

Abstract
The prolactin receptor (PRLR), its associated kinase Jak2, and Stat5 are essential for normal mammary gland development. Due to the upregulation of the PRLR and the local synthesis of its ligand in neoplastic cells, it has been proposed that PRL can act as a local growth factor in human breast cancers. This notion is supported by experimental evidence in transgenic mice that demonstrated that the mammary-specific expression of PRL contributes to carcinogenesis in vivo. To assess the importance of Jak2/Stat5 signaling during mammary cancer initiation and progression, we generated a PRL-induced mammary cancer model that allows the functional ablation of the Jak2 gene in the mammary epithelium prior to and after neoplastic transformation. Collectively, the results of this study show that the functional ablation of Jak2 protects against the onset of PRL-induced mammary tumorigenesis, suggesting that targeting this Janus kinase is a relevant strategy for mammary cancer prevention. Surprisingly, Jak2 deficiency did not affect the growth and survival of PRL-induced mammary cancer cells in culture and in vivo. Consequently, Jak2 cannot be a sole therapeutic target to treat the established disease. PRL-induced mammary cancers exhibited an upregulation of ErbB2 and other ErbB receptor tyrosine kinases that may supersede the functionality of PRLR signaling through Jak2.
Introduction
The peptide hormone prolactin (PRL) is known to play a pivotal role in the occurrence of breast cancer in humans and animal models. High circulating levels of PRL increases the risk of breast cancer in pre- and post-menopausal women (Hankinson et al., 1999; Tworoger and Hankinson, 2006), and the PRL receptor (PRLR) is overexpressed in greater than 95% of breast cancer cases (Clevenger et al., 1995; Ginsburg and Vonderhaar, 1995). The examination of transgenic mouse models provided experimental evidence that a sustained increase in the levels of circulating lactogenic hormones, in particular prolactin, causes mammary cancer (Tornell et al., 1991; Wennbo et al., 1997). More recently, it has been shown that human breast cancer cells are able to upregulate the local synthesis of prolactin, suggesting that this hormone can act in an autocrine manner to promote the proliferation of neoplastic cells (Clevenger et al., 1995; Ginsburg and Vonderhaar, 1995). This novel paradigm was verified in a transgenic model that overexpresses PRL in the mammary epithelium under regulation of the PRL- and estrogen-unresponsive, neu-related lipocalin (NRL) promoter (Rose-Hellekant et al., 2003). More than 65% of NRL-PRL transgenic females were reported to develop both estrogen receptor (ER)-positive and ER-negative lesions after a mean latency of 15 months.
In normal and neoplastic mammary epithelial cells, PRLR signaling synchronously activates multiple signaling cascades such as the Janus kinase 2 (Jak2), the signal transducer and transactivator of transcription 5 (Stat5a and Stat5b), the Src kinase, mitogen activated protein kinases (MAPKs), and the phosphoinositide 3 kinase (PI3K) pathway [for references see Wagner and Rui (2008)]. Jak2 is essential for the tyrosine phosphorylation and activation of Stat5 in response to PRL signaling (Shillingford et al., 2002; Wagner et al., 2004), and the phenotypic abnormalities observed in mammary glands of conventional and conditional knockout mice that lack PRL, its receptor, or both Stat5 isoforms (i.e. Stat5a and Stat5b) are strikingly similar (Horseman et al., 1997; Ormandy et al., 1997; Teglund et al., 1998; Wagner et al., 2004; Cui et al., 2004). This suggests that the Jak2/Stat5 signaling cascade is crucial for mediating the main biological responses that are being initiated by PRL. Collectively, the murine knockout models demonstrated that PRLR signaling though Jak2 and Stat5 is required for the proliferation and differentiation of alveolar progenitors during pregnancy and lactation.
Stat5 has been shown to be activated (i.e. phosphorylated on tyrosine residues 694 or 699 in Stat5a and Stat5b) in a subset of human best cancer cases (Cotarla et al., 2004; Nevalainen et al., 2004). Similar to an increase in circulating levels of PRL or stimulating the local synthesis of this hormone in the mammary gland, the overexpression of wildtype Stat5a as well as a constitutively active Jak2/Stat5 fusion protein is sufficient to induce neoplastic transformation of mammary epithelial cells in vivo (Iavnilovitch et al., 2002; Iavnilovitch et al., 2004). This finding supports the notion that a hyperactive Jak2/Stat5 signaling cascade might mediate the initiation of mammary cancer in response to elevated levels of PRL.
Despite the well-defined role of PRL signaling during the initiation of neoplastic transformation, the importance of this hormone, its receptor, as well as downstream signaling mediators has never been examined in established mammary cancers using genetic model systems (Wagner and Rui, 2008). To specifically assess the significance of Jak2/Stat5 signaling in PRL-induced mammary carcinogenesis, we generated a mouse model that overexpresses PRL in the mammary epithelium that is conditionally deficient in Jak2. Using Cre-mediated recombination, we deleted the Jak2 gene prior to or following neoplastic transformation. This unique approach allowed us to discriminate the role of the Jak2/Stat5 pathway during the initiation of mammary neoplasia as well as the growth and survival of fully neoplastic mammary cancer cells in vitro and in vivo.
Results
Jak2 deficiency prevents the initiation of PRL-induced mammary cancer
Jak2 and its downstream effector Stat5 are essential mediators for the main biological functions of PRL in normal mammary epithelial cells. To study whether Jak2 and active Stat5 are also required for the initiation of PRL-induced mammary cancer, we generated a mouse model that overexpresses PRL locally in the mammary epithelium of females that are conditionally deficient in Jak2 (NRL-PRL MMTV-Cre Jak2fl/fl). In this study, we utilized two NRL-PRL transgenic lines (1647-13 and 1655-8) that overexpress the rat PRL under the neu-related lipocalin (NRL) promoter, whose activation in the mammary epithelium is unresponsive to estrogen and PRL (Rose-Hellekant et al., 2003). We have shown previously that the MMTV-Cre transgene is expressed throughout the entire mammary ductal network in both luminal and myoepithelial cells prior to puberty (Wagner et al., 2001). The MMTV-Cre-mediated conditional deletion of the Jak2 gene does not significantly impair ductal elongation, but the lack of this Janus kinase prevents the specification of alveolar buds located at the terminal ends of the ductal tree in nulliparous females (Wagner et al., 2004). About one-third of all NRL-PRL MMTV-Cre Jak2fl/fl females and their littermate controls expressing Jak2 (NRL-PRL Jak2fl/fl) were bred at least once during their lifetime. As expected, females that were conditionally deficient in Jak2 were unable to nurse their offspring regardless of the local synthesis of exogenous PRL in the mammary epithelium. Experimental animals and their controls were monitored twice weekly over a period of 24 months for the growth of mammary tumors. As illustrated in Table 1, approximately 27% of all control females developed palpable lesions after a mean latency of 21.5 (± 2.1 SD) months. The reproductive status did not have a noticeable influence on the occurrence or latency of mammary cancer (not shown). In contrast to the controls, not a single Jak2 knockout female developed a palpable mammary tumor within the experimental time line. In addition to the primary mammary cancers in control mice, we collected mammary glands from Jak2 deficient and control females at the experimental end point, i.e. the day of tumor resection in a subset of control females or after 24 months in tumor free animals. The whole mount analysis of mammary glands from animals that did not develop palpable cancers revealed that approximately 34% of the control tissues of both NRL-PRL lines contained at least one atypical hyperplastic lesion (Fig. 1a, c, arrow). With the exception of two females of NRL-PRL line 1655-8, none of the Jak2 deficient mice exhibited any microscopic lesions (Fig. 1b, d). Interestingly, the two small hyperplastic lesions of approximately 3 mm in diameter that were observed in the Jak2 deficient mice were located in close proximity to the nipples and not within distant ducts (Suppl. Fig. S1). We previously reported that besides a lack of alveolar budding, Jak2-deficient mammary ducts were thinner and exhibited less tertiary branching (Wagner et al., 2004). These phenotypic abnormalities were also present in Jak2-deficient mammary glands overexpressing exogenous PRL in both NRL-PRL transgenic lines, suggesting that the lack of Jak2 effectively blocks the developmental effects of excess levels of locally synthesized PRL (Fig. 1b, d). This might explain why Jak2-deficient females did not develop any palpable tumors and significantly less hyperplastic lesions within epithelial ducts.
