any or several of these pathways may be implicated in the PRMT2-/- phenotype. However, these additional PRMT2 targets are not generally involved in energy regulation, and we are unaware of any evidence to implicate these factors as being contributory to the anti-obesity effect of PRMT2 deletion.
While the mechanism(s) remain to be clarified, our experiments suggest that PRMT2-mediated methylation regulates STAT3 tyrosine phosphorylation. Potentially related to this, an important modulator of STAT3 signaling, protein tyrosine phosphatase (PTP) 1B, is known to be a negative regulator of leptin signaling.37,38 Interestingly, mice lacking PTP1B display similar characteristics to PRMT2-/- mice, including reduced adiposity and obesity resistance.39 However, PTP1B is localized in the cytoplasmic fraction and it is known that leptin-activated JAK2, but not STAT3, is a substrate for PTP1B.40 Alternatively, it is known that T-cell (TC)-PTP, another member of PTP family, is ubiquitously expressed in various mammalian tissues including mouse brain.41 Analysis of TC-PTP-null MEFs revealed impaired dephosphorylation of nuclear STAT1 and STAT3, but not STAT5 or STAT6, suggesting TC-PTP may negatively regulate STAT3-mediated signaling.42 Furthermore, it is also known that inhibition of STAT1 arginine methylation results in decreased association of STAT1 with TC-PTP and delayed STAT1 tyrosine dephosphorylation.32 Thus, it appears plausible that PRMT2 deletion may result in a failure of STAT3 methylation at Arg31, decreasing the association of nuclear TC-PTP with STAT3, which in turn retards STAT3 dephosphorylation.
An unanticipated but provocative finding of this work is that while PRMT2 deletion was associated with decreased STAT3 methylation, we also identified that leptin administration reduces the extent of hypothalamic STAT3 methylation in wild-type but not PRMT2-/- animals. While this finding falls outside the purview of this study, it serves to underscore the importance of the leptin-PRMT2-STAT3 axis in energy regulation, implicating leptin as playing an upstream role in inhibiting the extent of PRMT2-mediated STAT3 methylation. This hypothesis is consistent with the known anorectic effects of both leptin administration and PRMT2 deletion, and is the subject of ongoing work in our laboratories.
Importantly, while almost all forms of human obesity involve leptin resistance, the administration of leptin is a generally inadequate treatment for the vast majority of obese subjects. Therefore our findings, along with the benign effects of PRMT2 genetic deletion, support the possibility that PRMT2 antagonism may represent an attractive therapeutic target for obesity management. In particular, the reduced body weight and favorable changes in glucose and lipid parameters observed in PRMT2-/- mice auger well for clinical utility.
In conclusion, our results demonstrate that PRMT2 is involved in the regulation of feeding, obesity and energy metabolism via a leptin-STAT3-dependent pathway. These data also show, we believe for the first time, that PRMT2 exhibits specific methyltransferase activity. Particularly given the looming global obesity epidemic and current lack of efficacious treatment options, we suggest that PRMT2 antagonism may hold promise as a therapeutic tool for the management of obesity.
Supplementary Material
Acknowledgments
We thank Dr. J. Darnell for the mouse STAT3 constructs, Ms. Robin Schwartzbeck for maintaining the PRMT2 mice colonies, Dr. Hong San for blood pressure measurements and members of the Nabel laboratory for helpful discussions. We acknowledge the professional skills and advice of Dr. Christian A. Combs and Dr. Daniela Malide (Light Microscopy Core Facility, National Heart, Lung and Blood Institute, National Institutes of Health).
Sources of Funding This work was supported by the Division of Intramural Research at the National Heart, Lung, and Blood Institute, the National Institutes of Health.
Non-Standard Abbreviations and Acronyms
- α-metR
antibody directed against both mono- and di-methyl arginine
- Ado-Met
S-adenosylmethionine
- ARC
Arcuate
- GST-STAT3
glutathione S-transferase-STAT3
- MEF
mouse embryonic fibroblast
- mSTAT3
methylated STAT3
- n.s.
not statistically significant
- POMC
pro-opiomelanocortin
- PRMT
protein N-arginine methyltransferase
- PTP
protein tyrosine phosphatase
- PVH
paraventricular hypothalamic
- qRT-PCR
quantitative real time-polymerase chain reaction
- TC-PTP
T-cell protein tyrosine phosphatase
- VMH
ventromedial hypothalamic
Novelty and Significance
What is known?
