

Abstract
We used stopped-flow calorimetry to measure the overall enthalpy change associated with template-directed nucleotide insertion and DNA extension. Specifically, we used families of hairpin self-priming templates in conjunction with an exonuclease-free DNA polymerase to study primer extension by one or more dA or dT residues. Our results reveal exothermic heats between –9.8 and –16.0 kcal/bp for template-directed enzymatic polymerization. These extension enthalpies depend on the identity of the inserting base, the primer terminus, and/or the preceding base. Despite the complexity of the overall process, the sign, magnitude, and sequence dependence of these insertion and extension enthalpies are consistent with nearest-neighbor data derived from DNA melting studies. We recognize that the overall process studied here involves contributions from a multitude of events, including dNTP to dNMP hydrolysis, phosphodiester bond formation, and enzyme conformational changes. It is therefore noteworthy that the overall enthalpic driving force per base pair is of a magnitude similar to that expected for addition of one base pair or base stack per insertion event, rather than that associated with the rupture and/or formation of covalent bonds, as occurs during this catalytic process. Our data suggest a constant sequence-independent background of compensating enthalpic contributions to the overall process of DNA synthesis, with discrimination expressed by differences in noncovalent interactions at the template–primer level. Such enthalpic discrimination underscores a model in which complex biological events are regulated by relatively modest energy balances involving weak interactions, thereby allowing subtle mechanisms of regulation.
Awealth of structural and biochemical information has provided significant insight into the mechanisms of DNA polymerase activity (1–8). Kinetic and structural studies on a number of polymerase systems (9–18) have furnished valuable information regarding the molecular mechanisms governing DNA replication. Despite the considerable efforts that have been undertaken to characterize the overall catalytic process, there is a paucity of thermodynamic data describing the energetic parameters associated with DNA chain elongation. Such studies are essential to elucidate the nature and magnitude of the forces that control the mechanisms of polymerase-mediated recognition and catalysis associated with template-directed nucleic acid synthesis.
Escherichia coli DNA polymerase I has been explored extensively in terms of its structural (9) and biochemical (3) properties. The Klenow fragment (KF) is a 68-kDa polypeptide catalytic domain of this enzyme that retains full polymerase and 3′–5′ exonuclease activity. To eliminate proofreading and editing that results in degradation of the DNA product, it is desirable to employ an exonuclease-deficient KF (KFexo–) in biochemical and biophysical studies. Such a system has been obtained by selective site-directed mutations (D355A and E357A), which effectively abrogate the 3′–5′ exonuclease activities (12) while retaining full polymerase activity (9). The availability of the KFexo– provides an ideal system to explore polymerase-catalyzed insertion and extension energetics because it permits characterization of both correct insertions and potential misincorporations while being devoid of concurrent reactions that preclude accurate determination of the relevant thermodynamic parameters.
Characterization of the energetics of DNA extension involves the partitioning of a multitude of molecular events. These events include the initial polymerase–DNA association, formation of the productive ternary complex with the incoming nucleotide, and the chemical processes culminating in extension of the oligonucleotide chain. In this multistep recognition and association process (3, 18), it has been proposed that KFexo–, in complex with the template–primer duplex, undergoes a conformational change on interaction with the incoming nucleotide, resulting in formation of the active closed ternary complex (2, 5, 17). After nucleotidyl transfer and phosphodiester bond formation, a second conformational change that results in relaxation of the complex and release of pyrophosphate has been reported (5). Structural insight into the KFexo– ternary complex has been reported (17), and the overall process has been reviewed in terms of transition-state mechanisms (19).
Numerous studies of polymerase-catalyzed template-directed DNA synthesis have evaluated the kinetic parameters for canonical extension, the preferences for misinsertions, and the characteristics of translesion synthesis. In the aggregate, these studies reveal canonical and noncanonical extension to be strongly dependent on the subtleties of the neighboring sequences comprising the template–primer region of the target DNA, as well as on the nature of the polymerase and the lesion (20–28). Consequently, the surrounding sequence context plays a critical role in the mutagenicity of a particular DNA lesion (26, 29, 30). Energetic characterization of the sequence-dependent extension events is needed to understand the nature and magnitude of the forces that dictate and control template-directed nucleic acid synthesis.
In recognition of this need, the energetics of nucleic acid synthesis has been the focus of intense interest, nearly concomitant with the discovery of the DNA helical structure. Among the pioneers in the field, Peller and coworkers (e.g., ref. 31) dedicated significant efforts toward elucidating the thermodynamic basis for nucleic acid synthesis by employing noncalorimetric techniques. A number of other studies have invoked thermodynamic models based on biochemical and kinetic approaches (32–34) to elucidate the origins of the high fidelity observed in DNA synthesis. The use of calorimetric techniques to assess enzyme kinetic and energetic parameters has long been proposed by Sturtevant and has been practiced since the early 1960s (35, 36). Only recently, however, have advances in calorimetric instrumentation (with sufficient sensitivity and resolution) and molecular biology and chemistry techniques allowed quantitative studies on the energetics of biomolecular synthesis and recognition events.
In this article, we report use of an ultrasensitive differential stopped-flow calorimeter to measure reaction heats associated with polymerase-mediated template-directed correct base insertion and extension. In addition to their intrinsic value, these data also provide a baseline for the dissection of the energetic landscape associated with both normal synthesis and patterns of mutagenesis and translesion synthesis.
