

Abstract
Riboswitches are noncoding RNA elements that are commonly found in the 5′-untranslated region of bacterial mRNA. Binding of a small-molecule metabolite to the riboswitch aptamer domain guides the folding of the downstream sequence into one of two mutually exclusive secondary structures that directs gene expression. The purine riboswitch family, which regulates aspects of purine biosynthesis and transport, contains three distinct classes that specifically recognize guanine/hypoxanthine, adenine, or 2′-deoxyguanosine (dG). Structural analysis of the guanine and adenine classes revealed a binding pocket that almost completely buries the nucleobase within the core of the folded RNA. Thus, it is somewhat surprising that this family of RNA elements also recognizes dG. We have used a combination of structural and biochemical techniques to understand how the guanine riboswitch could be converted into a dG binder and the structural basis for dG recognition. These studies reveal that a limited number of sequence changes to a guanine-sensing RNA are required to cause a specificity switch from guanine to 2′-deoxyguanosine, and to impart an altered structure for accommodating the additional deoxyribose sugar moiety.
Keywords: RNA–ligand interaction, riboswitch, X-ray crystallography, isothermal titration calorimetry, noncoding RNA
Introduction
Noncoding small RNAs and mRNA sequences play a central role in genetic regulation and are involved in virtually every aspect of the maintenance and transmission of genetic information.1,2 Riboswitches are a prevalent form of riboregulation in bacteria (reviewed by Winkler and Breaker3). They exert genetic control in a cis fashion by interacting with a cellular metabolite that directs the formation of one of two mutually exclusive mRNA secondary structures.4 Depending upon their placement within the mRNA, riboswitches control transcription or translation in many species of bacteria,5 and alternative splicing6 or 3′-end processing7 in plants and fungi. Currently, there are at least 20 identified distinct families of riboswitches that recognize a diverse set of metabolites, including nucleobases, sugars, vitamin cofactors, amino acids, and metal ions.8-10
The purine riboswitch family, containing the guanine (G)11 and adenine (A)12 classes, has emerged as an important model system for studying the mechanistic basis of gene expression regulation by these RNA elements (reviewed by Kim and Breaker13). Biochemical and phylogenetic analyses of this family indicated that the metabolite-binding aptamer domain folds into a three-way junction structure.11,12 Crystal and NMR structures of this domain bound to hypoxanthine or adenine revealed a compact fold in which the nucleobase is buried within the three-way junction.14-16 Discrimination between guanine and adenine is achieved through Watson–Crick pairing, with a pyrimidine at position 74 (Fig. 1a).9 Analysis of the binding pocket by NMR,17 fluorescence,18 and chemical probing techniques19 revealed a partially organized binding pocket in which a set of nucleotides in J2/3—one of the three strands of the three-way junction—acts as a flexible “lid” to completely enclose the ligand following its initial docking with pyrimidine 74.19,20 Thus, a limited RNA folding event results in complete encapsulation of the ligand and recognition of every functional group on the nucleobase by the RNA.
Fig. 1.
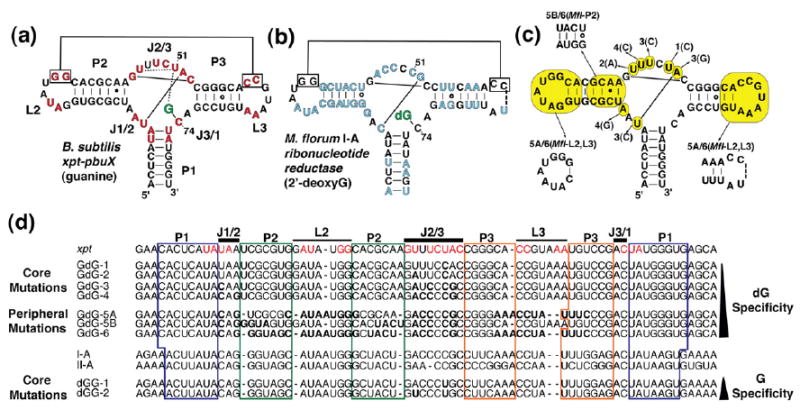
Secondary structure of riboswitch aptamer domains used in this study. (a) The secondary structure of the xpt riboswitch is shown with the ligand highlighted in green. Solid lines represent base-pairing interactions, while dotted lines denote non-Watson–Crick interactions. (b) The secondary structure of the M. florum I-A dG riboswitch aptamer domain. Nucleotide positions that diverge between the guanine and the dG riboswitch classes are shown in blue. (c) The secondary structure of the xpt–pbuX guanine riboswitch aptamer domain is shown. Nucleotides highlighted in yellow are positions that were mutated in the process of converting the native guanine aptamer into a dG-specific aptamer. The series begins with GdG-1, which contains the U51C mutation denoted by “1(C),” with a line pointing to the mutated position. Each subsequent mutation builds upon the previous mutant in the series, with each base change labeled. Seven constructs were made and are illustrated as described. (d) A sequence alignment of the B. subtilis xpt–pbuX G aptamer, the constructs used in this study, and the M. florum I-A and II-A dG aptamers is shown. In the xpt sequence, positions highlighted in red are >95% conserved among other known G/A riboswitches; these positions are also highlighted in (a). For constructs GdG-1 through GdG-6, nucleotides shown in bold are positions that have been mutated with respect to the xpt aptamer; for constructs dGG-1 and dGG-2, positions shown in bold are changed with respect to the I-A aptamer. Nucleotides that correspond to paired regions P1, P2, and P3 are boxed in blue, green, and orange, respectively. The J1/2, J2/3, J3/1, L2, and L3 regions are indicated with a black bar above the corresponding sequence positions.
In this light, the discovery of a third class of purine riboswitch in Mesoplasma florum that is responsive to 2′-deoxyguanosine (dG), which contains an additional sugar moiety, is surprising.21 Four dG riboswitches, comprising two subclasses (I-A/B and II-A/B), were demonstrated to control the expression of a ribonucleotide reductase gene (I-A) and a phosphate transporter (II-A). Although there are a number of sequence differences between the dG-binding class and the G/A binders, the secondary structure, as well as 64% of the most conserved nucleotides (defined as nucleotides whose sequence identity is 95% conserved within the guanine/adenine classes), is preserved among G/A and dG binders, strongly suggesting that these RNAs have a common three-dimensional structure (Fig. 1b and d).21 While both subclasses of dG riboswitches can bind dG with greater affinity than guanine, the I-A/B subclass displays greater selectivity (~100-fold) than the II-A/B subclass (~2-fold to 10-fold).21 The II-A/B subclass also displays weaker specificity for related compounds such as 3′-deoxyguanosine and guanosine, suggesting that these RNAs are more promiscuous in their binding than the II-A/B class.21
Some of the observed differences between the G/A and dG classes are likely not involved in generating changes in effector specificity. For instance, an invariant U22-A52 pair within the three-way junction of the guanine/adenine classes can be changed to a C-G pair, as observed in the dG class, without significant loss in ligand affinity or changes in RNA structure.22 Conversely, other differences—such as the identity changes of U47 and U51 (nucleotides essential for proper G/A nucleobase recognition)—are likely integral to the specificity switch. In part, these changes likely allow for structural differences between the classes that would accommodate the larger size of dG.