Table 1.
Tumor incidence in females of two NRL-PRL transgenic lines that are conditionally deficient in Jak2 (MMTV-Cre Jak2fl/fl) and their littermate controls expressing Jak2 (Jak2fl/fl).
| Genotype | Number of females | Incidence of proliferative lesions |
||
|---|---|---|---|---|
| Mammary Tumors | Atypical Hyperplasia | Total | ||
| Total | ||||
| Jak2fl/fl | 59 | 16 (27.1 %) | 20 (33.9 %) | 36 (61.0 %) |
| MMTV-Cre Jak2fl/fl | 28 | 0 | 2 (7.1 %) | 2 (7.1 %) |
| NRL-PRL (line 1647-13) | ||||
| Jak2fl/fl | 32 | 7 (21.9 %) | 13 (40.6 %) | 20 (62.5 %) |
| MMTV-Cre Jak2fl/fl | 14 | 0 | 0 | 0 |
| NRL-PRL (line 1655-8) | ||||
| Jak2fl/fl | 27 | 9 (33.3 %) | 7 (25.9 %) | 16 (59.3 %) |
| MMTV-Cre Jak2fl/fl | 14 | 0 | 2 (14.3 %) | 2 (14.3 %) |
Fig. 1. Inhibition of PRL-induced mammary tumorigenesis through deletion of Jak2.

Mammary gland whole-mounts from two NRL-PRL transgenic lines (1655-8 and 1647-13) that are Jak2-deficient (b, d) and their littermate wildtype controls (a, c). Arrows in panels a and c indicate the location of atypical hyperplastic lesions; LN lymph node; bar = 1mm.
It is generally known that the vast majority of genetically engineered mouse models for breast cancer that were generated over the last two decades develop estrogen receptor (ER) α negative mammary tumors. NRL-PRL transgenic females appear to be distinct from these earlier models since they were reported to give rise to both ERα-positive and ERα-negative mammary cancers (Rose-Hellekant et al., 2003). In addition to a histological examination, we stained 16 mammary tumors from both NRL-PRL lines [i.e. NRL-PRL (line 1647-13 or 1655-8) Jak2fl/fl control females] with an antibody against ERα to assess whether this steroid receptor was expressed (Table 2). We discriminated ERα negative mammary tumors from neoplasia that exhibited ERα staining in more than 10% of the cancer cells, which are typically defined as ERα positive tumors. To indicate the degree of intra-tumor heterogeneity between the various cancers, we further stratified tumors that exhibited a more homogeneous expression of ERα throughout the sections from those that were clearly comprised of both ERα positive and negative cancer cells. ERα was present within nearly all nuclei in only 2 out of 16 primary cancers, and these mammary lesions were comprised of neoplastic cells that appeared more differentiated (Fig. 2). Approximately 43% of all mammary cancers did not contain any epithelial cells that expressed ERα, and the remaining tumors (about 43%) exhibited mixed lesions that were comprised of ERα negative epithelial cells that contained regions that were clearly ERα positive. The ERα staining pattern among individual mammary tumors was indistinguishable between both NRL-PRL transgenic lines.
Table 2.
Expression of the estrogen receptor α (ERα) in PRL-induced mammary cancers
| Number of stained tumors | ERα positive | ERα negative | ||
|---|---|---|---|---|
| Homogeneous | Mixed | |||
| Total | 16 | 2 (12.5 %) | 7 (43.8 %) | 7 (43.8 %) |
| NRL-PRL (line 1647-13) | 7 | 1 (14.3 %) | 3 (42.9 %) | 3 (42.9 %) |
| NRL-PRL (line 1655-8) | 9 | 1 (11.1 %) | 4 (44.4 %) | 4 (44.4 %) |
Fig. 2. A subset of PRL-induced mammary cancers expresses the estrogen receptor α.

Immunohistochemistry of ERα in various mammary tumors (a-c) and the normal mammary epithelium (d); a, ERα-positive tumor, b, mixed lesion containing a subset of ERα-positive cells, c, ERα-negative cancer. Panel e shows a serial section of the mammary duct shown in panel d that was stained without the primary antibody against ERα. All slides were counterstained with hematoxylin (bar, 50 μm).
Jak2 is not required for the growth and survival of PRL-induced mammary cancer cells
Since both the PRL receptor and its ligand are upregulated in human breast cancers, targeting the main downstream mediators of this hormone receptor was suggested to be a suitable strategy to treat breast cancer patients. Since we have demonstrated that Jak2 is essential for the onset of PRL-induced mammary cancers, a rational objective of our subsequent work was to address whether Jak2 and active Stat5 are required for the growth and survival of PRL-induced mammary cancer cells. To experimentally address this issue, we derived neoplastic cells from primary tumors of NRL-PRL Jak2fl/fl control females, and deleted the Jak2 gene from these mammary cancer cells using an adenovirus expressing a Cre/GFP fusion protein (Fig. 3a). Following the fluorescence activated sorting of infected cells that were transiently positive for GFP, we obtained isogenic pairs of cancer cells with and without Jak2. The correct Cre-mediated excision of both floxed Jak2 alleles was verified by PCR and western blot analysis (Fig. 3b, 3c). Notably, the isogenic pair of PRL-induced mammary cancer cells with and without Jak2 shown in Fig. 3c expressed moderate levels of ERα. Despite the fact that the primary cancer cells were derived from PRL-induced tumors, they all lacked expression of active Stat5. This might suggest that most cancer cells had lost the PRL-mediated autocrine activation of its downstream mediator Stat5 in the progressing lesions (Fig. 3d). Therefore, the deletion of Jak2 had no noticeable effect on the growth and survival of these cancer cells. Stimulation of the primary cancer cells with PRL was sufficient to induce the phosphorylation of Stat5. This clearly showed that these cells were still responsive to exogenous PRL. The deletion of Jak2 led to a complete inhibition of Stat5 activation in these cells following PRL stimulation. In contrast to Stat5, loss of Jak2 had no significant effect on the expression and activation of Stat3, which was constitutively phosphorylated. Collectively, the successful generation of Jak2 deficient mammary cancer cells suggests that neither Jak2 nor active Stat5 are required for the growth and survival transformed mammary epithelial cells in culture.
