

Abstract
Mu and delta opioid receptors modulate the reinforcing effects of ethanol, however, their role in the subjective effects of ethanol is not well understood. This study evaluated the contribution of mu and delta opioid receptors to the subjective effects of ethanol using drug discrimination procedures. Monkeys were trained to discriminate ethanol from saline under a schedule of food delivery. In tests, ethanol engendered increases in drug-lever responding, reaching a maximum of >80%. The mu opioid receptor agonists fentanyl and buprenorphine and the delta opioid receptor agonists SNC 80 and SNC 162 did not substitute for the discriminative stimulus effects of ethanol. As pretreatments, the full agonists fentanyl and SNC 80 enhanced the effects of low doses of ethanol and fentanyl attenuated the effects of the ethanol training dose. Although the possibility of pharmacological antagonism of the effects of ethanol cannot be ruled out, a more likely alternative is that the diminished effects of ethanol were due to perceptual masking of the ethanol stimulus. In contrast, the partial agonists buprenorphine and SNC 162 did not alter ethanol’s effects. Finally, the discriminative stimulus effects of ethanol were attenuated following administration of presumably mu-selective doses of the antagonist naltrexone, but not after administration of the delta opioid receptor antagonist naltrindole. The ability of naltrexone to block the discriminative stimulus effects of ethanol likely reflects its capacity to attenuate ethanol-induced increases in endogenous opioids, in particular beta-endorphin, because attenuation of the ethanol stimulus was not accompanied by significant suppression of response rate.
Keywords: mu opioid receptors, delta opioid receptors, drug discrimination, monkey, beta-endorphin, perceptual masking
1. Introduction
Considerable evidence suggests that ethanol can modulate the endogenous opioid system and that this modulation contributes, at least in part, to ethanol’s reinforcing effects (for review see Oswald and Wand, 2004; Modesto-Lowe and Fritz, 2005). For example, both in vitro and in vivo, acute ethanol administration dose-dependently enhances the release of hypothalamic and pituitary beta-endorphin, an endogenous peptide that binds with approximately equal affinity to mu and delta opioid receptors (e.g., Gianoulakis, 1990; de Waele and Gianoulakis, 1993; Rasmussen et al., 1998). Moreover, in distinct brain areas, acute ethanol increases beta-endorphin gene expression, as well as gene expression levels of the enkephalins, endogenous peptides with approximately 20-fold greater affinity for delta compared to mu opioid receptors (Poplawski et al., 2005; Méndez and Morales-Mulia, 2006). These endogenous opioid peptides likely play a significant role in the addictive effects of ethanol given that beta-endorphin- and enkephalin-containing neurons project to various brain regions associated with reward mechanisms, including the ventral tegmental area and nucleus accumbens; and stimulation of these neurons by their respective endogenous ligands can result in increases in dopamine release, an important mechanism underlying reinforcement processes (Jamensky and Gianoulakis, 1999; Nestler, 2005).
Self-administration studies evaluating the effects of opioid antagonists have largely borne out the conclusions of gene expression and neurochemical studies that a link exists between the endogenous opioid system and the reinforcing effects of ethanol. Numerous studies conducted across several decades show that naloxone, naltrexone and nalmefene, relatively nonselective opioid antagonists, reduce ethanol consumption under a variety of schedules in rodents and primates (for review see Oswald and Wand, 2004; Modesto-Lowe and Fritz, 2005). In addition, studies with more selective antagonists suggest the involvement of both mu and delta opioid receptor mechanisms in ethanol reinforcement. For example, in rats selectively bred for ethanol preference (AA) and/or high alcohol drinking (HAD), administration of the selective mu opioid receptor antagonists D-Phe-Cys-Tyr-D-Trp-Orn-Thr-Pen-Thr-NH2 (CTOP) or beta-funaltrexamine produces significant decreases in ethanol consumption but not water consumption (Hyytiä, 1993; Krishnan-Sarin et al., 1998; Hyytiä and Kiianmaa, 2001). Likewise, the delta opioid receptor antagonists naltrindole, naltriben and ICI 174864 also significantly reduce ethanol intake in selectively bred rats (Krishnan-Sarin et al., 1995; June et al., 1999; Hyytiä and Kiianmaa, 2001; but see Hyytiä, 1993). However, this reduction is not always selective (i.e., antagonists also reduce intake of saccharin-containing solutions; Krishnan-Sarin et al., 1995). Collectively, these results support a role for both the beta-endorphin and enkephalin systems in maintaining ethanol consumption.