The extensive mutational analyses performed on G/A riboswitch classes22-25 provide a rationale for an exploration of sequence requirements that confer dG specificity. To achieve this, we have converted the Bacillus subtilis xpt–pbuX guanine riboswitch aptamer domain (henceforth referred to as xpt) into the dG aptamer domain from the M. florum I-A riboswitch by constructing a series of hybrid purine riboswitch aptamer domains. In these RNAs, sequence changes that are present in a naturally occurring dG aptamer have been introduced into the ligand-binding pocket and peripheral elements of the xpt aptamer. The affinity for guanine and dG was measured for each RNA in the series using isothermal titration calorimetry (ITC) to determine the basis for discrimination between these two compounds. Furthermore, their solution structure was chemically probed in the presence or in the absence of these compounds to examine whether each RNA adopts a conformation similar to those of wild-type G or dG riboswitches. This analysis shows that nucleotides responsible for inducing a specificity switch from G to dG are different from those responsible for imparting a high affinity for dG. Crystallographic analysis of one of these hybrid sequences, in complex with dG, provides a structural basis for how the RNA structure is altered to allow for specific recognition of the additional deoxyribose sugar. Together, these data suggest how the G and dG riboswitch classes could have easily diverged from a common ancestral sequence into distinct riboswitch classes via a small set of sequence changes.
Results
Mutations in a guanine sensor yield dG-binding RNAs
Comparison of the xpt G and M. florum dG riboswitches revealed more than 39 sequence differences between them scattered throughout their common secondary structure (Fig. 1a and b).21 While many of these differences lie in the paired regions and, therefore, are likely to have a minimal impact on ligand specificity and affinity, the terminal loops and three-way junction differ at many positions that are >95% conserved within the G/A classes. Of these, we reasoned that differences in nucleotides most proximal to the ligand have the greatest impact on ligand specificity.
To convert from G-specific to dG-specific binding, we changed the identity of nucleotides closest to the ligand in the three-way junction (constructs GdG-1 through GdG-4). Then, we included changes observed in the loop–loop interaction (GdG-5A), P2 helix (GdG-5B), or both (GdG-6) (Fig. 1c and d; Fig. S1). This strategy was further guided by previous mutational analyses of the xpt G riboswitch,22-24 along with a recent study showing that the loop–loop interaction is not essential for productive ligand binding.19 For example, the first altered sequence GdG-1 introduces the U51C mutation, a nucleotide directly responsible for ligand recognition in the guanine/adenine classes. This nucleotide was chosen to start the series, as its conformation in the crystal structure of the G-bound xpt RNA would have to be significantly altered to accommodate the 2′-deoxyribose moiety. Each successive change builds upon the last RNA construct (Fig. 1c and d) until the sequence is fully converted into the M. florum I-A riboswitch;21 the naming of each construct is in the numerical order of the change introduced.
A single-point mutation confers a specificity switch
Utilizing ITC, a technique that has been used extensively to analyze ligand binding to the purine riboswitch,19,20,22,26 we have measured the apparent equilibrium dissociation (Kd) constants associated with binding G or dG for each construct in the series (Table 1). Specificity is defined as the ratio of the apparent dissociation constant for G over dG (KD,G/KD,dG = Krel), and a higher selectivity for dG is observed when Krel has a value greater than 1. The xpt RNA has a Krel of 0.0004, reflecting a high specificity for G over dG, and the I-A aptamer has a Krel of 90, reflecting a modest selectivity for dG over G (Table 1). A single-base change in the xpt guanine riboswitch is already sufficient to impart a selectivity switch because the U51C mutation gives the RNA a 5-fold-higher affinity for dG over G—a value that is 12,500-fold higher than the Krel of xpt and 18-fold lower than the Krel of the I-A aptamer. Notably, this specificity swap is entirely driven by the loss of affinity for guanine (8400-fold), as both the xpt and the GdG-1 sequences have nearly identical affinities for dG. Although this point mutation results in an RNA construct that binds both ligands with relatively low affinities, a clear preference for dG is consistently observed. Thus, conversion between the two classes of purine riboswitch can be effected through a single-sequence change to the xpt G aptamer that significantly diminishes guanine affinity.
Table 1.
ITC data
| RNA | KD,dGa±errorb (μM) | KD,Ga±errorb (μM) | Krel (KD,G/KD,dG) |
|---|---|---|---|
| Wild-type G-box B. subtilis xpt–pbuX | 12±1 | 0.005±0.002 | 0.0004±0.0001 |
| xpt core mutations | |||
| GdG-1 | 9.0±0.1 | 42±9 | 5±1 |
| GdG-2 | 5.0±0.8 | 60±20 | 12±4 |
| GdG-3 | 39±1 | 1200±700 | 30±20 |
| GdG-4 | 54±4 | NDc | N/A |
| xpt peripheral mutations | |||
| GdG-5A | 87±4 | ND | N/A |
| GdG-5B | 0.20±0.02 | 40±6 | 200±20 |
| GdG-6 | 0.068±0.003 | 4.8±0.4 | 71±7 |
| Wild-type dG-box M. florum I-A | 0.06±0.01 | 5.3±0.5 | 90±20 |
| I-A core mutations | |||
| dGG-1 | 5.0±0.2 | 1.9±0.1 | 0.41±0.02 |
| dGG-2 | 4.0±0.5 | 0.43±0.06 | 0.11±0.01 |
Apparent dissociation constant.
Absolute error (calculated using the propagation-of-error method).
ND, no detectable binding.
As a further measure of binding specificity for dG versus G, the structures of several RNAs were chemically probed using N-methylisatoic anhydride (NMIA) in a method termed SHAPE chemistry.27-29 This compound reacts with the 2′-hydroxyl group of ribose sugars that adopt the C2′-endo conformation,30 which generally reflects the local dynamics of the backbone.29 The adduct created by reaction with NMIA is subsequently detected as a reverse transcriptase stop during primer extension, as visualized on a sequencing gel (Fig. 2). For each sequence examined, the RNA was probed at 25 °C with NMIA in the absence of ligand, 10 μM guanine, 100 μM dG, or 100 μM 2′-deoxyadenosine (as negative control). Additional reactions were performed for each construct in the absence of NMIA to control for modification-independent stops during reverse transcription. A complete unaltered gel image is shown in Fig. S2 (GdG-1 through GdG-3 and GdG-6 are shown in Fig. S1; data are not shown for GdG-4). For clarity, select regions corresponding to J1/2 and J2/3 that directly interact with the ligand are shown in Fig. 2.
Fig. 2.

SHAPE chemistry reveals ligand specificity and suggests an altered conformation in the binding pocket. RNA was incubated in the presence (+) or in the absence (−) of 130 mM NMIA, and in the presence or in the absence (−) of ligand [10 μM guanine (G), 100 μM dG (dG), or 100 μM 2′-deoxyadenosine (dA)] prior to the reverse transcription reaction. Bands corresponding to the J1/2 and J2/3 regions of the binding pocket are labeled to the right of the image. The sequencing reactions are labeled “A” or “C” for adenosine and cytosine sequencing, respectively. Note that the nucleotide position modified by NMIA is offset by one position in the 5′ direction with respect to the sequencing lanes. (a) The B. subtilis xpt–pbuX guanine riboswitch aptamer domain and two mutation variants (GdG-1 and GdG-3) are shown. (b) The M. florum dG riboswitch aptamer domain I-A is shown.