Fig. 3. PRL-induced mammary cancer cells that are deficient in Jak2 lack activation of Stat5.
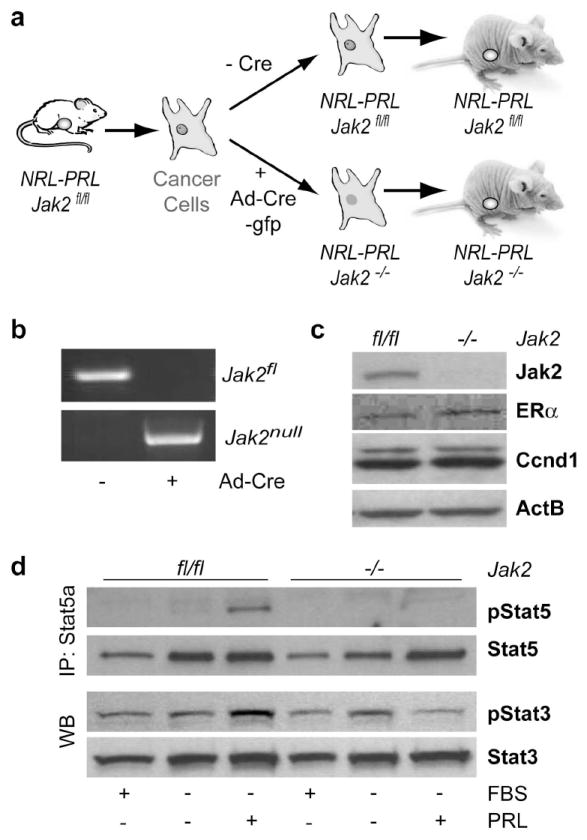
(a) Experimental design for the conditional deletion of Jak2 in PRL-induced mammary cancer cells. (b) PCR assay to verify the loss of both Jak2 floxed alleles (Jak2fl/fl) and the presence of Jak2 knockout alleles (Jak2-/-) following Cre-mediated recombination. (c) Expression analysis of Jak2, Estrogen Receptor α (ERα), and Cyclin D1 (Ccnd1) in Jak2-deficient cancer cells and their isogenic control cells. Beta-Actin (ActB) served as a loading control. (d) Immunoprecipitation (IP) and western blot (WB) analysis to assess the activation of Stat5 and Stat3 following PRL stimulation.
Jak2 is dispensable for engraftment and growth of cancer-initiating cells in vivo
The establishment of secondary cancers in recipient mice following transplantation serves as biological evidence that tumor-initiating cells that were cultured ex vivo still maintained their neoplastic characteristics. To address whether Jak2 is required in cancer-initiating cells in vivo, we transplanted 1 × 106 tumor cells with and without Jak2 into the collateral number four mammary glands of twenty Athymic nude mice (i.e. ten mice or twenty transplants per cell line) and obtained 19 palpable tumors in total (i.e. 10 with and 9 without Jak2). As shown in Fig. 4a, the functional ablation of Jak2 did not significantly alter the engraftment of cancer cells or the growth of the resulting tumor in recipient females. The subsequent analysis of genomic DNA from the Jak2-defficient tumors and their Jak2 expressing controls confirmed that all secondary lesions were comprised of the transplanted cancer cells carrying either two Jak2 null alleles or two unrecombined Jak2 floxed alleles in the controls (Fig. 4b). Therefore, all secondary tumors arose solely from Jak2-/- cancer-initiating cells, suggesting that Jak2 is dispensable for the survival and numeric expansion of this progenitor pool. A Jak2 wildtype allele served as an internal control in the PCR assay shown in Fig. 4b. This allele is not present in the transplanted cell types (i.e. Jak2fl/fl or Jak2-/-) and therefore originated from tumor-associated stromal cells and blood vessels of the wildtype host. The initial examination of secondary tumors revealed that, regardless whether Jak2 was expressed or not, cancer cells lacked expression of active Stat5 (Fig. 5a, upper panels). We therefore administered supraphysiological levels of PRL to a subset of recipient mice 30 min before collecting the tumor specimen to monitor the acute activation of Stat5 in response to PRL stimulation (Fig. 5a, lower panels). Although normal mammary epithelial cells within the thoracic #3 mammary glands of recipient mice showed more extensive nuclear Stat5 staining in both experimental groups (Fig. 5a, insets), the administration of PRL did not significantly increase the number of cells with active Stat5 in Jak2fl/fl control tumors, and nuclear Stat5 was not observed in Jak2-/- cancer cells. Collectively, these observations suggest that neither Jak2 nor active Stat5 are required for cancer cell growth and survival in vivo. Interestingly, the functional ablation of Jak2 did not affect the presence of ERα positive cells within secondary lesions (Fig. 5b). This might suggest that treatment of primary breast cancers with a Jak2 inhibitor will not lead to a selective expansion of ERα negative cancer-initiating cells.
Fig. 4. Jak2 deficiency does not affect the growth of PRL-induced mammary cancer cells in vivo.

(a) Growth of PRL-induced mammary cancer cells lacking Jak2 and their isogenic controls in an orthotropic transplant of Athymic nude mice. (b) PCR assay to verify the presence of Jak2 floxed alleles or Jak2 null alleles in resulting mammary tumors following orthotropic transplantation. Note that the Jak2 wildtype allele (Jak2wt), which was present in the PCR assay in both experimental groups, originated from stromal cells and blood vessels of the wildtype host. This allele was not present in the engrafted Jak2fl/fl or Jak2-/- cancer cells. NC: negative control.
Fig. 5. Jak2 deficiency leads to lack of Stat5 activation but not expression of ERα and Cyclin D1.

(a) Immunohistochemistry to assess the PRL-mediated activation of Stat5 in control tumors and cancers lacking Jak2. Insets show the PRL-induced activation of Stat5 in normal #3 mammary glands derived from the tumor-bearing, wildtype recipient females. (b) Immunohistochemistry to determine the continuous expression of ERα and Cyclin D1 (Ccnd1) in PRL-induced mammary cancers that lack Jak2 and their wildtype controls. All slides were counterstained with hematoxylin (bars, 50 μm).