Other self-administration studies using opioid agonists as pretreatments provide complementary evidence that stimulation of both mu and delta opioid receptors can modulate the reinforcing effects of ethanol. For example, both central and systemic administration of the mu opioid receptor agonists morphine or 14-methoxymetopon significantly increase ethanol self-administration under a two-bottle choice procedure in rodents (Hodge et al., 1995; Sabino et al., 2007; Barson et al., 2009, 2010). In monkeys, combinations of ethanol and the mu opioid receptor agonist methadone are preferred over ethanol alone under a fixed-ratio (FR) schedule of reinforcement when the work requirement is relatively high (i.e., FR 16 or 32; Shelton et al., 1998). Similar increases in ethanol drinking also occur following i.c.v. administration of the delta opioid receptor agonist D-Ala-Gly-Phe-Met-NH2 (DALA; Barson et al., 2009, 2010).
Although a role for mu and delta opioid receptors in the reinforcing effects of ethanol is clear, less is known about the role of these receptor subtypes in the interoceptive or subjective effects of ethanol. This question is significant given that a drug’s reinforcing effects likely depend, in part, on the ability of the drug to engender characteristic subjective effects. In a laboratory setting, the subjective effects of drugs are often studied using drug discrimination procedures in which the subject is trained to differentiate between at least two conditions (e.g., training dose vs. vehicle) based solely on interoceptive cues. Using these methods and selective opioid receptor agonists and antagonists, we systematically evaluated the contribution of mu and delta opioid receptor mechanisms to the discriminative stimulus effects of ethanol in squirrel monkeys.
2. Methods
2.1. Subjects and surgical procedure
Four male squirrel monkeys (Saimiri sciureus), weighing 750 g to 1050 g, were studied in daily experimental sessions (Monday to Friday). All subjects were experimentally naïve at the beginning of the study. Between sessions, monkeys lived in individual home cages where they had unlimited access to water. Monkeys were maintained at 90 to 95% of their free-feeding body weight by adjusting their access to food in the home cage (Teklad Monkey Diet, supplemented with fresh fruit). All animals were maintained in accordance with the guidelines of the Committee on Animals of the Harvard Medical School and the Guide for Care and Use of Laboratory Animals of the Institute of Laboratory Animal Resources, National Research Council, Department of Health, Education, and Welfare Publication No. National Institutes of Health 85-23, revised 1996. Research protocols were approved by the Harvard Medical School Institutional Animal Care and Use Committee.
Monkeys were prepared with a chronic indwelling venous catheter (polyvinyl chloride; i.d., 0.38 mm; o.d., 0.76 mm) using the general surgical procedures described by Platt et al. (2005). Under isoflurane anesthesia and aseptic conditions, one end of a catheter was passed to the level of the right atrium by way of a femoral or jugular vein. The distal end of the catheter was passed subcutaneously and exited in the mid-scapular region. Catheters were flushed daily with saline and were sealed with stainless steel obturators when not in use. Monkeys wore custom-made nylon-mesh jackets (Lomir Biomedical, Toronto, ON, Canada) at all times to protect the catheter.