The reactivity patterns for the xpt and GdG-1 RNAs are consistent with a specificity switch by the U51C sequence change. In the absence of a ligand, these two RNAs have identical patterns of chemical reactivity, indicating that the point mutation neither alters the secondary structure or the L2–L3 tertiary interaction nor causes the RNA to locally misfold in the three-way junction (Fig. 2a; Fig. S2). Consistent with previous studies,19 addition of 10 μM guanine induces a distinct protection pattern in J1/2 and J2/3 in the xpt guanine riboswitch along with a single enhancement in the reactivity of U48, which is flipped out into solvent in the bound form. No guanine-dependent protection pattern is observed for GdG-1, consistent with a KD, G of 5.0 μM observed with ITC. On the other hand, both RNAs are able to bind dG, as evidenced by a similar protection pattern in J1/2 and J2/3 when modified with NMIA in the presence of 100 μM dG. Notably, in comparison to J1/2, the J2/3 region of both RNAs remains moderately reactive in the presence of dG, suggesting that this loop does not adopt the same conformation as observed in the xpt-guanine/hypoxanthine crystal structures,14,16 likely reflecting its accommodation of the 2′-deoxyribose ring. Both RNAs show no change in reactivity upon addition of 100 μM 2′-deoxyadenosine, indicating that the G- and dG-dependent changes reflect specific binding by these compounds.
To determine whether this specificity switch is an evolutionary two-way street, a limited set of sequence changes was introduced into the binding pocket of the M. florum I-A dG aptamer. The C51U change in the wild-type I-A sequence (construct dGG-1; Fig. 1d) results in a Krel of 0.41 using ITC (Table 1). Thus, a complementary point mutation at position 51 is sufficient to confer a modest selectivity switch from dG to G in the I-A aptamer. Like the GdG-1 sequence, the selectivity switch in dGG-1 is almost entirely driven by a substantial drop in affinity for the wild-type ligand (G and dG, respectively). However, a second change in this sequence, A47U, is required to fully restore the direct hydrogen-bonding pattern to guanine observed in the xpt sequence.14-16 We constructed this RNA (construct dGG-2; Fig. 1d) and determined a Krel of 0.11, which is almost entirely driven by a fourfold increase in G binding affinity (Table 1). Thus, the specificity switch between G and dG in the purine riboswitch can be effected solely by a change in pyrimidine identity at position 51 in either the xpt G riboswitch or the M. florum 1-A dG riboswitch.
Further changes in the ligand-binding pocket do not improve dG affinity
A point mutation at position 51 in either the xpt G aptamer or the I-A dG aptamer is sufficient to impart a ligand specificity switch. However, just as the C51U mutation in the I-A aptamer does not produce the ligand binding characteristics observed for the xpt G aptamer, the U51C mutation does not account for the high affinity for dG and the specificity for dG versus G that are observed in two out of the four naturally occurring M. florum dG riboswitches (variants I-A/B).21 Thus, other differences in the ligand-binding pocket between the G/A and dG riboswitches are responsible for achieving the higher specificity and affinity observed for the wild-type I-A riboswitch. Base changes were introduced into GdG-1 in order to convert the ligand-binding pocket of the guanine riboswitch class into that of the M. florum dG riboswitch (GdG-2, GdG-3, and GdG-4; Fig. 1a and d; Fig. S1). Examination of these constructs by ITC and SHAPE chemistry revealed that the wild-type M. florum dG binding characteristics are not restored, even when the entire sequence of the ligand-binding pocket is converted (GdG-4) (Table 1; SHAPE data not shown). In fact, as an increasing number of changes are introduced, the binding affinity for both G and dG sharply drops, while the specificity for dG only moderately increases (Table 1). This behavior is somewhat surprising in that several changes were introduced into the RNA where they had a negligible effect on ligand-binding affinity and on the structure of the xpt riboswitch.22 Therefore, the added binding affinity of the M. florum I-A aptamer must also require differences in the peripheral elements of the riboswitch.
Increased ligand binding affinity stems from changes observed in peripheral elements
The M. florum I-A riboswitch differs from the B. subtilis xpt riboswitch with regard to the length and sequence composition of the P2 and P3 helices along with the terminal loops21 (Fig. 1a, b, and d). The lengths of the P2 and P3 helices are 7 and 6 bp, respectively, in xpt, whereas this pattern is reversed (P2, 6 bp; P3, 7 bp) in the I-A aptamer. This change in helical length may impact the association of the L2 and L3 terminal loops, which are critical for high-affinity binding of the ligand to the G/A classes.11,14,31 Moreover, the sequence composition of the P2 and P3 helices has only a 31% sequence homology in these regions between the xpt and I-A aptamers.21 Finally, the terminal loops differ such that highly conserved residues in L2 and L3 that are critical for establishing the loop–loop interaction in the G/A classes are altered in all four of the M. florum dG riboswitches21 (Fig. 1a, b, and d). Given that the NMIA reactivity pattern for these two loops is extremely different between xpt and M. florum I-A (Fig. S2), an alternative L2–L3 interaction is likely. In particular, L2 from M. florum I-A is highly reactive to NMIA, suggesting a weakened interaction with L3. Taken together, these changes have the potential to greatly influence the ligand binding affinity.
We constructed two new RNAs built upon GdG-4 in which changes were introduced to the terminal loops and helix lengths (GdG-5A) or only to the base of the P2 helix (GdG-5B). An additional RNA, in which mutations in both GdG-5A and GdG-5B were combined to yield GdG-6, was constructed (Fig. 1c and d; Fig. S1). GdG-5A has ligand binding characteristics very similar to those of GdG-4, as it shows a modest drop (1.6-fold) in dG binding affinity and no detectable improvement in G binding affinity (Table 1). Therefore, changing the lengths of the P2 and P3 helices, as well as replacing the L2 and L3 loops of the GdG-4 RNA with those found in the I-A aptamer, does not account for the heightened improvement in dG binding affinity observed in the I-A aptamer. Conversely, GdG-5B shows a dramatic improvement in ligand binding characteristics in comparison to all of the previously described constructs in our series, as it binds dG with a Kd of ~200 nM (compared to ~60 nM for the M. florum I-A riboswitch) and has a ~200-fold-greater specificity for dG over guanine (versus a Krel of 90 observed in the M. florum I-A aptamer) (Table 1). Thus, the sequence composition of the first four bases in the P2 helix has a great affect on ligand binding affinity.