PRL-induced mammary cancers overexpress ErbB family members, which may supersede the functionality of Jak2
We have reported previously that the functional ablation of Jak2 in normal mammary epithelial cells and Jak2 conditional knockout mice results in a decrease in active Akt1 as well as expression and nuclear accumulation of Cyclin D1 (Sakamoto et al., 2007). In contrast to our earlier studies in untransformed cells, Jak2 deficiency had no significant effect on the expression of Akt1 and Cyclin D1 in PRL-induced mammary cancer cells in vitro (Fig. 3c) and in vivo (Fig. 5b, lower panel; Suppl. Fig. S2). Since Stat5 is not phosphorylated in these mammary cancer cells by receptor tyrosine kinases, cytokine receptor-associated Janus kinases other than Jak2, or cytoplasmic kinases, it is evident that compensatory signaling networks are being activated in neoplastic cells that supersede the function of Jak2 in regulating the expression of Akt1 and Cyclin D1. ErbB receptor tyrosine kinases, which are upregulated or constitutively active in a significant subset of human breast cancer cases, are known to activate Akt1 and thereby maintain the nuclear retention of Cyclin D1 through inactivation of GSK3β (Diehl et al., 1998). We therefore assessed by western blot analysis and immunohistochemistry whether selected ErbB receptor tyrosine kinases were upregulated in PRL-induced mammary tumors. It is evident from the results shown in Table 3, Fig. 6a as well as supplemental Fig. S2, that PRL-induced mammary cancers predominantly expressed ErbB2 compared to normal mammary gland tissues. In addition, other ErbB family members were upregulated in selective tumor specimens. In some cases, we detected a significantly elevated expression of all four ErbB family members (for example, Fig. 6a, tumor #1). The ErbB2 receptor and its heterodimeric partner ErbB3 have been suggested to function as an oncogenic unit in breast cancer (Holbro et al., 2003; Kim et al., 2005). Despite variable expression of both receptors in individual PRL-induced mammary tumors, ErbB2 and ErbB3 engaged in heterodimer formation and were tyrosine phosphorylated (Fig. 6b). Since only Jak2 expressing females developed PRL-induced mammary cancer (i.e. NRL-PRL Jak2fl/fl), it is apparent that the selective upregulation of these receptor tyrosine kinases occurs independently of Jak2/Stat5 signaling during or following neoplastic transformation. Also, the reduction in the functionality of Jak2 and therefore loss of nuclear Stat5 during disease progression seems not to have an effect on the elevated expression levels of ErbB kinases (Fig. 6c). Interestingly, Jak2 deficiency in normal mammary epithelial cells in culture already seems to favor the selection of cells with an upregulated expression of the EGFR (Fig. 6c), and it is therefore possible that epithelial subtypes with high ErbB activity might be the cancer-initiating population in NRL-PRL transgenic females. Collectively, the results of this study show that the upregulation of ErbB receptor tyrosine kinase might be sufficient to supersede the importance of Jak2/Stat5 signaling in cancer cells. This might explain why targeting Jak2 alone is not sufficient to halt the growth and survival of mammary cancer cells in culture and in vivo.
Table 3.
Expression of the ErbB family members in PRL-induced mammary cancers.
| Number of stained tumors | EGFR | ErbB2 | ErbB3 | ErbB4 | |
|---|---|---|---|---|---|
| Total | 16 | 4 (25.0 %) | 16 (100 %) | 7 (43.8%) | 9 (56.3%) |
| NRL-PRL (line 1647-13) | 7 | 3 (42.9 %) | 7 (100 %) | 4 (42.9 %) | 4 (57.1 %) |
| NRL-PRL (line 1655-8) | 9 | 1 (11.1 %) | 9 (100 %) | 3 (33.3 %) | 5 (55.5 %) |
Fig. 6. PRL-induced mammary cancers exhibit an upregulation of ErbB receptor tyrosine kinases.

(a) Western blot analysis to determine the expression of all four ErbB family members in NRL-PRL-induced mammary cancers as well as normal mammary glands (MG) from lactating NRL-PRL females. (b) Immunoprecipitation (IP) and western blot (WB) analysis to assess the tyrosine phosphorylation and heterodimer formation of ErbB2 and ErbB3. (c) Western blot analysis to assess the expression of ErbB receptors in untransformed and neoplastic mammary epithelial cells that lack Jak2 and their isogenic controls expressing Jak2. Beta-Actin (ActB) served as a loading control in both panels.
Discussion
Essential biological functions of the peptide hormone PRL during normal mammary gland development are mediated by Jak2 and its downstream effector Stat5. The functional ablation of Jak2 in the normal mammary epithelium prior to tumorigenesis clearly demonstrates that this receptor-associated Janus kinase is equally important for mammary carcinogenesis in response to an increase in PRL signaling. Among various epithelial subtypes that originate during mammary gland development, alveolar progenitors are highly responsive to PRL, and it can be assumed that these cells are also the cellular targets for PRL-induced neoplastic transformation. We have previously reported that alveolar progenitors in nulliparous mice and parity-induced mammary epithelial cells (PI-MECs) in parous females facilitate mammary tumorigenesis in transgenic mice overexpressing wildtype ErbB2 (Henry et al., 2004). These cell types are located within lobular units, and they are pregnancy-hormone-responsive. Although PI-MECs have a limited role as alveolar progenitors in multiparous females, these cells retain characteristics of multipotent stem cells such as self-renewal and contribution to ductal and alveolar morphogenesis upon transplantation (Wagner et al., 2002; Boulanger et al., 2005; Matulka et al., 2007). Given the fact that the genesis of PI-MECs during pregnancy depends on PRL signaling, we anticipated that targeting Jak2 or Stat5 might be a feasible strategy to prevent the onset of PRL-mediated mammary cancer (Wagner and Rui, 2008). Indirect confirmation for this assumption was previously provided by studies that utilized mice that overexpress TGFα and the SV40 large T-antigen. In these models, Stat5a or PRLR deficiency caused a delay or reduced incidence in mammary tumorigenesis (Humphreys and Hennighausen, 1999; Ren et al., 2002; Oakes et al., 2007; Miermont et al., 2010). The complete absence of PRL-induced mammary cancer in females lacking Jak2 now provides direct experimental evidence that targeting Jak2 might be a suitable strategy for the prevention of breast cancer subtypes that originate from luminal cells within terminal ducts and alveolar units that are PRL responsive. Specifically, individuals with hyperprolactinemia or women that are at risk of developing pregnancy-associated breast cancers might benefit from such a preventive regimen.