Protein methylation is an important step in the post-translational modification and functioning of mammalian proteins.
Protein methylation is catalyzed by Protein Methyltransferase enzymes (PRMT 1 – 8), although the ability of PRMT2 to perform this function had not been convincingly shown in vivo.
STAT family members may undergo methylation, and STAT3 is of importance in hypothalamic feeding and energy regulation.
What new information does this article contribute?
We show that PRMT2 methylates STAT3 in vivo.
Genetic deletion of PRMT2 in mice decreases hypothalamic STAT3 methylation and prolongs STAT3 phosphorylation.
Altered hypothalamic leptin-STAT3 signaling appears responsible for the phenotype of mice with PRMT2 genetic deletion, which have reduced body weight and food intake, resistance to food-dependent obesity and increased sensitivity to exogenous leptin administration.
Summary
Obesity is a major cause of health-related morbidity and mortality, and new insights and treatment modalities to combat this condition are urgently required. Here we show that genetic deletion of PRMT2, a member of the protein methyltransferase family of enzymes, is associated with a lean, leptin-hypersensitive and ‘anti-diabetes-like’ phenotype in mice. PRMT2 co-localizes with STAT3 in hypothalamic nuclei, where it binds and methylates STAT3. Absence of PRMT2 is associated with reduced methylation and prolonged phosphorylation of hypothalamic STAT3, likely contributing to enhanced downstream anorexigenic signaling. These findings identify a molecular pathway linking arginine methylation of STAT3 by PRMT2 to the regulation of body weight and energy metabolism, suggesting a potential role for PRMT2 antagonism in the treatment of diabetes and obesity.
Footnotes
Disclosures None.
References
- 1.Katsanis N, Yaspo ML, Fisher EM. Identification and mapping of a novel human gene, HRMT1L1, homologous to the rat protein arginine N-methyltransferase 1 (PRMT1) gene. Mamm Genome. 1997;8:526–9. doi: 10.1007/s003359900491. [DOI] [PubMed] [Google Scholar]
- 2.Scott HS, Antonarakis SE, Lalioti MD, Rossier C, Silver PA, Henry MF. Identification and characterization of two putative human arginine methyltransferases (HRMT1L1 and HRMT1L2) Genomics. 1998;48:330–40. doi: 10.1006/geno.1997.5190. [DOI] [PubMed] [Google Scholar]
- 3.Qi C, Chang J, Zhu Y, Yeldandi AV, Rao SM, Zhu YJ. Identification of protein arginine methyltransferase 2 as a coactivator for estrogen receptor alpha. J Biol Chem. 2002;277:28624–30. doi: 10.1074/jbc.M201053200. [DOI] [PubMed] [Google Scholar]
- 4.Ganesh L, Yoshimoto T, Moorthy NC, Akahata W, Boehm M, Nabel EG, Nabel GJ. Protein methyltransferase 2 inhibits NF-kappaB function and promotes apoptosis. Mol Cell Biol. 2006;26:3864–74. doi: 10.1128/MCB.26.10.3864-3874.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yoshimoto T, Boehm M, Olive M, Crook MF, San H, Langenickel T, Nabel EG. The arginine methyltransferase PRMT2 binds RB and regulates E2F function. Exp Cell Res. 2006;312:2040–53. doi: 10.1016/j.yexcr.2006.03.001. [DOI] [PubMed] [Google Scholar]
- 6.Meyer R, Wolf SS, Obendorf M. PRMT2, a member of the protein arginine methyltransferase family, is a coactivator of the androgen receptor. J Steroid Biochem Mol Biol. 2007;107:1–14. doi: 10.1016/j.jsbmb.2007.05.006. [DOI] [PubMed] [Google Scholar]