Materials and Methods
DNA Polymerase. The KFexo– of E. coli DNA polymerase I (37) has been overexpressed and purified as described (9). The lack of detectable exonuclease activity (over incubation periods as long as 48 h) has been ensured by gel electrophoresis analysis of KFexo– preparations with DNA. The integrity of the enzyme preparations in terms of both activity and stability was verified under the solution conditions selected for calorimetric measurement of template-directed DNA synthesis.
Deoxyoligonucleotide Templates. Oligonucleotides designed to form hairpin structures containing an overhang of one to five nucleotide repeats (Table 1) have been synthesized by conventional phosphoramidite chemistry and solid-state methods by using an automated DNA synthesizer (Expedite 8909, Applied Biosystems). The oligonucleotides have been purified by reverse-phase chromatographic methods to isolate the trityl-on and trityl-off forms by using ammonium bicarbonate as the mobile phase and a linear gradient of acetonitrile. Extinction coefficients of the deoxyoligonucleotides were determined by complete enzymatic digestion, followed by colorimetric phosphate analysis (38).
Table 1. Self-priming template hairpin sequences and their designations.
| Deoxyoligonucleotide sequence | Template sequence | Hairpin designation |
|---|---|---|
| G·CH(A)n series | ||
| AGCTACTTTCCTTTTTGGAAAGTAGC | 1A | G·CH1A |
| AAGCTACTTTCCTTTTTGGAAAGTAGC | 2A | G·CH2A |
| AAAGCTACTTTCCTTTTTGGAAAGTAGC | 3A | G·CH3A |
| AAAAGCTACTTTCCTTTTTGGAAAGTAGC | 4A | G·CH4A |
| AAAAAGCTACTTTCCTTTTTGGAAAGTAGC | 5A | G·CH5A |
| G·CH(A·T)n series | ||
| AAAAAGCTACTTTCCTTTTTGGAAAGTAGCT | 4A | G·CH5A·1T |
| AAAAAGCTACTTTCCTTTTTGGAAAGTAGCTT | 3A | G·CH5A·2T |
| AAAAAGCTACTTTCCTTTTTGGAAAGTAGCTTT | 2A | G·CH5A·3T |
| AAAAAGCTACTTTCCTTTTTGGAAAGTAGCTTTT | 1A | G·CH5A·4T |
| T·AH | ||
| AAAAATCTACTTTCCTTTTTGGAAAGTAGA | 5A | T·AH5A |
| G·CH(T)1,5 | ||
| TTTTTGCTACTTTCCTTTTTGGAAAGTAGC | 5T | G·CH5T |
| TGCTACTTTCCTTTTTGGAAAGTAGC | 1T | G·CH1T |
| Terminated | ||
| AAAAAGCTACTTTCCTTTTTGGAAAGTAGCdT | NE | G·CH5A·dT |
| Fully extended | ||
| AAAAAGCTACTTTCCTTTTTGGAAAGTAGCTTTTT | NE | G·CH5A·5T |
The standard stem—loop structure of the template—primer hairpins is comprised of the sequence (from 5′ to 3′) 5′-GCTACTTTCC(T)5GGAAAGTAGC-3′, in which the template—primer termini consist of G·C base pairs (except for T·AH5A, in which the template—primer terminus is a T·A). The loop consists of 5Ts with a C·G closing base pair, and the underlined segments comprise the base-paired stem region. The bases in bold (within the sequences) correspond to the 5′ single-strand template region. NE, not extendable. The following guidelines have been employed for the hairpin designations. The first two letters (e.g., G·C) and an H (hairpin) refer to the template-primer terminal junction, followed by the number and identity of the template single-strand bases (e.g., 5A). In several hairpins, the stem region is extended with additional base pairs [i.e., G·CH(A·T)n series].
Nucleoside Triphosphates. The nucleoside triphosphates were purified by ion-exchange chromatography by applying a gradient of ammonium bicarbonate (0–500 mM). The purified stocks were stored at –20°C, and the residual ammonium bicarbonate was removed by extensive lyophilization involving multiple resuspensions in water. This purification/lyophilization protocol effectively inhibits disproportionation of the nucleotides before calorimetric measurement.
Temperature-Dependent UV Spectroscopy. An aliquot of each oligonucleotide stock solution was diluted in the standard reagent buffer, composed of 50 mM Pipes and 5.0 mM MgCl2 (pH 7.4), to a concentration of 10 μM. Thermal melting profiles were monitored at 260 nm in an AVIV 14DS UV/Vis spectrophotometer (Aviv Biomedical, Lakewood, NJ) over the temperature range of 0–100°C by employing a 10-sec integration period at 0.5°C increments. Repetitive optical profiles revealed monophasic concentration-independent melting behavior, consistent with the thermally induced disruption of hairpin structures.
Differential Stopped-Flow Calorimetry. Heats associated with template-directed DNA synthesis were measured in a differential stopped-flow heat conduction calorimeter (Commonwealth Technology, Alexandria, VA), which is described in detail elsewhere (39). The superior sensitivity and resolution of this instrument reside in the differential measurement scheme that incorporates the use of parallel tantalum mixing chambers housed within a thermostated adiabatic chamber. The heat generated from each extension reaction is detected by thermopiles situated on all six faces of the two mixing chambers. Integration of the area beneath the heat flow-versus-time profile determines the total heat evolved for a single extension reaction.