Finally, GdG-6 shows that all ligand binding characteristics are restored to those observed in the I-A aptamer when the helix length, the sequence composition of the terminal loops, and the base of the P2 helix are changed to represent M. florum I-A. In this construct, we observe a ligand binding affinity of ~68 nM and ~71-fold-higher specificity for dG over guanine—values that are almost identical with those of the M. florum I-A riboswitch (Table 1). Notably, GdG-6 shows an almost 10-fold improvement in G affinity, leading to a slightly lowered specificity as compared to GdG-5B. Thus, the precise helical lengths and terminal loop sequences confer a modest increase in ligand binding affinity for dG only in the context of the M. florum I-A P2 helix, and these peripheral elements act together to achieve the wild-type I-A binding characteristics. Taken together, our data show that alterations in P2 proximal to the three-way junction appear to exert the greatest effect on dG binding affinity, while the alternative loop–loop interaction only provides a small improvement. Thus, while the U51C change in the three-way junction is sufficient to confer specificity to dG, further changes in P2 are needed to enhance affinity and specificity.
Structural characterization of the purine riboswitch bound to dG reveals a conformational change in J2/3
To further our understanding of how dG is accommodated in the binding pocket and how specific sequence alterations in the xpt guanine riboswitch facilitate specific recognition of dG, we sought to crystallize this ligand in complex with the xpt aptamer or one of the GdG RNAs. The rationale for using these RNAs is that they might still be able to crystallize under the same conditions as the original xpt guanine riboswitch by preserving the identity of nucleotides that make key lattice contacts, particularly around the L2–L3 interaction.14 It is important to note that these RNAs do not represent a definitive model of the ligand-bound wild-type dG riboswitch. Principally, this is because the peripheral elements of natural G and dG aptamers are markedly different, and our binding data clearly indicate that they influence the RNA’s ligand binding properties. Thus, failing to obtain a structural model depicting either the altered loop–loop interaction present in naturally occurring dG aptamers or the altered P2 helix sequence that clearly improves affinity for dG. Instead, a structure of any one of these RNAs bound to dG informs us about how the purine riboswitch accommodates the 2′-deoxyribose moiety and how the dG riboswitch discriminates between dG and G.
Screening each of these RNAs in the presence of dG revealed that GdG-1, GdG-3, and GdG-4 crystallized under the previously established conditions.14 The GdG-3 complex yielded crystals that diffracted X-rays to 1.85 Å, but the space group changed from C2 to P1, with two molecules per asymmetric unit. Using the xpt RNA bound to hypoxanthine (Protein Data Bank ID 1U8D) as a molecular replacement search model, we were able to calculate an interpretable electron density map and to independently build the two protomers, each containing a bound dG molecule in the asymmetric unit. The data collection and refinement statistics for the final model are presented in Table 2.
Table 2.
Crystallographic statistics
| Data collection | |
| Space group | P1 |
| a, b, c (Å) | 35.14, 41.83, 64.81 |
| α, β, γ (°) | 86.55, 81.16, 89.64 |
| Wavelength (Å) | 1.5418 |
| Resolutiona Resolutiona (Å) | 20–1.85 (1.92–1.85) |
| % Completeness | 94.0 (91.4) |
| Average redundancy | 3.56 (3.57) |
| ⟨I⟩/⟨σ(I)⟩ | 8.4 (3.4) |
| Rmergeb (%) | 7.5 (29.7) |
| Refinement | |
| Resolution (Å) | 20–1.85 (1.92–1.85) |
| Reflections [n (%)] | |
| Total | 29,005 (93.5)c |
| Working | 26,111 (84.1) |
| Test set | 2894 (9.3) |
| Rxtald | 20.0 (26.8) |
| Rfree | 25.9 (35.2) |
| r.m.s.d. bonds (Å) | 0.0209 |
| r.m.s.d. angles (°) | 2.047 |
| Luzzati coordinate errore (Å) | 0.31 |
| Sigma-a coordinate errore (Å) | 0.23 |
| Average B-factor (Å2) | 34.41 |
Numbers in parenthesis correspond to the highest-resolution shell.
Rmerge = Σ | I−⟨I⟩ |/ΣI, where I is the observed intensity, and ⟨I⟩ is the average intensity of multiple measurements of symmetry-related reflections.
Percentage of the theoretical maximum number of reflections.
Rxtal = Σ | | Fo | − | Fχ | |/Σ | Fo |, where Rxtal is from the working set and Rfree is from the test set.
Cross-validated.
While the global fold of the complex is highly similar to that of the xpt–hypoxanthine complex (r.m.s.d.=0.94 Å),32 there are significant differences within the three-way junction (Fig. 3; Fig. S3). The most important difference is the positioning of C51, which is shifted relative to the U51 of the xpt–guanine complex (Fig. 4a). C51 interacts with the minor groove face of dG via hydrogen bonds between C51(N4) and dG(N3), and between C51(N3) and dG(N2). Because the wild-type xpt riboswitch can also bind dG, albeit with highly reduced affinity, we postulate that U51 can bind dG in a tautomeric (enol) form, such that its O4 and N3 atoms can provide the necessary hydrogen-bonding pattern for productive binding.
Fig. 3.
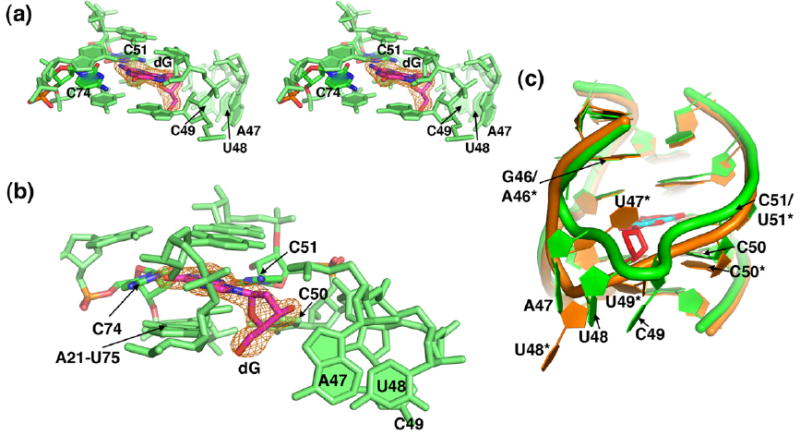
X-ray crystal structure of the binding pocket of GdG-3 bound to dG. In (a)–(c), the GdG-3 aptamer is shown in green; in (c), the xpt riboswitch is shown in orange. Molecular model images of GdG-3 shown in (a)–(c) are from protomer A of the asymmetric unit. In (a) and (b), images are taken from a simulated annealing omit map contoured to 1σ and calculated with the dG ligand omitted. The dG ligand is shown in magenta; oxygen atoms from dG, C74 (forming a Watson–Crick pair with dG), and C51 (contacting the minor groove face of dG) are shown in blue; and nitrogen atoms are shown in red. (a) A stereo view of the ligand-binding pocket. (b) The image shown in (a) is rotated ~90° to the left. (c) An overlay of GdG-3 bound to dG (cyan), with the xpt aptamer bound to hypoxanthine (red), is shown. All images were prepared in PyMOL.33
Fig. 4.