- 7.Herrmann F, Pably P, Eckerich C, Bedford MT, Fackelmayer FO. Human protein arginine methyltransferases in vivo - distinct properties of eight canonical members of the PRMT family. J Cell Sci. 2009;122:667–77. doi: 10.1242/jcs.039933. [DOI] [PubMed] [Google Scholar]
- 8.Chen D, Ma H, Hong H, Koh SS, Huang SM, Schurter BT, Aswad DW, Stallcup MR. Regulation of transcription by a protein methyltransferase. Science. 1999;284:2174–7. doi: 10.1126/science.284.5423.2174. [DOI] [PubMed] [Google Scholar]
- 9.Mowen KA, Tang J, Zhu W, Schurter BT, Shuai K, Herschman HR, David M. Arginine methylation of STAT1 modulates IFNalpha/beta-induced transcription. Cell. 2001;104:731–41. doi: 10.1016/s0092-8674(01)00269-0. [DOI] [PubMed] [Google Scholar]
- 10.Chen W, Daines MO, Hershey GK. Methylation of STAT6 modulates STAT6 phosphorylation, nuclear translocation, and DNA-binding activity. J Immunol. 2004;172:6744–50. doi: 10.4049/jimmunol.172.11.6744. [DOI] [PubMed] [Google Scholar]
- 11.Rho J, Choi S, Seong YR, Choi J, Im DS. The arginine-1493 residue in QRRGRTGR1493G motif IV of the hepatitis C virus NS3 helicase domain is essential for NS3 protein methylation by the protein arginine methyltransferase 1. J Virol. 2001;75:8031–44. doi: 10.1128/JVI.75.17.8031-8044.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vaisse C, Halaas JL, Horvath CM, Darnell JE, Jr, Stoffel M, Friedman JM. Leptin activation of Stat3 in the hypothalamus of wild-type and ob/ob mice but not db/db mice. Nat Genet. 1996;14:95–7. doi: 10.1038/ng0996-95. [DOI] [PubMed] [Google Scholar]
- 13.Kloek C, Haq AK, Dunn SL, Lavery HJ, Banks AS, Myers MG., Jr Regulation of Jak kinases by intracellular leptin receptor sequences. J Biol Chem. 2002;277:41547–55. doi: 10.1074/jbc.M205148200. [DOI] [PubMed] [Google Scholar]
- 14.Banks AS, Davis SM, Bates SH, Myers MG., Jr Activation of downstream signals by the long form of the leptin receptor. J Biol Chem. 2000;275:14563–72. doi: 10.1074/jbc.275.19.14563. [DOI] [PubMed] [Google Scholar]
- 15.Bates SH, Stearns WH, Dundon TA, Schubert M, Tso AW, Wang Y, Banks AS, Lavery HJ, Haq AK, Maratos-Flier E, Neel BG, Schwartz MW, Myers MG., Jr STAT3 signalling is required for leptin regulation of energy balance but not reproduction. Nature. 2003;421:856–9. doi: 10.1038/nature01388. [DOI] [PubMed] [Google Scholar]
- 16.Hakansson ML, Brown H, Ghilardi N, Skoda RC, Meister B. Leptin receptor immunoreactivity in chemically defined target neurons of the hypothalamus. J Neurosci. 1998;18:559–72. doi: 10.1523/JNEUROSCI.18-01-00559.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Elias CF, Aschkenasi C, Lee C, Kelly J, Ahima RS, Bjorbaek C, Flier JS, Saper CB, Elmquist JK. Leptin differentially regulates NPY and POMC neurons projecting to the lateral hypothalamic area. Neuron. 1999;23:775–86. doi: 10.1016/s0896-6273(01)80035-0. [DOI] [PubMed] [Google Scholar]
- 18.Elias CF, Lee C, Kelly J, Aschkenasi C, Ahima RS, Couceyro PR, Kuhar MJ, Saper CB, Elmquist JK. Leptin activates hypothalamic CART neurons projecting to the spinal cord. Neuron. 1998;21:1375–85. doi: 10.1016/s0896-6273(00)80656-x. [DOI] [PubMed] [Google Scholar]
- 19.Hakansson ML, Meister B. Transcription factor STAT3 in leptin target neurons of the rat hypothalamus. Neuroendocrinology. 1998;68:420–7. doi: 10.1159/000054392. [DOI] [PubMed] [Google Scholar]