Primer Extension Analysis by Gel Electrophoresis. Gel electrophoresis assays of the 32P-labeled reaction mixtures were performed in parallel with calorimetric measurements. Samples for gel analysis contained appropriate amounts of each hairpin in association with the enzyme in the standard reagent buffer described above. A 10-fold ratio of the correct dNTP was added to each sample vial, the reaction was allowed to incubate for 300 sec at 25°C, and the reaction was subsequently terminated by heating to 95°Cfor5min. Gel-mobility analysis of the 32P-labeled reaction mixtures revealed complete extension of the DNA substrates under the experimental conditions used in the calorimetric studies.
Results and Discussion
The Experimental System: Self-Priming Hairpins. The template–primer deoxyoligonucleotides used in the extension experiments are designed to form hairpins resulting in self-priming templates. The oligonucleotide sequences and their respective abbreviated designations are listed in Table 1. We selected a monomolecular design to reduce concentration-dependent effects and to stabilize the DNA duplex substrate. The length of the stem structure (10 bp) is sufficient to encompass a single enzyme footprint (40), and the 5′-dangling ends comprising one to five bases results in one to five incorporations, as illustrated in Fig. 1. The basic T5-loop and a closing C·G base pair provide greater stability for these hairpins (41). We designed template single strands composed of either all A or all T residues, thereby focusing our initial studies on demonstrating the feasibility of the experimental design and evaluating the energetic contributions of insertion and extension associated with one class of canonical interactions.
Fig. 1.

Gel analysis of the DNA substrates (–) and products (+). The migration distances of the KFexo–-catalyzed reaction products of G·CH1A through G·CH5A are indicated to the right of the gel, yielding G·CH1A·1T through G·CH5A·5T as extension products.
The Experimental Measurement: Energetics of Nucleotide Insertion and DNA Extension. In a typical differential stopped-flow calorimetry experiment, one of the reagent syringes contains a preformed KF·DNA complex [prepared by combining KFexo– (2.5 μM) with a corresponding 2-fold concentration of template hairpin (5.0 μM)], and the other reagent syringe contains an excess of a nucleoside triphosphate solution (50 μM). These concentrations ensure detection of the reaction heats and complete extension of the template hairpin with respect to the reported Kd of 20 μM for insertion of the correct dNTP (1).
A representative differential stopped-flow calorimetry experiment at 25°C for the KFexo–-catalyzed extension reaction of the self-priming hairpin C·GH5A with dTTP is illustrated in the thermogram presented in Fig. 2. The reported enthalpies are the average of 25–30 consecutive mixing profiles per template–primer extension. Each data point in Fig. 2 Inset represents a separate extension experiment for one of the G·CH5(A)1–5 template–primer hairpins. These values have been corrected for mixing artifacts by conducting control experiments in which different combinations of the sample reagents (DNA, KFexo–, and dNTP) are diluted 1:1 into dialysate. Use of an excess of either KFexo– or DNA substrate results in essentially identical reaction heats on a molar basis (data not shown), thereby indicating that neither product inhibition nor incomplete catalysis occurs under our experimental conditions. Additional control experiments have used fully extended (G·CH5A·5T) and terminated (G·CH5A·dT) hairpins complexed to KFexo– and challenged with excess dNTP. In the sections that follow, we discuss the calorimetric data in terms of the measured extension enthalpies, considering how the additivity of single base insertions and sequence context-dependent variations influence the total apparent enthalpy (ΔHapp) observed.
Fig. 2.

Thermogram depicting the profile of heat generated from a single injection of the protein–DNA complex and a solution of the nucleoside triphosphate in a typical mixing experiment. (Inset) Average apparent enthalpies for extension of the G·CH(A)1–5 hairpin family. The total heat per mole of template–primer is presented as a function of the number of bases in the template region.
Energetics of Template-Directed DNA Synthesis: The Insertion and Extension Enthalpies Reflect Noncovalent Interactions. We have calorimetrically monitored the insertion of dT residues into a series of G·CH(A)1–5 hairpins containing one to five dangling A residues in the template strand. The measured insertion and extension enthalpies for each hairpin in this series, expressed as the total heat and the enthalpy per base pair, are listed in Table 2 (columns 2 and 3, respectively). Parallel analysis of the hairpin extension products has been performed by gel electrophoresis of the 5′-32P-labeled products (Fig. 1), revealing that full extension is achieved in each reaction. Compilation of the results for insertion of dTMP into the (A)n templates (n = 1, 2, 3, 4, or 5) yields a range of enthalpies from –9.8 to –61.4 kcal/mol template. As expected, the reaction heat increases with increasing numbers of insertion and extension events. In addition, the average ΔHapp per base pair added is –12.3 kcal/mol. The sign (exothermic) and magnitude of this value for insertion and extension by one base pairing or base stacking interaction is of similar magnitude to that expected from nearest-neighbor data derived from nucleic acid melting studies (42–44). We recognize, of course, that the insertion and extension reaction studied here involves contributions from a multitude of other events, including dTTP to dTMP hydrolysis, phosphodiester bond formation, enzyme conformational changes, etc., as well as factors such as solution conditions and heat capacity effects. It is therefore noteworthy that the overall enthalpic driving force per base pair is of a magnitude similar to that expected for the addition of one base pair or base stack per insertion event rather than of the magnitude associated with the rupture and/or formation of covalent bonds, as occurs during the catalytic process studied here. Furthermore, we find these insertion and extension enthalpies to be dependent on sequence context in a manner consistent with available nearest-neighbor data (as discussed below). These observations may reflect a constant background of large and compensating enthalpic contributions to the overall process of DNA synthesis, with the discrimination expressed at the level of differences in the noncovalent nearest-neighbor interactions at the template–primer level. Such discrimination would underscore a model in which complex biological events involving covalent bond rupture and formation can be regulated by relatively modest energy balances involving weak interactions, thereby allowing subtle mechanisms of regulation.