2.2. Apparatus
Experimental sessions were conducted in ventilated and sound-attenuated chambers. Monkeys were seated in primate chairs with two response levers mounted on the panel in front of the monkey. Each press of a lever with a minimum downward force of approximately 0.25 N produced an audible click and was recorded as a response. Colored lights mounted above the levers could be illuminated to serve as visual stimuli. Food pellets (Bioserve Precision pellets, Formula 0069, 190 mg; Bioserve, Frenchtown, NJ) could be delivered to a tray located between the levers.
2.3. Ethanol discrimination procedure
Monkeys initially were trained to respond on each of two levers under a 10-response FR schedule of food reinforcement. Once consistent lever pressing was established, the monkeys were implanted with intravenous catheters, and drug discrimination training was started one week after recovery from surgery. The training dose of ethanol was 1.0 g/kg administered intravenously from a 0.4 g/ml stock solution of ethanol. The i.v. route of administration was chosen to avoid taste cues, which can interfere with the stimulus control of behavior by pharmacological cues (Duka et al., 1999). After an i.v. injection of ethanol, 10 consecutive responses on one lever produced a food pellet, whereas after an i.v. injection of saline, 10 consecutive responses on the other lever produced a pellet. For two of the monkeys, responding on the right lever after an injection of ethanol resulted in pellet delivery. For the other monkeys, responding on the left lever after injection of ethanol was reinforced. Delivery of each pellet was followed by a 10-s timeout period. Responses on the incorrect lever (e.g., the saline-appropriate lever after ethanol injection) reset the FR requirement.
Training sessions consisted of a variable number of components (n = 1 – 3) of the FR schedule. The number of components per session was randomized from day-to-day with the restriction that each number occurred equally often within a block of 20 sessions. Each component ended after 10 food pellets had been delivered or after 5 min had elapsed, whichever occurred first. A 10-min timeout period, during which the lights were off and responses had no programmed consequences, preceded each component. During most training sessions, saline was injected during timeout periods preceding the first n − 1 components, and ethanol was injected before the nth component of the session. Periodically, saline was injected before all components of a training session to prevent an invariant association between the last component and ethanol injection. Injections of ethanol or saline were administered from outside the chamber via a catheter extension during the 5th min of the 10-min timeout periods. Each injection was followed by a 2-ml infusion of saline to flush the catheter of any residual drug solution.
2.4. Drug testing procedure
Once consistent stimulus control was achieved, drug test sessions were conducted once or twice per week with training sessions scheduled on intervening days. Test sessions were conducted only if ≥ 80% of responses were made on the injection-appropriate lever during at least four of the preceding five training sessions. In general, test sessions consisted of three FR components, each preceded by a 10-min timeout period. During each component, completion of 10 consecutive responses on either lever produced food. Dose-response functions were determined for test drugs using a cumulative dosing procedure. The drugs studied using this procedure were ethanol, the high efficacy mu opioid receptor agonist fentanyl (0.0001 – 0.001 mg/kg), the mu opioid receptor partial agonist buprenorphine (0.0003 – 0.01 mg/kg), the high efficacy delta opioid receptor agonist SNC 80 (0.003 – 0.03 mg/kg), and the delta opioid receptor partial agonist SNC 162 (0.01 – 0.3 mg/kg). Under the cumulative dosing procedure, incremental doses of each drug (1/4 – 1/2 log increments) were injected i.v. during timeout periods that preceded sequential FR components, permitting a three-point cumulative dose-response function to be determined in a single session. When warranted, four or more different doses of a drug were studied by administering overlapping ranges of cumulative doses during test sessions on different days. The effects of most doses were determined twice, although low doses that were found to be inactive and high doses that produced adverse effects usually were studied only once in each subject.
Subsequently, drug combination studies were carried out with ethanol and the opioid agonists. Agonists (or vehicle) were administered i.v. as pretreatments immediately upon being seated in the primate chair. Five-min after agonist administration, monkeys received the first dose in an ethanol dose-response function (0.1 g/kg). Subsequent doses of ethanol were administered cumulatively as described above. Pretreatment doses of the opioid agonists were chosen to be the highest doses that did not significantly alter response rate or induce untoward side effects in initial studies.