The 2′-deoxyribose moiety participates in hydrogen-bonding interactions with the P1 helix. In all figures, the left panel shows a model of xpt–pbuX guanine riboswitch bound to guanine,16 and the right panel shows a model of dG bound to the GdG-3 construct. The ligand is shown in red, and nucleotides from the J1/2, J2/3, J3/1, and P1 regions are shown in cyan, green, yellow, and black, respectively. The nitrogen atoms in all nucleotides are shown in blue, while the oxygen atoms are shown in salmon. Atoms within hydrogen-bonding distance are denoted with a dashed black line. In (a), the ligand-binding pocket is shown as viewed from above the RNA. In (b) and (c), the ligand-binding pocket is shown as viewed from below the RNA, and only the 2′-deoxyribose moiety of dG is highlighted in thick red lines. (a) In order to accommodate the 2′-deoxyribose moiety, C51 is shifted toward the exocyclic amine of the purine ring, and A47 is rotated away from the binding pocket as compared to the positions of U51 and U47 in the guanine-bound structure. (b) The 2′-deoxyribose moiety participates in stabilizing the C50·(U75-A21) base triple immediately below the binding pocket. (c) The 5′-OH group of dG participates in stabilizing the A76-U20 pair of the P1 helix and replaces pyrimidine 49, which is rotated out of the binding pocket as compared to its position in the guanine-bound aptamer. All images were prepared in PyMOL.33
To accommodate the sugar moiety of dG, residues A47, U48, and C49 in J2/3 are rotated out of the binding pocket relative to what is observed in the xpt structure (Figs. 3b and c and 4b and c; Fig. S4).14,16 The positions of these three nucleotides are different in each of the two protomers. In molecule A, the electron density around these nucleotides is weakly defined, and the bases make a series of hydrogen-bonding contacts to a neighboring molecule in the lattice (Fig. S4a). The electron density around these residues in protomer B is absent, indicating that these nucleotides are conformationally disordered (Fig. S4b). The apparent conformational flexibility of nucleotides 47–49 in J2/3 in the dG bound state is consistent with the NMIA chemical probing pattern (Fig. 2a).
This structure reveals that changes in the positions of nucleotides 47–49 and 51 in J2/3 create a space within the three-way junction to accommodate the 2′-deoxyribose moiety (Figs. 3b and 4b and c). The result of this spatial reorganization is that part of the extensive hydrogen-bonding network between J2/3 and P1 observed surrounding the ligand in the G/A classes is lost; only the base triple between C50 and the A21-U75 base pair at the top of the P1 helix is preserved (Figs. 3b and 4b and c). Part of this network is restored through interactions between the RNA and the 2′-deoxyribose sugar. Specifically, to replace the xpt U49·(U20-A76) base triple, the 5′-hydroxyl group of the 2′-deoxyribose sugar sits within hydrogen-bonding distance of the N3 from A21 and A76 (2.7 and 3.0 Å, respectively) (Fig. 4c). A further interaction is made between 2′-deoxyribose O4′ and C50(N3), but the 3′-hydroxyl group of the sugar appears not to be directly contacted (Fig. 4b).
Discussion
The biochemical and structural studies presented reveal that the purine riboswitch requires a change in the identity of the nucleotide at position 51 from uridine to cytidine (U51C) to cause a selectivity switch from guanine to dG. A similar but weaker switch is observed for the C51U mutation in the M. florum dG riboswitch. The single-point change in the xpt G riboswitch, however, does not confer the observed specificity for dG in the M. florum I-A/B sequences; rather, this reflects the weaker specificities of the II-A/B class.21 When other sequence differences within the three-way junction between the G/A and dG classes are introduced into the GdG series, the RNA does not converge upon the wild-type M. florum I-A behavior. While there is a moderate increase in selectivity, the affinity for both guanine and 2′-deoxyguanosine decreases significantly. Therefore, the three-way junction is the primary—but not the sole—determinant for ligand binding affinity or specificity.
The peripheral region of this RNA—specifically the identities of pairs in P2 proximal to the three-way junction—plays a key role in influencing affinity and specificity. The likely mechanism for the effect of the P2 helix is the increased stability of pairing interactions in this part of the RNA. We observed that in previous NMIA probing of the xpt riboswitch, bases 25–27 and 45 in the unbound xpt riboswitch aptamer domain (also seen in GdG-1 through GdG-4) show enhanced NMIA modification in comparison to the other paired regions.19 In contrast, these nucleotides are not reactive to NMIA in either the M. florum I-A RNA or GdG-6.
We hypothesize that alteration of the first two pairs of the P2 helix to a more stable pairing arrangement serves to further preorganize the unbound three-way junction such that it pays a lower entropic penalty for ligand binding, translating into higher affinity. In fact, we found that in the context of the GdG-5A construct, dG binding affinity increased by approximately 45-fold when the C27-A44 mismatch was stabilized with an A44G point mutation (data not shown). This effect was also clearly observed in an NMR study of the unbound xpt RNA, in which the A44G point mutation was necessary to sufficiently stabilize the three-way junction for subsequent structural analysis.17 Conversely, the peripheral loop–loop interaction has only a moderate influence on improving dG binding with respect to the base of the P2 helix. This may result from the loop–loop interaction forming an alternative conformation with respect to that of the xpt RNA, as demonstrated by chemical probing with NMIA (Fig. S2). This provides further evidence that the loop–loop interaction plays a role in affinity but is not absolutely required for a productive ligand–RNA interaction. Given that the L2–L3 interaction provides a significant amount of binding energy in the xpt–ligand interaction,19 it was unexpected to find that the L2–L3 tertiary contact is arranged disparately and may be poorly organized in the dG riboswitch.
From the GdG-3 structure, it is not clear how the M. florum I-A and I-B sequences discriminate between guanosine and dG. Modeling a ribose sugar with a C3′-endo sugar pucker does not appear to cause a steric clash between the 2′-hydroxyl group and any part of the RNA (data not shown). However, the GdG-3 RNA does appear to be slightly more specific to dG with respect to guanosine, as observed by ITC data revealing a threefold higher binding affinity for dG versus guanosine (KD,rG = 121±8 μM). Thus, the GdG-3 structure may be more representative of the M. florum II-A and II-B aptamers that exhibit reduced discrimination between these two compounds, compared to the I-A and I-B aptamers.21
The role of a conformationally flexible J2/3
Previous studies have shown that the J2/3 linker acts as a flexible lid that encapsulates the ligand only after it has initially docked with the three-way junction.19,20 It is possible that nature exploited this property to evolve a dG aptamer in which the highly dynamic nucleotides in the J2/3 region of the RNA-bound form adopt a conformation different from that of the G/A classes in order to accommodate the bulky 2′-deoxyribose moiety. Although our structural model of GdG-3 bound to dG does not fully represent a wild-type M. florum dG aptamer due to differences in peripheral elements, such as the sequence composition of P2, it may represent an evolutionary intermediate between the G/A classes and the dG class. The inherent flexibility of J2/3 reported here may give the purine riboswitch family the flexibility to bind diverse compounds through minor sequence changes using the basic three-way junction structural scaffold.
Flexible regions in protein and RNA receptors are widely recognized as important functional elements because of their essential role in the induced-fit mechanism of ligand recognition.34,35 Moreover, flexible regions of proteins have become increasingly targeted during structure-based drug design projects. For instance, the specificity pocket of aldose reductase—an enzyme central to late-onset diabetic disorders—houses several amino acid residues with flexible side chains capable of accommodating inhibitory compounds with a wide array of shapes and sizes.36,37 Analogously, the flexibility of J2/3 could be exploited in the search for antimicrobial compounds that target the purine riboswitch,38 as this region can clearly change in conformation when challenged with different compounds.