Table 2. Sequence-context dependence of the insertion and extension enthalpies: Comparison of the G·CH(A)1–5 versus G·CH(A·T)1–4 family of template primers.
| G·CH(A)n template—primers
|
G·CH(A·T)n template—primers
|
|||||
|---|---|---|---|---|---|---|
| -ΔHapp*
|
-ΔHapp†
|
|||||
| Hairpin | kcal·mol-1 | kcal·mol-1·bp-1 | Addition | Hairpin | kcal·mol-1 | kcal·mol-1·bp-1 |
| G·CH1A | 9.8 | 9.8 | 1 | G·CH5A·4T | 12.9 | 12.9 |
| G·CH2A | 22.6 | 11.3 | 2 | G·CH5A·3T | 26.0 | 13.0 |
| G·CH3A | 35.8 | 11.9 | 3 | G·CH5A·2T | 38.5 | 12.8 |
| G·CH4A | 48.5 | 12.1 | 4 | G·CH5A·1T | 51.6 | 12.9 |
| G·CH5A | 61.4 | 12.3 | 5 | |||
The data represent the heat generated per mole of template and correspond to the total ΔHapp on extension of 1-5 dangling A repeats by dTTP.
Total heat after addition of dTTP against the indicated number of single-strand A repeats (1-5) within the G·CH(A)1-5 family per mole of extended DNA (kcal·mol-1) or calculated per base pair (kcal·mol-1·bp-1).
Extensions of one to four A bases of template—primer hairpins in the G·CH(A·T)1-4 family, such that the primer terminus is a T.
Extension Enthalpies Are a Function of Base Identity. We also have measured the reaction heat associated with extension by five dA residues of the 3′ end of the corresponding self-priming hairpin used in dT extension studies. The data listed in the first two rows of Table 3 summarize and compare the enthalpies that we measured for dT extension of the G·CH5A hairpin and dA extension of the G·CH5T hairpin. Inspection of these data reveals significant enthalpic differences between insertion of a dA residue and insertion of a dT residue into the corresponding T- and A-containing templates. In short, the measured extension enthalpies are dependent on the identity of the newly inserted base, which, consistent with kinetic studies, suggests asymmetric behavior in template-directed nucleic acid synthesis. Without taking into account all possible nearest-neighbor effects, the enthalpy of dT insertion (ΔHapp =–12.3 kcal·mol–1·bp–1) is less favorable than the enthalpy of dA insertion (ΔHapp = –15.1 kcal·mol–1·bp–1), with a resultant enthalpic discrimination (ΔΔH) of ≈2.8 kcal·mol–1·bp–1.
Table 3. Base identity and sequence context effects on the insertion and extension enthapies (ΔHapp) for template—primer hairpins.
| -ΔHapp
|
||||
|---|---|---|---|---|
| Hairpin primer | Incoming nucleotide | Insertion/extension nearest neighbor(s) | kcal·mol-1 | kcal·mol-1·bp-1* |
| G·CH5A | dTTP | 1 CT + 4 TT | 61.4 | 12.3 |
| G·CH5T | dATP | 1 CA + 4 AA | 75.6 | 15.1 |
| T·AH5A | dTTP | 1 AT + 4 TT | 63.7 | 12.7 |
| G·CH1A | dTTP | 1 CT | 9.8 | 9.8 |
| G·CH1T | dATP | 1 CA | 11.6 | 11.6 |
| G·CH5A·4T | dTTP | 1 TT | 12.9 | 12.9 |
Resultant nearest-neighbor insertion enthalpies (-ΔHapp, kcal·mol-1·bp-1) for AA, TT, AT, CA, and CT are 16.0, 12.9, 12.1, 11.6, and 9.8, respectively. Values for AA and AT are calculated from the difference between the total heat measured for the fully extended five-base template—primer and the enthalpic contribution for the corresponding single-base insertion(s).
Average enthalpy of insertion/extension calculated per mole of DNA base pair.
Extension Enthalpies Are a Function of the Neighboring Base at the Template–Primer Junction. To assess the impact of the neighboring base on the extension enthalpies, we replaced the template–primer terminus from a G·C base pair (in the G·CH5A hairpin) with a T·A base pair (in the T·AH5A hairpin). Inspection of the data in rows one and three in Table 3 reveals that this alteration in neighboring base pair results in an extension enthalpy that is 2.3 kcal·mol–1 more favorable. These data demonstrate that both the identity of the incoming nucleotide and that of the preceding 3′-terminus base pair are critical determinants of the extension enthalpy measured for a single base insertion.