- 20.Cowley MA, Smart JL, Rubinstein M, Cerdan MG, Diano S, Horvath TL, Cone RD, Low MJ. Leptin activates anorexigenic POMC neurons through a neural network in the arcuate nucleus. Nature. 2001;411:480–4. doi: 10.1038/35078085. [DOI] [PubMed] [Google Scholar]
- 21.Bousquet C, Zatelli MC, Melmed S. Direct regulation of pituitary proopiomelanocortin by STAT3 provides a novel mechanism for immuno-neuroendocrine interfacing. J Clin Invest. 2000;106:1417–25. doi: 10.1172/JCI11182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xu AW, Ste-Marie L, Kaelin CB, Barsh GS. Inactivation of signal transducer and activator of transcription 3 in proopiomelanocortin (Pomc) neurons causes decreased pomc expression, mild obesity, and defects in compensatory refeeding. Endocrinology. 2007;148:72–80. doi: 10.1210/en.2006-1119. [DOI] [PubMed] [Google Scholar]
- 23.Challis BG, Coll AP, Yeo GS, Pinnock SB, Dickson SL, Thresher RR, Dixon J, Zahn D, Rochford JJ, White A, Oliver RL, Millington G, Aparicio SA, Colledge WH, Russ AP, Carlton MB, O’Rahilly S. Mice lacking pro-opiomelanocortin are sensitive to high-fat feeding but respond normally to the acute anorectic effects of peptide-YY(3-36) Proc Natl Acad Sci U S A. 2004;101:4695–700. doi: 10.1073/pnas.0306931101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cowley MA, Pronchuk N, Fan W, Dinulescu DM, Colmers WF, Cone RD. Integration of NPY, AGRP, and melanocortin signals in the hypothalamic paraventricular nucleus: evidence of a cellular basis for the adipostat. Neuron. 1999;24:155–63. doi: 10.1016/s0896-6273(00)80829-6. [DOI] [PubMed] [Google Scholar]
- 25.Yaswen L, Diehl N, Brennan MB, Hochgeschwender U. Obesity in the mouse model of pro-opiomelanocortin deficiency responds to peripheral melanocortin. Nat Med. 1999;5:1066–70. doi: 10.1038/12506. [DOI] [PubMed] [Google Scholar]
- 26.Fan W, Boston BA, Kesterson RA, Hruby VJ, Cone RD. Role of melanocortinergic neurons in feeding and the agouti obesity syndrome. Nature. 1997;385:165–8. doi: 10.1038/385165a0. [DOI] [PubMed] [Google Scholar]
- 27.Gao Q, Wolfgang MJ, Neschen S, Morino K, Horvath TL, Shulman GI, Fu XY. Disruption of neural signal transducer and activator of transcription 3 causes obesity, diabetes, infertility, and thermal dysregulation. Proc Natl Acad Sci U S A. 2004;101:4661–6. doi: 10.1073/pnas.0303992101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Piper ML, Unger EK, Myers MG, Jr, Xu AW. Specific physiological roles for signal transducer and activator of transcription 3 in leptin receptor-expressing neurons. Mol Endocrinol. 2008;22:751–9. doi: 10.1210/me.2007-0389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Axen KV, Dikeakos A, Sclafani A. High dietary fat promotes syndrome X in nonobese rats. J Nutr. 2003;133:2244–9. doi: 10.1093/jn/133.7.2244. [DOI] [PubMed] [Google Scholar]
- 30.El-Haschimi K, Pierroz DD, Hileman SM, Bjorbaek C, Flier JS. Two defects contribute to hypothalamic leptin resistance in mice with diet-induced obesity. J Clin Invest. 2000;105:1827–32. doi: 10.1172/JCI9842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pelleymounter MA, Cullen MJ, Baker MB, Hecht R, Winters D, Boone T, Collins F. Effects of the obese gene product on body weight regulation in ob/ob mice. Science. 1995;269:540–3. doi: 10.1126/science.7624776. [DOI] [PubMed] [Google Scholar]