Although the findings from various genetically engineered mouse models including Jak2 deficient mice suggest that PRL signaling through the Jak2/Stat5 cascade can contribute to mammary cancer initiation, in vivo model systems were missing that specifically address whether inhibiting the activation of the PRLR or its downstream mediators is also a therapeutically relevant strategy to treat the established disease. Multiple Jak2 tyrosine kinase inhibitors are currently being developed and tested to treat myeloproliferative disorders and other hematopoietic malignancies [for a review, see Pardanani, 2007], and it is therefore feasible that these drugs could also be utilized in therapeutic regimens to combat advanced breast cancer. Prior to such clinical studies, the availability of a Jak2 conditional knockout model provides a unique opportunity to examine the significance of the Jak2/Stat5 signaling cascade in fully neoplastic mammary cancer cells that were transformed in response to an increase in PRL signaling. Although Jak2 is essential for the onset of PRL-induced mammary tumorigenesis, the deletion of Jak2 following neoplastic transformation had no significant impact on the survival and growth of mammary cancer cells in culture and in vivo. These observations clearly suggest that Jak2 cannot be the sole target to treat advanced breast cancers that are of luminal origin and that express the PRLR. Our findings are in line with observations in primary human breast cancer cases that show that transcriptionally active Stat5 is not required for cancer progression and metastasis. Nuclear Stat5 was reported to contribute to a more differentiated phenotype of breast cancer cells, and it might therefore serve as a favorable prognostic marker, in particular for lymph node-negative breast cancers (Nevalainen et al., 2004). Among both Stat5 isoforms, Stat5a was suggested to play the predominant role in promoting the differentiation and in suppressing the motility of cancer cells, and this effect might be facilitated through suppression of Bcl6 (Tang et al., 2010; Tran et al., 2010).
At this point, we cannot exclude that PRLR signaling is still capable of promoting breast cancer progression and invasion through Jak2/Stat5-independent pathways such as c-Src, FAK, and MAP kinases. We and others have demonstrated recently that the activity of c-Src does not depend on the functionality of Jak2 as previously suggested (Dominguez-Caceres et al., 2004; Sakamoto et al., 2007; Garcia-Martinez et al., 2010). The contribution of these signal transducers during PRL-induced mammary tumor progression needs to be examined in more detail using novel in vivo model systems that allow a conditional inactivation of signal transducers such as c-Src. It is evident that conventional knockout models (e.g. Cyclin D1, c-Src, or Akt1 deficient mice) in combination with oncogene-expressing transgenics are not suitable to specifically address whether the targeted inhibition of signal transducers is therapeutically relevant (Matulka and Wagner, 2005). Many of these animals lack expression of putative therapeutic targets from the day they were conceived and never develop mammary cancer. Consequently, they can only serve as models for cancer prevention and not for therapy. Our studies in females that are conditionally deficient in Jak2 clearly shows that the significance of particular signal transducers can shift dramatically following neoplastic transformation. Cancer cells frequently upregulate or acquire mutations within receptors or associated kinases that mediate self-sufficiency in growth signals and evasion from apoptosis. In particular, receptor tyrosine kinases such as ErbB2 are upregulated in a significant subset of human breast cancers (Slamon et al., 1989), and these kinases phosphorylate the same downstream signaling mediators that are being activated by multiple cytokine receptors including the PRLR. This might explain why PRL potentiates signals downstream of other growth factor receptors and why the overexpression of ErbB receptors and their ligands such as TGFα are able to cooperate with PRLR signaling during tumor initiation (Arendt et al., 2006; Arendt and Schuler, 2008). In this study, we observed that PRL-induced mammary tumors exhibited an upregulation of ErbB2 and selected other members of the ErbB family, and these molecular changes may function independently from Jak2/Stat5 signaling. We propose that the gain-of-function of receptor tyrosine kinases during neoplastic transformation is able to supersede the importance of Jak2/Stat5 signaling in regulating downstream effectors following mammary cancer initiation. Although the PRL-induced activation of Jak2 and Stat5 is able to regulate the levels of active Akt1 and nuclear accumulation of Cyclin D1 in normal cells (Brockman et al., 2002; Brockman and Schuler, 2005; Sakamoto et al., 2007), we have demonstrated recently that the expression of constitutively active ErbB2 was sufficient to elevate the expression of these downstream mediators independently of Jak2. Similar to PRL overexpressing mice, Jak2 plays a role in the initiation of ErbB2-associated mammary tumorigenesis, but Jak2 is dispensable for the maintenance of ErbB2-expressing mammary cancer cells (Sakamoto et al., 2009). Although Jak2 may not be a sole target to treat the established disease, targeting this kinase to prevent the initiation of breast cancer might be more broadly applicable since Jak2 is required for the proliferation of luminal progenitor cells located within terminal ducts and alveolar units that are prime targets for growth factor induced neoplasia (Wellings et al., 1975; Cardiff, 1998). Also, it has been proposed recently that luminal progenitors are the likely source of basal-like breast tumors in humans (Lim et al., 2009). Future studies will show whether Jak2 is also required for the genesis of breast cancer subtypes that express basal epithelial cell markers such Brca1-associated mammary tumors (Triplett et al., 2008).
Materials and Methods
Mouse models
MMTV-Cre (Wagner et al., 1997a) transgenics was crossed with Jak2 conditional knockout mice. The generation of genetically engineered animals with Jak2 conditional knockout alleles (Jak2fl/fl) and the PCR protocols to determine the presence of Jak2 floxed, Jak2 recombined/null, and Jak2 wildtype alleles have been described previously (Krempler et al., 2004; Wagner et al., 2004). In this study, we used two transgenic lines that overexpress PRL in the mammary gland under the control of the neu-related lipocalin promoter [NRL-PRL, lines 1647-13 and 1655-8] (Rose-Hellekant et al., 2003). All targeted alleles and transgenes were carried in a predominantly FvB background (>50-75%). To establish orthotopic transplant models, 1.0 × 106 mammary cancer cells deficient in Jak2 and isogenic wildtype control cells were injected into the number four inguinal mammary glands of Athymic nude females (NCr strain, National Cancer Institute). Tumor volumes were measured every two weeks using a caliper. To estimate the tumor volume, we used the following equation as described previously (aoui-Jamali et al., 2003): volume = π/6 (length × width2). Thirty minutes prior to the experimental end point and harvesting the tumors, a subset of mice was injected intraperitoneally with ovine PRL (AFP-10692C; 5 μg per g of body weight) or saline as a control to assess the PRLR/Jak2-dependent activation of Stat5. All animals were treated humanely and in accordance with institutional guidelines and federal regulations.