The Extension Enthalpies Depend on Sequence Context. Further inspection of the data in Table 2 yields insight into the dependence of the insertion enthalpies on sequence context. Note that the total measured heat of –61.4 kcal·mol–1 for dT insertions into the G·CH(A)1–5 hairpin series is not shared equally among the five nucleotides. This feature is apparent when comparing the estimated value for a single dT base insertion in the G·CH1A hairpin (–9.8 kcal·mol–1·bp–1) versus the average of five dT base insertions in the G·CH5A hairpin (–12.3 kcal·mol–1·bp–1). Note that the least favorable extension enthalpy is observed when the dT residue initially is inserted adjacent to the G·C base pair, whereas the extension enthalpies are increasingly more favorable as dT extension continues with neighboring A·T base pairs.
To assess further this apparent sequence context/positional effect, we measured dT insertion enthalpies for a G·CH(A·T)1–4 series of hairpins. In this construct, the template–primer terminus is invariant (A·T), such that all dT insertions are adjacent to A·T base pairs. Given this constant sequence context, one might anticipate little difference in extension enthalpies as we systematically alter the template length by single A·T base pair increments. The results summarized in Table 2 confirm this expectation in that the values for single base insertions are nearly identical (ΔH ≈ –12.9 kcal·mol–1·bp–1) for each of the four G·CH(A·T)1–4 hairpins. Based on the differences we observe for the enthalpy of dT insertion in these two families of hairpins, we conclude that sequence-context and positional effects manifest primarily through the preceding base pair at the 3′ terminus, with minimal longer range sequence-dependent influences.
Comparison of Insertion Enthalpies and Nearest-Neighbor Data. The data obtained on the two families of hairpins noted above reveal that the insertion of a dT residue following an A·T terminal base pair is enthalpically more favorable by ≈–3.1 kcal·mol–1 than the corresponding dT insertion after a G·C base pair. This result is qualitatively consistent with established nearest-neighbor enthalpy data in which the stacking of two A·T base pairs is enthalpically more favorable than the stacking of an A·T and a G·C base pair (42–44). Analysis of all of the data listed in Table 3 reveals a remarkable correspondence between the sequence dependence of the insertion and extension enthalpies and the sequence dependence of published nearest-neighbor stacking enthalpies. Both data sets exhibit parallel enthalpy trends, with the exothermicity for pairwise interactions decreasing in the order AA > TT > AT > CA > CT, as reflected in the data listed at the bottom of Table 3. It is intriguing that the enthalpy differences for DNA synthesis measured on our complex systems are consistent with those differences derived from studies on isolated oligonucleotides. This empirical correspondence, in terms of sign, magnitude, and sequence dependence suggests that stacking and pairing interactions between the incoming base and the primer terminus provide the enthalpic discrimination observed in the extension enthalpies measured here. In such an interpretation, we are assuming that the multitude of other interactions (including bond rupture and formation) effectively cancel one another to produce a sequence-independent common background contribution.
Resolving the ΔHapp into Its Multistep Components. The overall results of control experiments (data not shown) reveal that, under the solution conditions used, the enthalpic contribution arising from binding of the incoming dNTP in the absence of catalysis is indistinguishable from the heats associated with nucleotide dilution into dialysate. Moreover, there is no evidence for detectable heats of association between the template–primer and nucleoside triphosphate, or between the latter and the enzyme (data not shown). Considering that the interactions between KFexo– and the DNA substrate might be expected to contribute a significant enthalpic component to the measured ΔHapp, these observations may seem counterintuitive. Nevertheless, the addition of a preformed mixture of DNA–dNTP to the enzyme yielded reaction heats similar to those obtained by addition of dNTP to the enzyme–DNA complex. One plausible explanation is suggested by analysis of the KFexo––DNA calorimetric binding profile (C.A.S.A.M., unpublished data), in which compensatory events at the selected temperature and protein/DNA ratio result in an enthalpic term that is essentially indistinguishable from nucleotide dilution heats. The net ΔHapp may also include additional contributions from protonation events due to the selected pH (7.4) and heat of ionization for Pipes (ΔHion = 2.7 kcal·mol–1). Therefore, the measured enthalpy for base insertion and incorporation represents the sum of multiple events, with contributions arising from the catalytic processes and from the energetics associated with base stacking and hydrogen bonding in the template and the extended product, including the impact of single-stranded stacking. In this regard, the enthalpic differential (ΔΔH) of 3.1 kcal·mol–1 between AA and TT neighboring bases noted at the bottom of Table 3 (both part of an AA·TT doublet) is consistent with preexisting single-strand stacking (45–48) in the dA template bases within the G·CH(A)1–5 hairpin family. Assuming that the enthalpic contributions from the catalytic events are sequence independent and that the hydrogen bonding and base pairing interactions are relatively constant (by selection of As and Ts as templates), the sequence-dependent extension enthalpies are likely to reflect differences in stacking interactions between the incoming base and the primer terminus.
Thermodynamic Driving Forces of the Overall Synthesis Process. The present study has been designed to assess the feasibility of quantitatively measuring the enthalpies of DNA chain elongation with the eventual goal of partitioning the ΔHapp into its respective components. Enthalpic characterization of an enzyme-mediated reaction furnishes important information regarding the driving forces for a particular catalytic process. Nevertheless, such a characterization does not yield a complete thermodynamic profile of the process. As discussed below, several studies have indirectly provided limited information that may permit estimation of the free energy (ΔG) of the overall process.