- 32.Zhu W, Mustelin T, David M. Arginine methylation of STAT1 regulates its dephosphorylation by T cell protein tyrosine phosphatase. J Biol Chem. 2002;277:35787–90. doi: 10.1074/jbc.C200346200. [DOI] [PubMed] [Google Scholar]
- 33.Shuai K, Liao J, Song MM. Enhancement of antiproliferative activity of gamma interferon by the specific inhibition of tyrosine dephosphorylation of Stat1. Mol Cell Biol. 1996;16:4932–41. doi: 10.1128/mcb.16.9.4932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lakowski TM, Frankel A. Kinetic analysis of human protein arginine N-methyltransferase 2: formation of monomethyl- and asymmetric dimethyl-arginine residues on histone H4. Biochem J. 2009;421:253–61. doi: 10.1042/BJ20090268. [DOI] [PubMed] [Google Scholar]
- 35.Ernst MB, Wunderlich CM, Hess S, Paehler M, Mesaros A, Koralov SB, Kleinridders A, Husch A, Munzberg H, Hampel B, Alber J, Kloppenburg P, Bruning JC, Wunderlich FT. Enhanced STAT3 activation in POMC neurons provokes negative feedback inhibition of leptin and insulin signaling in obesity. J Neurosci. 2009;29:11582–11593. doi: 10.1523/JNEUROSCI.5712-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kzhyshkowska J, Schutt H, Liss M, Kremmer E, Stauber R, Wolf H, Dobner T. Heterogeneous nuclear ribonucleoprotein E1B-AP5 is methylated in its Arg-Gly-Gly (RGG) box and interacts with human arginine methyltransferase HRMT1L1. Biochem J. 2001;358:305–14. doi: 10.1042/0264-6021:3580305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kaszubska W, Falls HD, Schaefer VG, Haasch D, Frost L, Hessler P, Kroeger PE, White DW, Jirousek MR, Trevillyan JM. Protein tyrosine phosphatase 1B negatively regulates leptin signaling in a hypothalamic cell line. Mol Cell Endocrinol. 2002;195:109–18. doi: 10.1016/s0303-7207(02)00178-8. [DOI] [PubMed] [Google Scholar]
- 38.Lund IK, Hansen JA, Andersen HS, Moller NP, Billestrup N. Mechanism of protein tyrosine phosphatase 1B-mediated inhibition of leptin signalling. J Mol Endocrinol. 2005;34:339–51. doi: 10.1677/jme.1.01694. [DOI] [PubMed] [Google Scholar]
- 39.Elchebly M, Payette P, Michaliszyn E, Cromlish W, Collins S, Loy AL, Normandin D, Cheng A, Himms-Hagen J, Chan CC, Ramachandran C, Gresser MJ, Tremblay ML, Kennedy BP. Increased insulin sensitivity and obesity resistance in mice lacking the protein tyrosine phosphatase-1B gene. Science. 1999;283:1544–8. doi: 10.1126/science.283.5407.1544. [DOI] [PubMed] [Google Scholar]
- 40.Cheng A, Uetani N, Simoncic PD, Chaubey VP, Lee-Loy A, McGlade CJ, Kennedy BP, Tremblay ML. Attenuation of leptin action and regulation of obesity by protein tyrosine phosphatase 1B. Dev Cell. 2002;2:497–503. doi: 10.1016/s1534-5807(02)00149-1. [DOI] [PubMed] [Google Scholar]
- 41.Mosinger B, Jr, Tillmann U, Westphal H, Tremblay ML. Cloning and characterization of a mouse cDNA encoding a cytoplasmic protein-tyrosine-phosphatase. Proc Natl Acad Sci U S A. 1992;89:499–503. doi: 10.1073/pnas.89.2.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.ten Hoeve J, de Jesus Ibarra-Sanchez M, Fu Y, Zhu W, Tremblay M, David M, Shuai K. Identification of a nuclear Stat1 protein tyrosine phosphatase. Mol Cell Biol. 2002;22:5662–8. doi: 10.1128/MCB.22.16.5662-5668.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.