Implications for the evolution of novel RNAs
The similar secondary structures of the guanine, adenine, and dG riboswitch classes strongly suggest that these RNAs are evolutionarily related paralogs. Only one base change (C74U) is required to swap specificity from G to A.11,12,20 In this study, we have analogously shown that the pyrimidine at position 51 is the principal determinant of specificity for G versus dG. Therefore, from an evolutionary standpoint, there is a small barrier between the three classes in the purine riboswitch family. Thus, we hypothesize that a small number of base changes occurred in an ancestral RNA sequence that resulted in the emergence of the guanine and dG riboswitch classes. Specifically, the identity of position 51 (U or C) is the minimum requirement to impart specificity for G or dG. In the context of C51, a small number of further changes at the base of P2 and nucleotides involved in the loop–loop interaction were required to optimize dG binding affinity. Other observed changes likely accumulated through neutral genetic drift or provided only a modest functional gain.
This model is analogous to the proposed evolutionary ties between other RNAs. For example, the GIR1 branching ribozyme39 may have derived from the group I splicing intron through a small number of base changes that slightly altered the core structure, leading to strand exchange and formation of a double pseudoknot.40 In fact, like the ligand specificity of the purine riboswitch, the branching activity of GIR1 can be changed into splicing activity upon mutation of only one base at the catalytic center.40 Another more dramatic example was illustrated by the discovery of a sequence that can adopt two dramatically different folds and functions: that of the class I ligase and that of the HδV ribozymes.41 This RNA exists at a position in sequence space such that mutating only two of four key residues is sufficient to completely commit the ribozyme to function either as a ligase or as an endonuclease. It is very possible that we will see other instances of riboswitch classes evolving new classes of specificity via small changes in their core. Thus, like proteins, a limited set of structural scaffolds can be used to generate RNAs of diverse functions.
Materials and Methods
RNA synthesis and purification
The RNA constructs used in this study were synthesized by in vitro transcription with T7 RNA polymerase and purified using previously described methods.20,42 Briefly, RNAs were transcribed in reactions containing 40 mM Tris–HCl (pH 8.0), 10 mM DTT, 0.01% Triton X-100, 2 mM spermidine–HCl, 4 mM each NTP, 36 mM MgCl2, 0.25 mg/mL T7 RNA polymerase, 1.0 mL of 0.5 μM double-stranded DNA template, and 1 U/mL inorganic pyrophosphatase. The product RNA was ethanol-precipitated and purified on a 12% denaturing polyacrylamide gel. The RNA was eluted from the gel using the crush-and-soak method into 10 mM K+-Hepes (pH 7.5) buffer. Each sample was concentrated with a 10,000-molecular weight cutoff centrifugal filter device (Ultra-15; Amicon) to a final volume of approximately 500 μL. The concentration was calculated from the absorbance at 260 nm and the calculated molar extinction coefficient.
Isothermal titration calorimetry
The affinity of each RNA for guanine and dG was measured using previously described methods.20,22,42 RNA samples were prepared for ITC by dialyzing an appropriate amount of RNA in 20 mM MgCl2, 100 mM KCl, and 50 mM K+-Hepes (pH7.5) for approximately 16 h at 4 °C. Following dialysis, dG (Sigma-Aldrich) or guanine (Fluka) was dissolved in an aliquot of dialysis buffer. All samples were then degassed at 25 °C for 10 min and loaded into the calorimeter syringe or sample cell. Experiments measuring the interaction between RNA and dG were set up with ~50 μM RNA in the sample cell and with ~500 μM dG in the syringe to yield a c-value ranging from 0.5 to 140.43-45 Similarly, experiments measuring the RNA–guanine interaction were set with 5–15 μM guanine in the sample cell (due to the poor solubility of guanine) and 100 μM RNA in the syringe; the c-value for these experiments ranged from 0.07 to 1100. Each experiment was performed at a temperature of 30 °C, a reference power of 5 μcal/s, and a titration of either thirteen 20-μL injections (for titrations of dG into RNA) or ten 25-μL injections (for titrations of RNA into guanine) at a rate of 0.5 μL/s. Data were fitted with Origin ITC software (Microcal Software, Inc.) to a single-site binding model to determine the apparent association constant Ka.
Chemical probing with NMIA
RNA samples were prepared as described previously with the addition of 5′ and 3′ structure cassettes flanking the RNA sequence, and the NMIA modification reaction was carried out following established protocols.27 RNA (2.0 pmol) in 12 μL of 0.5× TE (10 mM Tris-HCl, 0.1 mM EDTA, pH 8.0) buffer was incubated for 2 min at 90 °C, immediately placed on ice, and incubated for 5 min. Following RNA refolding, 6.0 μL of 3.3× folding buffer [333 mM K+-Hepes (pH 8.0), 20 mM MgCl2, and 333 mM NaCl] or 6.0 μL of 3.3× folding buffer containing 10 μM guanine, 333 μM dG, or 333 μM 2′-deoxyadenosine was added to each sample, and the RNA was incubated at room temperature for approximately 10 min. After this time, each reaction was split into two 9 μL aliquots in thin-walled PCR tubes and incubated at 30 °C for 2.0 min. One microliter of 130 mM NMIA in DMSO or neat DMSO was then added to each aliquot, and the samples were incubated for five NMIA half-lives. Because the xpt RNA has an extraordinarily high binding affinity for guanine with respect to the other RNA constructs examined, it was necessary to account for the possibility that the dG sample is contaminated with trace amounts of guanine. Therefore, prior to folding the RNA, 2.0 pmol of the xpt RNA harboring the 5′ and 3′ structure cassettes was supplemented with 60.0 pmol of xpt RNA lacking the structure cassettes.
Immediately following modification, the samples were subjected to a reverse transcription reaction. Three microliters of 32P 5′-end-labeled DNA oligomer was added to each modified RNA sample. Reaction mixtures were incubated at 65 °C for 5 min, 35 °C for 5 min, and, finally, 52 °C for 1 min to prepare for reverse transcription. Each sample was mixed with 6.0 μL of enzyme reaction buffer containing 250 mM KCl, 167 mM Tris–HCl (pH 8.3), 16.7 mM DTT, 1.67 mM each dNTP, and 0.33 U of Superscript III reverse transcriptase (Invitrogen, Inc.). The reverse transcription reaction was incubated at 52 °C for 5 min and stopped by the addition of 1.0 μL of 4.0 M NaOH, followed by a 5-min incubation at 90 °C. The samples were then quenched with 29.0 μL of acid stop mix and incubated at 90 °C for 5 min. The reactions were resolved on a 12% denaturing polyacrylamide gel, electro-phoresed at 55 W for 4 h, and imaged using a Typhoon PhosphorImager (Molecular Dynamics).