It is well established that biosynthetic processes involving peptide, phosphodiester, or glycosidic bond formation are thermodynamically unfavorable unless they are coupled to “high-energy” phosphoanhydride bond cleavage from nucleoside triphosphates, the hydrolysis of which is energetically favorable (49). To rationalize DNA biosynthesis in terms of the overall ΔG, the global process may be represented by
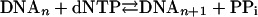 |
The current estimated standard ΔG for NTP (and conceivably dNTP) hydrolysis to form NMP (and dNMP) and PPi is –10.9 kcal·mol–1 (50), a magnitude that is considered sufficient to drive most biosynthesis processes within the cell. Likewise, nucleotidyl transfer during base insertion into an elongating DNA chain should provide much of the driving force for the successful incorporation reaction. The ΔG of phosphodiester bond formation recently has been estimated by determining the equilibrium constant (Keq) and the corresponding ΔG for the ligation reaction of nicked DNA (49). The net ΔG of –6.3 kcal·mol·1 calculated for DNA ligation is the sum of the endergonic phosphodiester bond formation (ΔG ≈+5.3 kcal·mol–1) coupled with the highly exergonic ATP hydrolysis (ΔG = –11.6 kcal·mol–1). Although DNA polymerase and DNA ligase reactions are similar in that both involve formation of a phosphodiester bond at the expense of hydrolyzing an α,β-phosphoanhydride bridge (49), the two synthetic processes (ligation versus template-directed synthesis) are not thermodynamically equivalent. The newly inserted base has reduced conformational mobility in the DNA chain, conceivably adding to the overall entropic penalty. In other words, hydrolysis of the incoming deoxynucleoside triphosphate in template-directed DNA synthesis is coupled to its own immobilization into the elongating chain, a mechanism that is likely governed by distinct thermodynamic driving forces.
Biological Significance. A number of studies have revealed that translesion synthesis and lesion-induced mutagenesis are dependent on the subtleties of the neighboring sequence (26, 29). Despite extensive genetic, kinetic, and structural investigations, the origins for these sequence-dependent effects have yet to be determined. Reviews on the mechanisms of DNA polymerase fidelity have stressed the importance of reconciling structural and biochemical evidence with the inherent thermodynamic driving forces governing these processes (33, 34). Investigations on the mechanisms of DNA polymerase fidelity employing kinetic approaches have inspired the development of models that address the thermodynamics of DNA synthesis fidelity (32–34). These models attempt to decipher the relative contributions of hydrogen bonding and base stacking to the accuracy of DNA synthesis (34). These thermodynamic models can be evaluated empirically by using our experimental approach for directly measuring the relative insertion enthalpies of a correct and potentially incorrect or damaged base. Dissecting the overall exothermic enthalpies of a correct base insertion in terms of its chemical and enzymatic components versus those contributions originating from additional base stacking and hydrogen bonding interactions in the elongated chain should enable one to address the biologically relevant process of DNA synthesis fidelity. In particular, misincorporation and misinsertion preferences against both canonical and damaged or modified bases can be assessed.
Over the past two decades, a number of kinetic investigations in conjunction with DNA melting thermodynamic studies (32) have attempted to correlate polymerase misinsertion rates with the energetics of mismatch-containing DNA duplexes. These findings have led to the development of interesting thermodynamic models aimed at reconciling the relatively modest differences in the thermodynamic stability of mismatches or modified bases (ΔΔG ≈ 0.3–1 kcal·mol–1) (51) with the extremely low misincorporation frequencies (≈10–4 to 10–5). Among possible working hypotheses, these authors have suggested that the common phenomena of enthalpy–entropy compensation reported for DNA (42), proteins (52), and DNA-binding drugs (53) in aqueous solution would not be observed within the polymerase site.
Several of these working hypotheses have been reviewed by Goodman and Fygenson (34). The first model assumes that the lack of hydration in the polymerase cleft facilitates base–base hydrogen bonding in the absence of counteracting water–base hydrogen bonding interactions. Despite the small differences in thermodynamic stability for mismatch-containing duplexes in solution, the high fidelity of a polymerase (ranging from 10–9 to 10–10) to discriminate between correct versus incorrect nucleotides is therefore based on the property of excluding water. This possibility has been challenged recently by the demonstration that apolar nucleotide analogues that lack the ability to form Watson–Crick hydrogen bonding interactions are incorporated efficiently by polymerases (54–56).
An alternative possibility proposed by Tinoco and coworkers (32) is that enthalpy–entropy compensation mechanisms observed in solution do not occur in the environment of the enzyme pocket, and thus the differences in ΔG that account for the fidelity mechanisms might originate from large differences in the enthalpic term. In other words, the relatively substantial discrimination free energies at the polymerase site might result from significant enthalpic differences (ΔΔH) without compensatory entropic contributions (ΔΔS) for correct versus incorrect incorporation, a proposal that implies that the free-energy differences within the polymerase site might be amplified relative to solution conditions. This represents an attractive hypothesis to explain the energetic origins of fidelity. Studies evaluating the impact of a mismatch at the template–primer terminus on the overall insertion and extension enthalpies are possible. Our experimental strategy applied to studies of misincorporation and mismatch extension may possibly provide additional insight into the energetic origins underlying fidelity, thereby allowing these hypotheses to be tested.