Cell culture
Mammary cancer cells were derived from tumor bearing NRL-PRL Jak2fl/fl females and cultured according to a protocol published by Medina and Kittrell (2000). To delete both Jak2 conditional knockout alleles, primary tumor cell were infected with an adenoviral vector expressing a fusion protein of Cre recombinase and GFP (Vector Biolabs) and fractionated using fluorescence activated cell sorting. Recombinant mouse PRL (AFP306C) was kindly provided by Dr. A. F. Parlow under the sponsorship of the National Hormone and Pituitary Program, NIDDK (National Institute of Health). Mammary cancer cells were treated with 10nM of PRL for 20 minutes to induce the activation of Stat5.
Immunoprecipitation and western blot analysis
The preparation of whole cell extracts of clarified cell lysates and tissue homogenates as well as the experimental procedures for immunoprecipitation (IP) and western blot analysis were described in detail elsewhere (Sakamoto et al., 2007). The following antibodies were used for immunoblotting: α-β-Actin (I-19; 1;2000 dilution), α-Cyclin D1 (72G-13; 1:1000 dilution), α-Estrogen Receptor α (MC-20; 1:1000 dilution), α-ErbB3 (C-17; 1:1000 dilution), α-ErbB4 (C-18; 1:1000 dilution) from Santa Cruz Biotechnology; α-Stat5 (2:250 dilution) from BD Biosciences; α-pAkt (Ser473; 1:1000 dilution), α-Akt (1:1000 dilution), α-pStat3 (Tyr705; 58E12; 1:1000 dilution), α-Stat3 (124H6; 1:1000 dilution) from Cell Signaling Technology; α-EGFR (ab2430; 1:200 dilution) from Abcam; α-Jak2 antibody (691R5; 1:2000 dilution) from Biosource; α-ErbB2/c-Neu (Ab-3; 1:1000 dilution) from Calbiochem. The α-phospho-Stat5a/b (Y694/9) antibody (AX1; 2 μg/ml; Advantex Bioreagents) was kindly provided from Dr. H. Rui (Thomas Jefferson University). The polyclonal α-Stat5a antiserum was a gift from Dr. L. Hennighausen (National Institute of Health).
Whole mount staining and immunohistochemistry
Whole mounts were prepared and stained in carmine alum as described previously (Wagner et al., 1997b). Basic protocols for immunohistochemistry on paraffin-embedded mammary gland specimens were described elsewhere (Wagner et al., 2004). The following antibodies were used for immunohistochemistry: α-pStat5A/B (Tyr694/699) antibody (1:100 dilution) from Upstate Biotechnology; α-Estrogen Receptor α, ERα (1115-1; 1:100 dilution) from Epitomics; α-Cyclin D1 antibody (Ab-4, 1:250 dilution) from NeoMarkers; α-EGFR (ab2430; 1:200 dilution) from Abcam; α-HER2/ErbB2 (1:25 dilution) from Cell Signaling Technology; α-ErbB3 (C-17; 1:100 dilution), α-ErbB4 (C-18; 1:100 dilution) from Santa Cruz Biotechnology; α-CK14 antibody (1:1000 dilution) from Convance; α-CK8 antibody (TROMA-I; 1:250 dilution) from the Developmental Studies Hybridoma Bank at the University of Iowa. For visualization of the specific targets, we used corresponding biotinylated secondary antibodies and Vectastain Elite ABC kit (Vector Laboratories).
Supplementary Material
(a) H&E staining (bar, 100 μm). (b, c) Immunostaining for cytokeratin (CK) 8 and 14. Slides were counterstained with DAPI (bar, 30 μm). (d -h) Immunohistochemistry for ERα and all four ErbB family members. Slides were counterstained with hematoxylin (bar, 50 μm).
Western blot analysis to determine the expression of phosphorylated and total levels of Akt1 as well as Cyclin D1 in PRL-induced mammary tumors lacking Jak2 and their wildtype controls. α-Tubulin was used as a loading control.
Control panels on the right side show serial sections that were stained without the corresponding primary antibodies. All slides were counterstained with hematoxylin (bar, 50 μm).
Acknowledgments
This work was supported by the Public Health Service grant CA117930 (to K.U.W.) as well as grant CA78312 (to L.A.S.) from the National Cancer Institute. Additional financial support provided to K.U.W. by the Nebraska Cancer and Smoking Disease Research Program (NE DHHS LB506 2009-45) was imperative to finance the maintenance of the Jak2 conditional knockout model. K.S. received a postdoctoral fellowship from the Susan G. Komen Breast Cancer Foundation (PDF0600835).
Footnotes
Disclosure statement: The authors have nothing to disclose
References
- aoui-Jamali MA, Song DJ, Benlimame N, Yen L, Deng X, Hernandez-Perez M, et al. Regulation of multiple tumor microenvironment markers by overexpression of single or paired combinations of ErbB receptors. Cancer Res. 2003;63:3764–3774. [PubMed] [Google Scholar]
- Arendt LM, Rose-Hellekant TA, Sandgren EP, Schuler LA. Prolactin potentiates transforming growth factor alpha induction of mammary neoplasia in transgenic mice. Am J Pathol. 2006;168:1365–1374. doi: 10.2353/ajpath.2006.050861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arendt LM, Schuler LA. Prolactin drives estrogen receptor-alpha-dependent ductal expansion and synergizes with transforming growth factor-alpha to induce mammary tumors in males. Am J Pathol. 2008;172:194–202. doi: 10.2353/ajpath.2008.070597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulanger CA, Wagner KU, Smith GH. Parity-induced mouse mammary epithelial cells are pluripotent, self-renewing and sensitive to TGF-beta1 expression. Oncogene. 2005;24:552–560. doi: 10.1038/sj.onc.1208185. [DOI] [PubMed] [Google Scholar]
- Brockman JL, Schroeder MD, Schuler LA. PRL activates the cyclin D1 promoter via the Jak2/Stat pathway. Mol Endocrinol. 2002;16:774–784. doi: 10.1210/mend.16.4.0817. [DOI] [PubMed] [Google Scholar]
- Brockman JL, Schuler LA. Prolactin signals via Stat5 and Oct-1 to the proximal cyclin D1 promoter. Mol Cell Endocrinol. 2005;239:45–53. doi: 10.1016/j.mce.2005.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardiff RD. Are the TDLU of the human the same as the LA of mice? J Mammary Gland Biol Neoplasia. 1998;3:3–5. doi: 10.1023/a:1018714016205. [DOI] [PubMed] [Google Scholar]
- Clevenger CV, Chang WP, Ngo W, Pasha TL, Montone KT, Tomaszewski JE. Expression of prolactin and prolactin receptor in human breast carcinoma. Evidence for an autocrine/paracrine loop. Am J Pathol. 1995;146:695–705. [PMC free article] [PubMed] [Google Scholar]