X-ray crystallography
GdG RNA constructs were prepared as described previously and exchanged into 10 mM K+-Hepes (pH 7.5) buffer containing 2.0 mM dG. Crystallization trials were set up using the hanging-drop vapor-diffusion method in which 2.0 μL of the complex was mixed with 2.0 μL of mother liquor and incubated at 25 °C. The GdG-3 complex crystallized with 10 mM K+-Hepes (pH7.5), 11.9 mM cobalt (III)hexamine, 22% polyethylene glycol 2000, and 660 mM ammonium acetate. For cryoprotection, 30% 2-methyl-2,4-pentanediol was added to the mother liquor, the crystal soaked for 5 min, and flash-frozen in liquid nitrogen. Diffraction data were collected in-house up to 1.85 Å resolution using an R-AXIS IV++ detector on an RU-200/confocal blue optic source (Rigaku MSC). Data collection statistics are shown in Table 1.
Data were indexed, integrated, and scaled using D*TREK46 according to space group P1 and a resolution limit of 1.85 Å. An electron density map was calculated with the molecular replacement method in CNS47 using the guanine riboswitch RNA (Protein Data Bank ID 1U8D) as search model, in which hypoxanthine and solvent molecules were removed. To achieve a clearly interpretable 2Fo−Fc map, two protomers had to be placed in the unit cell, at which time clear density was observed for the majority of the RNA for each molecule. After several rounds of simulated annealing from 5000 K in 25-K steps to minimize model bias, a number of nucleotides in J2/3 (46–51) were removed from the model because no clear electron density was observed in that region of the 2Fo−Fc map. These nucleotides, along with the ligand and solvent molecules, were rebuilt for each of the two protomers using CNS and PyMOL graphical interface for manual adjustments. The model was further refined by combined energy minimization/simulated annealing and B-factor refinement in CNS until the Rfree no longer decreased. The final model has a working crystallographic R-factor of 20.0% and an Rfree of 25.9% (refinement statistics are shown in Table 2).
Accession codes
The coordinates and structure factors have been deposited in the RCSB database under the identifier 3DS7.
Supplementary Material
Acknowledgments
We thank members of the Batey Laboratory, Jeffrey Kieft, and Jennifer Pfingsten for careful reading of, and helpful discussion on, the contents of this manuscript. This work was supported by a grant from the National Institutes of Health (GM 073850; to R.T.B.) and a National Institutes of Health Predoctoral Training Grant (T32 GM-065103; to A.L.E.).
Abbreviations used
- A
adenine
- dG
2′-deoxyguanosine
- G
guanine
- ITC
isothermal titration calorimetry
- NMIA
N-methylisatoic anhydride
Footnotes
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j. jmb.2008.10.074
References
- 1.Gottesman S. Micros for microbes: non-coding regulatory RNAs in bacteria. Trends Genet. 2005;21:399–404. doi: 10.1016/j.tig.2005.05.008. [DOI] [PubMed] [Google Scholar]
- 2.Mattick JS, Makunin IV. Non-coding RNA. Hum Mol Genet. 2006;15(Spec. No. 1):R17–R29. doi: 10.1093/hmg/ddl046. [DOI] [PubMed] [Google Scholar]
- 3.Winkler WC, Breaker RR. Regulation of bacterial gene expression by riboswitches. Annu Rev Microbiol. 2005;59:487–517. doi: 10.1146/annurev.micro.59.030804.121336. [DOI] [PubMed] [Google Scholar]
- 4.Irnov I, Kertsburg A, Winkler WC. Genetic control by cis-acting regulatory RNAs in Bacillus subtilis: general principles and prospects for discovery. Cold Spring Harbor Symp Quant Biol. 2006;71:239–249. doi: 10.1101/sqb.2006.71.021. [DOI] [PubMed] [Google Scholar]
- 5.Nudler E, Mironov AS. The riboswitch control of bacterial metabolism. Trends Biochem Sci. 2004;29:11–17. doi: 10.1016/j.tibs.2003.11.004. [DOI] [PubMed] [Google Scholar]
- 6.Cheah MT, Wachter A, Sudarsan N, Breaker RR. Control of alternative RNA splicing and gene expression by eukaryotic riboswitches. Nature. 2007;447:497–500. doi: 10.1038/nature05769. [DOI] [PubMed] [Google Scholar]
- 7.Wachter A, Tunc-Ozdemir M, Grove BC, Green PJ, Shintani DK, Breaker RR. Riboswitch control of gene expression in plants by splicing and alternative 3′ end processing of mRNAs. Plant Cell. 2007;19:3437–3450. doi: 10.1105/tpc.107.053645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Weinberg Z, Barrick JE, Yao Z, Roth A, Kim JN, Gore J, et al. Identification of 22 candidate structured RNAs in bacteria using the CMfinder comparative genomics pipeline. Nucleic Acids Res. 2007;35:4809–4819. doi: 10.1093/nar/gkm487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barrick JE, Breaker RR. The distributions, mechanisms, and structures of metabolite-binding riboswitches. Genome Biol. 2007;8:R239. doi: 10.1186/gb-2007-8-11-r239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barrick JE, Corbino KA, Winkler WC, Nahvi A, Mandal M, Collins J, et al. New RNA motifs suggest an expanded scope for riboswitches in bacterial genetic control. Proc Natl Acad Sci USA. 2004;101:6421–6426. doi: 10.1073/pnas.0308014101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mandal M, Boese B, Barrick JE, Winkler WC, Breaker RR. Riboswitches control fundamental biochemical pathways in Bacillus subtilis and other bacteria. Cell. 2003;113:577–586. doi: 10.1016/s0092-8674(03)00391-x. [DOI] [PubMed] [Google Scholar]
- 12.Mandal M, Breaker RR. Adenine riboswitches and gene activation by disruption of a transcription terminator. Nat Struct Mol Biol. 2004;11:29–35. doi: 10.1038/nsmb710. [DOI] [PubMed] [Google Scholar]
- 13.Kim JN, Breaker RR. Purine sensing by riboswitches. Biol Cell. 2008;100:1–11. doi: 10.1042/BC20070088. [DOI] [PubMed] [Google Scholar]
- 14.Batey RT, Gilbert SD, Montange RK. Structure of a natural guanine-responsive riboswitch complexed with the metabolite hypoxanthine. Nature. 2004;432:411–415. doi: 10.1038/nature03037. [DOI] [PubMed] [Google Scholar]
- 15.Noeske J, Richter C, Grundl MA, Nasiri HR, Schwalbe H, Wohnert J. An intermolecular base triple as the basis of ligand specificity and affinity in the guanine- and adenine-sensing riboswitch RNAs. Proc Natl Acad Sci USA. 2005;102:1372–1377. doi: 10.1073/pnas.0406347102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Serganov A, Yuan YR, Pikovskaya O, Polonskaia A, Malinina L, Phan AT, et al. Structural basis for discriminative regulation of gene expression by adenine- and guanine-sensing mRNAs. Chem Biol. 2004;11:1729–1741. doi: 10.1016/j.chembiol.2004.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ottink OM, Rampersad SM, Tessari M, Zaman GJ, Heus HA, Wijmenga SS. Ligand-induced folding of the guanine-sensing riboswitch is controlled by a combined predetermined induced fit mechanism. RNA. 2007;13:2202–2212. doi: 10.1261/rna.635307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rieder R, Lang K, Graber D, Micura R. Ligand-induced folding of the adenosine deaminase A-riboswitch and implications on riboswitch translational control. ChemBioChem. 2007;8:896–902. doi: 10.1002/cbic.200700057. [DOI] [PubMed] [Google Scholar]