Concluding Remarks. Although the mechanisms underlying mutagenesis have been explored in detail by biochemical and structural approaches, characterization of the energetics involved in this process remains elusive. In the present study, we have demonstrated the usefulness of employing calorimetric techniques for assessing the energetics of template-directed DNA synthesis and for determining sequence-dependent insertion and extension enthalpies. This approach establishes an experimental framework for constructing the energetic database needed to evaluate the enthalpic origins of DNA polymerase fidelity. In this regard, a wealth of evidence suggests that the high fidelity of polymerases may not be rationalized solely in terms of the energetic impact of a mismatch relative to a Watson–Crick base pair nor be explained solely on the basis of structural constraints imposed during insertion of an incorrect, damaged, or modified nucleotide (32–34, 57). Consequently, characterizations resulting from the combined efforts of structural, kinetic, and energetic approaches are needed to gain further insight into the fundamentally important mechanisms of replication and mutagenesis.
Acknowledgments
This work was supported by National Institutes of Health Grants GM23509, GM34469 (to K.J.B.), and CA47995 (to K.J.B. and A.P.G.).
Abbreviations: KF, Klenow fragment; KFexo–, exonuclease-deficient KF.
References
- 1.Kuchta, R. D., Mizrahi, V., Benkovic, P. A., Johnson, K. A. & Benkovic, S. J. (1987) Biochemistry 26, 8410–8417. [DOI] [PubMed] [Google Scholar]
- 2.Kuchta, R. D., Benkovic, P. & Benkovic, S. J. (1988) Biochemistry 27, 6716–6725. [DOI] [PubMed] [Google Scholar]
- 3.Kuchta, R. D., Cowart, M., Allen, D. & Benkovic, S. J. (1988) Biochem. Soc. Trans. 16, 947–949. [DOI] [PubMed] [Google Scholar]
- 4.Carroll, S. S., Cowart, M. & Benkovic, S. J. (1991) Biochemistry 30, 804–813. [DOI] [PubMed] [Google Scholar]
- 5.Dahlberg, M. E. & Benkovic, S. J. (1991) Biochemistry 30, 4835–4843. [DOI] [PubMed] [Google Scholar]
- 6.Eger, B. T., Kuchta, R. D., Carroll, S. S., Benkovic, P. A., Dahlberg, M. E., Joyce, C. M. & Benkovic, S. J. (1991) Biochemistry 30, 1441–1448. [DOI] [PubMed] [Google Scholar]
- 7.Polesky, A. H., Dahlberg, M. E., Benkovic, S. J., Grindley, N. D. & Joyce, C. M. (1992) J. Biol. Chem. 267, 8417–8428. [PubMed] [Google Scholar]
- 8.Benkovic, S. J. & Cameron, C. E. (1995) Methods Enzymol. 262, 257–269. [DOI] [PubMed] [Google Scholar]
- 9.Joyce, C. M. & Grindley, N. D. (1983) Proc. Natl. Acad. Sci. USA 80, 1830–1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Steitz, T. A., Freemont, P. S., Ollis, D. L., Joyce, C. M. & Grindley, J. M. (1986) Biochem. Soc. Trans. 14, 205–207. [DOI] [PubMed] [Google Scholar]
- 11.Steitz, T. A., Beese, L., Freemont, P. S., Friedman, J. M. & Sanderson, M. R. (1987) Cold Spring Harbor Symp. Quant. Biol. 52, 465–471. [DOI] [PubMed] [Google Scholar]
- 12.Derbyshire, V., Freemont, P. S., Sanderson, M. R., Beese, L., Friedman, J. M., Joyce, C. M. & Steitz, T. A. (1988) Science 240, 199–201. [DOI] [PubMed] [Google Scholar]
- 13.Joyce, C. M. (1989) J. Biol. Chem. 264, 10858–10866. [PubMed] [Google Scholar]
- 14.Beese, L. S. & Steitz, T. A. (1991) EMBO J. 10, 25–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Beese, L. S., Derbyshire, V. & Steitz, T. A. (1993) Science 260, 352–325. [DOI] [PubMed] [Google Scholar]
- 16.Beese, L. S., Friedman, J. M. & Steitz, T. A. (1993) Biochemistry 32, 14095–14101. [DOI] [PubMed] [Google Scholar]
- 17.Astatke, M., Grindley, N. D. & Joyce, C. M. (1998) J. Mol. Biol. 278, 147–165. [DOI] [PubMed] [Google Scholar]
- 18.Spratt, T. E. (2001) Biochemistry 40, 2647–2652. [DOI] [PubMed] [Google Scholar]
- 19.Showalter, A. & Tsai, M. (2002) Biochemistry 41, 10571–15076. [DOI] [PubMed] [Google Scholar]
- 20.Clark, J. M. & Beardsley, G. P. (1989) Biochemistry 28, 775–779. [DOI] [PubMed] [Google Scholar]
- 21.Dosanjh, M. K., Galeros, G., Goodman, M. F. & Singer, B. (1991) Biochemistry 30, 11595–11599. [DOI] [PubMed] [Google Scholar]
- 22.Maldonado-Rodriguez, R., Espinosa-Lara, M. & Beattie, K. L. (1991) Mutat. Res. 251, 217–226. [DOI] [PubMed] [Google Scholar]
- 23.Wang, F. J. & Ripley, L. S. (1994) Genetics 136, 709–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hashim, M. F. & Marnett, L. J. (1996) J. Biol. Chem. 271, 9160–9165. [DOI] [PubMed] [Google Scholar]