- Cotarla I, Ren S, Zhang Y, Gehan E, Singh B, Furth PA. Stat5a is tyrosine phosphorylated and nuclear localized in a high proportion of human breast cancers. Int J Cancer. 2004;108:665–671. doi: 10.1002/ijc.11619. [DOI] [PubMed] [Google Scholar]
- Cui Y, Riedlinger G, Miyoshi K, Tang W, Li C, Deng CX, et al. Inactivation of Stat5 in mouse mammary epithelium during pregnancy reveals distinct functions in cell proliferation, survival, and differentiation. Mol Cell Biol. 2004;24:8037–8047. doi: 10.1128/MCB.24.18.8037-8047.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diehl JA, Cheng M, Roussel MF, Sherr CJ. Glycogen synthase kinase-3beta regulates cyclin D1 proteolysis and subcellular localization. Genes Dev. 1998;12:3499–3511. doi: 10.1101/gad.12.22.3499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez-Caceres MA, Garcia-Martinez JM, Calcabrini A, Gonzalez L, Porque PG, Leon J, et al. Prolactin induces c-Myc expression and cell survival through activation of Src/Akt pathway in lymphoid cells. Oncogene. 2004;23:7378–7390. doi: 10.1038/sj.onc.1208002. [DOI] [PubMed] [Google Scholar]
- Garcia-Martinez JM, Calcabrini A, Gonzalez L, Martin-Forero E, gullo-Ortuno MT, Simon V, et al. A non-catalytic function of the Src family tyrosine kinases controls prolactin-induced Jak2 signaling. Cell Signal. 2010;22:415–426. doi: 10.1016/j.cellsig.2009.10.013. [DOI] [PubMed] [Google Scholar]
- Ginsburg E, Vonderhaar BK. Prolactin Synthesis and Secretion by Human Breast Cancer Cells. Cancer Res. 1995;55:2591–2595. [PubMed] [Google Scholar]
- Hankinson SE, Willett WC, Michaud DS, Manson JE, Colditz GA, Longcope C, et al. Plasma Prolactin Levels and Subsequent Risk of Breast Cancer in Postmenopausal Women. J Natl Cancer Inst. 1999;91:629–634. doi: 10.1093/jnci/91.7.629. [DOI] [PubMed] [Google Scholar]
- Henry MD, Triplett AA, Oh KB, Smith GH, Wagner KU. Parity-induced mammary epithelial cells facilitate tumorigenesis in MMTV-neu transgenic mice. Oncogene. 2004;23:6980–6985. doi: 10.1038/sj.onc.1207827. [DOI] [PubMed] [Google Scholar]
- Holbro T, Beerli RR, Maurer F, Koziczak M, Barbas CF, Hynes NE. The ErbB2/ErbB3 heterodimer functions as an oncogenic unit: ErbB2 requires ErbB3 to drive breast tumor cell proliferation. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:8933–8938. doi: 10.1073/pnas.1537685100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horseman ND, Zhao W, Montecino-Rodriguez E, Tanaka M, Nakashima K, Engle SJ, et al. Defective mammopoiesis, but normal hematopoiesis, in mice with a targeted disruption of the prolactin gene. EMBO J. 1997;16:6926–6935. doi: 10.1093/emboj/16.23.6926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphreys RC, Hennighausen L. Signal transducer and activator of transcription 5a influences mammary epithelial cell survival and tumorigenesis. Cell Growth Differ. 1999;10:685–694. [PubMed] [Google Scholar]
- Iavnilovitch E, Cardiff RD, Groner B, Barash I. Deregulation of Stat5 expression and activation causes mammary tumors in transgenic mice. Int J Cancer. 2004;112:607–619. doi: 10.1002/ijc.20484. [DOI] [PubMed] [Google Scholar]
- Iavnilovitch E, Groner B, Barash I. Overexpression and forced activation of stat5 in mammary gland of transgenic mice promotes cellular proliferation, enhances differentiation, and delays postlactational apoptosis. Mol Cancer Res. 2002;1:32–47. [PubMed] [Google Scholar]
- Kim A, Liu B, Ordonez-Ercan D, Alvarez K, Jones L, McKimmey C, et al. Functional interaction between mouse erbB3 and wild-type rat c-neu in transgenic mouse mammary tumor cells. Breast Cancer Research. 2005;7:R708–R718. doi: 10.1186/bcr1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krempler A, Qi Y, Triplett AA, Zhu J, Rui H, Wagner KU. Generation of a conditional knockout allele for the Janus kinase 2 (Jak2) gene in mice. Genesis. 2004;40:52–57. doi: 10.1002/gene.20063. [DOI] [PubMed] [Google Scholar]
- Lim E, Vaillant F, Wu D, Forrest NC, Pal B, Hart AH, et al. Aberrant luminal progenitors as the candidate target population for basal tumor development in BRCA1 mutation carriers. Nat Med. 2009;15:907–913. doi: 10.1038/nm.2000. [DOI] [PubMed] [Google Scholar]
- Matulka LA, Triplett AA, Wagner KU. Parity-induced mammary epithelial cells are multipotent and express cell surface markers associated with stem cells. Dev Biol. 2007;303:29–44. doi: 10.1016/j.ydbio.2006.12.017. [DOI] [PubMed] [Google Scholar]
- Matulka LA, Wagner KU. Models of Breast Cancer. Drug Discov Today Dis Models. 2005;2:1–6. [Google Scholar]
- Medina D, Kittrell FS. Establishment of mouse mammary cell lines. In: Ip MM, Ash BB, editors. Methods in mammary gland biology and breast cancer. Chapter 13. New York: Kluwer Academic/Plenum Publishers; 2000. Nov 1, 2000. pp. 137–145. [Google Scholar]
- Miermont AM, Parrish AR, Furth PA. Role of ER{alpha} in the differential response of Stat5a loss in susceptibility to mammary preneoplasia and DMBA-induced carcinogenesis. Carcinogenesis. 2010 doi: 10.1093/carcin/bgq048. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nevalainen MT, Xie J, Torhorst J, Bubendorf L, Haas P, Kononen J, et al. Signal transducer and activator of transcription-5 activation and breast cancer prognosis. J Clin Oncol. 2004;22:2053–2060. doi: 10.1200/JCO.2004.11.046. [DOI] [PubMed] [Google Scholar]
- Oakes SR, Robertson FG, Kench JG, Gardiner-Garden M, Wand MP, Green JE, et al. Loss of mammary epithelial prolactin receptor delays tumor formation by reducing cell proliferation in low-grade preinvasive lesions. Oncogene. 2007;26:543–553. doi: 10.1038/sj.onc.1209838. [DOI] [PubMed] [Google Scholar]
- Ormandy CJ, Camus A, Barra J, Damotte D, Lucas B, Buteau H, et al. Null mutation of the prolactin receptor gene produces multiple reproductive defects in the mouse. Genes Dev. 1997;11:167–178. doi: 10.1101/gad.11.2.167. [DOI] [PubMed] [Google Scholar]