- 19.Stoddard CD, Gilbert SD, Batey RT. Ligand-dependent folding of the three-way junction in the purine riboswitch. RNA. 2008;14:675–684. doi: 10.1261/rna.736908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gilbert SD, Stoddard CD, Wise SJ, Batey RT. Thermodynamic and kinetic characterization of ligand binding to the purine riboswitch aptamer domain. J Mol Biol. 2006;359:754–768. doi: 10.1016/j.jmb.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 21.Kim JN, Roth A, Breaker RR. Guanine riboswitch variants from Mesoplasma florum selectively recognize 2′-deoxyguanosine. Proc Natl Acad Sci USA. 2007;104:16092–16097. doi: 10.1073/pnas.0705884104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gilbert SD, Love CE, Edwards AL, Batey RT. Mutational analysis of the purine riboswitch aptamer domain. Biochemistry. 2007;46:13297–13309. doi: 10.1021/bi700410g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mulhbacher J, Lafontaine DA. Ligand recognition determinants of guanine riboswitches. Nucleic Acids Res. 2007;35:5568–5580. doi: 10.1093/nar/gkm572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lemay JF, Lafontaine DA. Core requirements of the adenine riboswitch aptamer for ligand binding. RNA. 2007;13:339–350. doi: 10.1261/rna.142007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lemay JF, Penedo JC, Tremblay R, Lilley DMJ, Lafontaine DA. Folding of the adenine riboswitch. Chem Biol. 2006;13:857–868. doi: 10.1016/j.chembiol.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 26.Gilbert SD, Mediatore SJ, Batey RT. Modified pyrimidines specifically bind the purine riboswitch. J Am Chem Soc. 2006;128:14214–14215. doi: 10.1021/ja063645t. [DOI] [PubMed] [Google Scholar]
- 27.Wilkinson KA, Merino EJ, Weeks KM. Selective 2′-hydroxyl acylation analyzed by primer extension (SHAPE): quantitative RNA structure analysis at single nucleotide resolution. Nat Protoc. 2006;1:1610–1616. doi: 10.1038/nprot.2006.249. [DOI] [PubMed] [Google Scholar]
- 28.Wilkinson KA, Merino EJ, Weeks KM. RNA SHAPE chemistry reveals nonhierarchical interactions dominate equilibrium structural transitions in tRNA(Asp) transcripts. J Am Chem Soc. 2005;127:4659–4667. doi: 10.1021/ja0436749. [DOI] [PubMed] [Google Scholar]
- 29.Merino EJ, Wilkinson KA, Coughlan JL, Weeks KM. RNA structure analysis at single nucleotide resolution by selective 2′-hydroxyl acylation and primer extension (SHAPE) J Am Chem Soc. 2005;127:4223–4231. doi: 10.1021/ja043822v. [DOI] [PubMed] [Google Scholar]
- 30.Vicens Q, Gooding AR, Laederach A, Cech TR. Local RNA structural changes induced by crystallization are revealed by SHAPE. RNA. 2007;13:536–548. doi: 10.1261/rna.400207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lemay JF, Penedo JC, Tremblay R, Lilley DM, Lafontaine DA. Folding of the adenine riboswitch. Chem Biol. 2006;13:857–868. doi: 10.1016/j.chembiol.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 32.Kleywegt GJ. Use of non-crystallographic symmetry in protein structure refinement. Acta Crystallogr Sect D. 1996;52:842–857. doi: 10.1107/S0907444995016477. [DOI] [PubMed] [Google Scholar]
- 33.DeLano WL. The PyMOL Molecular Graphics System, 1.0 Ed. DeLano Scientific; Palo Alto, CA: 2002. [Google Scholar]
- 34.Leulliot N, Varani G. Current topics in RNA–protein recognition: control of specificity and biological function through induced fit and conformational capture. Biochemistry. 2001;40:7947–7956. doi: 10.1021/bi010680y. [DOI] [PubMed] [Google Scholar]
- 35.Williamson JR. Induced fit in RNA–protein recognition. Nat Struct Biol. 2000;7:834–837. doi: 10.1038/79575. [DOI] [PubMed] [Google Scholar]
- 36.Klebe G, Kramer O, Sotriffer C. Strategies for the design of inhibitors of aldose reductase, an enzyme showing pronounced induced-fit adaptations. Cell Mol Life Sci. 2004;61:783–793. doi: 10.1007/s00018-003-3406-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Steuber H, Zentgraf M, La Motta C, Sartini S, Heine A, Klebe G. Evidence for a novel binding site conformer of aldose reductase in ligand-bound state. J Mol Biol. 2007;369:186–197. doi: 10.1016/j.jmb.2007.03.021. [DOI] [PubMed] [Google Scholar]
- 38.Blount KF, Breaker RR. Riboswitches as antibacterial drug targets. Nat Biotechnol. 2006;24:1558–1564. doi: 10.1038/nbt1268. [DOI] [PubMed] [Google Scholar]
- 39.Nielsen H, Westhof E, Johansen S. An mRNA is capped by a 2′,5′ lariat catalyzed by a group I-like ribozyme. Science. 2005;309:1584–1587. doi: 10.1126/science.1113645. [DOI] [PubMed] [Google Scholar]
- 40.Beckert B, Nielsen H, Einvik C, Johansen SD, Westhof E, Masquida B. Molecular modelling of the GIR1 branching ribozyme gives new insight into evolution of structurally related ribozymes. EMBO J. 2008;27:667–678. doi: 10.1038/emboj.2008.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schultes EA, Bartel DP. One sequence, two ribozymes: implications for the emergence of new ribozyme folds. Science. 2000;289:448–452. doi: 10.1126/science.289.5478.448. [DOI] [PubMed] [Google Scholar]
- 42.Doudna JA. Preparation of homogeneous ribozyme RNA for crystallization. Methods Mol Biol. 1997;74:365–370. doi: 10.1385/0-89603-389-9:365. [DOI] [PubMed] [Google Scholar]
- 43.Tellinghuisen J. Optimizing experimental parameters in isothermal titration calorimetry: variable volume procedures. J Phys Chem B. 2007;111:11531–11537. doi: 10.1021/jp074515p. [DOI] [PubMed] [Google Scholar]
- 44.Turnbull WB, Daranas AH. On the value of c: can low affinity systems be studied by isothermal titration calorimetry? J Am Chem Soc. 2003;125:14859–14866. doi: 10.1021/ja036166s. [DOI] [PubMed] [Google Scholar]
- 45.Wiseman T, Williston S, Brandts JF, Lin LN. Rapid measurement of binding constants and heats of binding using a new titration calorimeter. Anal Biochem. 1989;179:131–137. doi: 10.1016/0003-2697(89)90213-3. [DOI] [PubMed] [Google Scholar]
- 46.Pflugrath JW. The finer things in X-ray diffraction data collection. Acta Crystallogr Sect D. 1999;55:1718–1725. doi: 10.1107/s090744499900935x. [DOI] [PubMed] [Google Scholar]
- 47.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, et al. Crystallography and NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr Sect D. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.