- 25.Kamiya, H. & Kasai, H. (1996) FEBS Lett. 391, 113–116. [DOI] [PubMed] [Google Scholar]
- 26.Miller, H. & Grollman, A. P. (1997) Biochemistry 36, 15336–15342. [DOI] [PubMed] [Google Scholar]
- 27.Burnouf, D. Y., Miturski, R. & Fuchs, R. P. (1999) Chem. Res. Toxicol. 12, 144–150. [DOI] [PubMed] [Google Scholar]
- 28.Zhuang, P., Kolbanovskiy, A., Amin, S. & Geacintov, N. E. (2001) Biochemistry 40, 6660–6669. [DOI] [PubMed] [Google Scholar]
- 29.Shibutani, S. & Grollman, A. P. (1993) J. Biol. Chem. 268, 11703–11710. [PubMed] [Google Scholar]
- 30.Maccabee, M., Evans, J., Glackin, M., Hatahet, Z. & Wallace, S. (1994) J. Mol. Biol. 236, 514–530. [DOI] [PubMed] [Google Scholar]
- 31.Peller, L. (1976) Biochemistry 15, 141–146. [DOI] [PubMed] [Google Scholar]
- 32.Petruska, J., Goodman, M. F., Boosalis, M. S., Sowers, L. C., Cheong, C. & Tinoco, I. J., Jr. (1988) Proc. Natl. Acad. Sci. USA 85, 6252–6256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Goodman, M. F. (1988) Mutat. Res. 200, 11–20. [DOI] [PubMed] [Google Scholar]
- 34.Goodman, M. F. & Fygenson, K. D. (1998) Genetics 148, 1475–1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Inagami, T. & Sturtevant, J. M. (1963) J. Biol. Chem. 238, 3499–3501. [PubMed] [Google Scholar]
- 36.Sturtevant, J. M. (1972) J. Biol. Chem. 247, 968–969. [PubMed] [Google Scholar]
- 37.Derbyshire, V., Grindley, N. D. & Joyce, C. M. (1991) EMBO J. 10, 17–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Snell, F. D. & Snell, C. T. (1972) Colorimetric Methods of Analysis, Including Some Turbidimetric and Nephelometric Methods (Van Nostrand Reinhold, New York).
- 39.Remeta, D. P., Mudd, C. P., Berger, R. L. & Breslauer, K. J. (1991) Biochemistry 30, 9799–9809. [DOI] [PubMed] [Google Scholar]
- 40.Freemont, P., Ollis, D., Steitz, T. & Joyce, C. (1986) Proteins 1, 66–73. [DOI] [PubMed] [Google Scholar]
- 41.Senior, M. M., Jones, R. A. & Breslauer, K. J. (1988) Proc. Natl. Acad. Sci. USA 85, 6242–6246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Breslauer, K. J., Frank, R., Blocker, H. & Marky, L. A. (1986) Proc. Natl. Acad. Sci. USA 83, 3746–3750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.SantaLucia, J., Jr. (1998) Proc. Natl. Acad. Sci. USA 95, 1460–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sugimoto, N., Nakano, S., Yoneyama, M. & Honda, K. (1996) Nucleic Acids Res. 24, 4501–4505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Breslauer, K. J. & Sturtevant, J. M. (1977) Biophys. Chem. 7, 205–209. [DOI] [PubMed] [Google Scholar]
- 46.Suurkuusk, J., Alvarez, J., Freire, E. & Biltonen, R. (1977) Biopolymers 16, 2641–2652. [DOI] [PubMed] [Google Scholar]
- 47.Filimonov, V. V. & Privalov, P. L. (1978) J. Mol. Biol. 122, 465–470. [DOI] [PubMed] [Google Scholar]
- 48.Freier, S. M., Hill, K. O., Dewey, T. G., Marky, L. A., Breslauer, K. J. & Turner, D. H. (1981) Biochemistry 20, 1419–1426. [DOI] [PubMed] [Google Scholar]
- 49.Dickson, K. S., Burns, C. M. & Richardson, J. P. (2000) J. Biol. Chem. 275, 15828–15831. [DOI] [PubMed] [Google Scholar]
- 50.Frey, P. A. & Arabshahi, A. (1995) Biochemistry 34, 11307–11310. [DOI] [PubMed] [Google Scholar]
- 51.Law, S. M., Erijita, R., Goodman, M. F. & Breslauer, K. J. (1996) Biochemistry 35, 12329–12337. [DOI] [PubMed] [Google Scholar]
- 52.Lumry, R. & Rajenders, S. (1970) Biopolymers 9, 1125–1227. [DOI] [PubMed] [Google Scholar]
- 53.Breslauer, K. J., Remeta, D. P., Chou, W. Y., Ferrante, R., Curry, J., Zaunczkowski, D., Snyder, J. G. & Marky, L. A. (1987) Proc. Natl. Acad. Sci. USA 84, 8922–8926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Moran, S., Ren, R. X. & Kool, E. T. (1997) Proc. Natl. Acad. Sci. USA 94, 10506–10511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dzantiev, L., Alekseyev, Y. O., Morales, J. C., Kool, E. T. & Romano, L. J. (2001) Biochemistry 40, 3215–3221. [DOI] [PubMed] [Google Scholar]
- 56.O'Neill, B. M., Ratto, J. E., Good, K. L., Tahmassebi, D. C., Helquist, S. A., Morales, J. C. & Kool, E. T. (2002) J. Org. Chem. 67, 5869–5875. [DOI] [PubMed] [Google Scholar]
- 57.Florian, J., Goodman, M. F. & Warshel, A. (2003) Biopolymers 68, 286–299. [DOI] [PubMed] [Google Scholar]