- Pardanani A. JAK2 inhibitor therapy in myeloproliferative disorders: rationale, preclinical studies and ongoing clinical trials. Leukemia. 2007;22:23–30. doi: 10.1038/sj.leu.2404948. [DOI] [PubMed] [Google Scholar]
- Ren S, Cai HR, Li M, Furth PA. Loss of Stat5a delays mammary cancer progression in a mouse model. Oncogene. 2002;21:4335–4339. doi: 10.1038/sj.onc.1205484. [DOI] [PubMed] [Google Scholar]
- Rose-Hellekant TA, Arendt LM, Schroeder MD, Gilchrist K, Sandgren EP, Schuler LA. Prolactin induces ERalpha-positive and ERalpha-negative mammary cancer in transgenic mice. Oncogene. 2003;22:4664–4674. doi: 10.1038/sj.onc.1206619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto K, Creamer BA, Triplett AA, Wagner KU. The Janus kinase 2 is required for expression and nuclear accumulation of cyclin D1 in proliferating mammary epithelial cells. Mol Endocrinol. 2007;21:1877–1892. doi: 10.1210/me.2006-0316. [DOI] [PubMed] [Google Scholar]
- Sakamoto K, Lin WC, Triplett AA, Wagner KU. Targeting janus kinase 2 in Her2/neu-expressing mammary cancer: Implications for cancer prevention and therapy. Cancer Res. 2009;69:6642–6650. doi: 10.1158/0008-5472.CAN-09-0746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shillingford JM, Miyoshi K, Robinson GW, Grimm SL, Rosen JM, Neubauer H, et al. Jak2 is an essential tyrosine kinase involved in pregnancy-mediated development of mammary secretory epithelium. Mol Endocrinol. 2002;16:563–570. doi: 10.1210/mend.16.3.0805. [DOI] [PubMed] [Google Scholar]
- Slamon DJ, Godolphin W, Jones LA, Holt JA, Wong SG, Keith DE, et al. Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science. 1989;244:707–712. doi: 10.1126/science.2470152. [DOI] [PubMed] [Google Scholar]
- Tang JZ, Zuo ZH, Kong XJ, Steiner M, Yin Z, Perry JK, et al. Signal Transducer and Activator of Transcription (STAT)-5A and STAT5B Differentially Regulate Human Mammary Carcinoma Cell Behavior. Endocrinology. 2010;151:43–55. doi: 10.1210/en.2009-0651. [DOI] [PubMed] [Google Scholar]
- Teglund S, McKay C, Schuetz E, van Deursen JM, Stravopodis D, Wang D, et al. Stat5a and Stat5b proteins have essential and nonessential, or redundant, roles in cytokine responses. Cell. 1998;93:841–850. doi: 10.1016/s0092-8674(00)81444-0. [DOI] [PubMed] [Google Scholar]
- Tornell J, Rymo L, Isaksson OG. Induction of mammary adenocarcinomas in metallothionein promoter-human growth hormone transgenic mice. Int J Cancer. 1991;49:114–117. doi: 10.1002/ijc.2910490121. [DOI] [PubMed] [Google Scholar]
- Tran TH, Utama FE, Lin J, Yang N, Sjolund AB, Ryder A, et al. Prolactin Inhibits BCL6 Expression in Breast Cancer through a Stat5a-Dependent Mechanism. Cancer Res. 2010;70:1711–1721. doi: 10.1158/0008-5472.CAN-09-2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Triplett AA, Montagna C, Wagner KU. A mammary-specific, long-range deletion on mouse chromosome 11 accelerates Brca1-associated mammary tumorigenesis. Neoplasia. 2008;10:1325–1334. doi: 10.1593/neo.08524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tworoger SS, Hankinson SE. Prolactin and breast cancer risk. Cancer Letters. 2006;243:160–169. doi: 10.1016/j.canlet.2006.01.032. [DOI] [PubMed] [Google Scholar]
- Wagner KU, Boulanger CA, Henry MD, Sgagias M, Hennighausen L, Smith GH. An adjunct mammary epithelial cell population in parous females: its role in functional adaptation and tissue renewal. Development. 2002;129:1377–1386. doi: 10.1242/dev.129.6.1377. [DOI] [PubMed] [Google Scholar]
- Wagner KU, Krempler A, Triplett AA, Qi Y, George NM, Zhu J, et al. Impaired alveologenesis and maintenance of secretory mammary epithelial cells in Jak2 conditional knockout mice. Mol Cell Biol. 2004;24:5510–5520. doi: 10.1128/MCB.24.12.5510-5520.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner KU, McAllister K, Ward T, Davis B, Wiseman R, Hennighausen L. Spatial and temporal expression of the Cre gene under the control of the MMTV-LTR in different lines of transgenic mice. Transgenic Res. 2001;10:545–553. doi: 10.1023/a:1013063514007. [DOI] [PubMed] [Google Scholar]
- Wagner KU, Wall RJ, St-Onge L, Gruss P, Wynshaw-Boris A, Garrett L, et al. Cre-mediated gene deletion in the mammary gland. Nucleic Acids Res. 1997a;25:4323–4330. doi: 10.1093/nar/25.21.4323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner KU, Young WS, III, Liu X, Ginns EI, Li M, Furth PA, et al. Oxytocin and milk removal are required for post-partum mammary-gland development. Genes Funct. 1997b;1:233–244. doi: 10.1046/j.1365-4624.1997.00024.x. [DOI] [PubMed] [Google Scholar]
- Wagner KU, Rui H. Jak2/Stat5 Signaling in Mammogenesis, Breast Cancer Initiation and Progression. Journal of Mammary Gland Biology and Neoplasia. 2008;13:93–103. doi: 10.1007/s10911-008-9062-z. [DOI] [PubMed] [Google Scholar]
- Wellings SR, Jensen HM, Marcum RG. An atlas of subgross pathology of the human breast with special reference to possible precancerous lesions. J Natl Cancer Inst. 1975;55:231–273. [PubMed] [Google Scholar]
- Wennbo H, Gebre-Medhin M, Gritli-Linde A, Ohlsson C, Isaksson OG, Tornell J. Activation of the prolactin receptor but not the growth hormone receptor is important for induction of mammary tumors in transgenic mice. J Clin Invest. 1997;100:2744–2751. doi: 10.1172/JCI119820. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(a) H&E staining (bar, 100 μm). (b, c) Immunostaining for cytokeratin (CK) 8 and 14. Slides were counterstained with DAPI (bar, 30 μm). (d -h) Immunohistochemistry for ERα and all four ErbB family members. Slides were counterstained with hematoxylin (bar, 50 μm).
Western blot analysis to determine the expression of phosphorylated and total levels of Akt1 as well as Cyclin D1 in PRL-induced mammary tumors lacking Jak2 and their wildtype controls. α-Tubulin was used as a loading control.
Control panels on the right side show serial sections that were stained without the corresponding primary antibodies. All slides were counterstained with hematoxylin (bar, 50 μm).