Finally, antagonism studies were carried out with the mu opioid receptor antagonist naltrexone (0.01 – 0.1 mg/kg) and the delta opioid receptor antagonist naltrindole (0.1 – 3 mg/kg). The training dose of ethanol (1.0 g/kg) was administered in the first component of a three component test session, followed by saline or cumulative doses of the opioid receptor antagonist. Three or more different doses of the antagonists were studied by administering overlapping ranges of cumulative doses during test sessions on different days.
2.5. Analysis of drug effects
Percentage of ethanol-lever responding was computed for individual subjects in each component of a test session by dividing the number of responses on the ethanol lever by the total number of responses on both levers and multiplying by 100. Percentage of ethanol-lever responding was calculated for an individual monkey only if the response rate was > 0.1 responses/s during the component. Mean percentage of ethanol-lever responding and S.E.M. were then calculated for the group of monkeys at each dose. A drug was considered to substitute fully for ethanol if the maximum percentage of drug-lever responding was ≥ 80%. The overall rate of responding in each component was computed by dividing the total number of responses in a component (regardless of lever) by the total component duration. Mean response rate (percentage of control ± S.E.M.) was then calculated for the group at each dose.
The effects of each drug on response rate, as well as the ability of the opioid antagonists to reduce ethanol-lever responding, were analyzed by separate repeated measures ANOVAs. Further analysis was performed using Bonferroni t-tests comparing the effects of different doses of each drug to a vehicle control or comparing the effects of different doses of the antagonist combined with ethanol to ethanol alone. The capacity of the opioid agonists to alter the ethanol dose-response function was evaluated using separate repeated measures two-way ANOVAs followed by Bonferroni t-tests. The alpha level for all statistical tests was P<0.05.
2.6. Drugs
95% ethanol was purchased from Pharmco Products (Brookfield, CT) and diluted with saline to create a 0.4 g/ml ethanol stock solution. Fentanyl citrate salt, buprenorphine HCl, SNC 80 ((+)-4-[(αR)-α-((2S,5R)-4-allyl-2,5-dimethyl-1-piperazinyl)-3-methoxybenzyl]-N,N-diethylbenzamide), naltrexone HCl and naltrindole HCl were purchased from Sigma RBI (St. Louis, MO). SNC 162 (4-[(S)-((2S,5R)-2,5-dimethyl-4-(2-propenyl)-1-piperazinyl)phenylmethyl]-N,N-diethylbenzamide) was purchased from Tocris Bioscience (Ellisville, MO). Naltrexone was dissolved in 0.9% saline solution and fentanyl and buprenorphine were dissolved in sterile water. All other drugs were dissolved in small amounts of 0.1N HCl as required and then diluted to the desired concentrations in either saline or sterile water.
3. Results
3.1. Ethanol discrimination
During training sessions on days immediately before test sessions, individual monkeys made an average of 99 (± 1) responses on the ethanol-paired lever after injections of ethanol and 3 (± 2) responses on the ethanol-paired lever after injections of saline. Rates of responding during training sessions were 0.67 (± 0.06) responses/s after injections of ethanol and 0.80 (± 0.05) responses/s after injections of saline. Under test conditions, increasing cumulative doses of ethanol (0.1 – 1.8 g/kg) engendered dose-dependent increases in the percentage of responses on the ethanol-paired lever (Fig. 1, left). Low doses of ethanol (0.1 – 0.3 g/kg) engendered little or no responding on the ethanol-paired lever, whereas doses of ethanol ≥1.0 g/kg elicited virtually exclusive responding on the ethanol-paired lever. As shown in the right panel of Figure 1, the average response rate decreased with administration of increasing doses of ethanol. However, no dose of ethanol reliably reduced rates of responding compared to vehicle.
Figure 1.
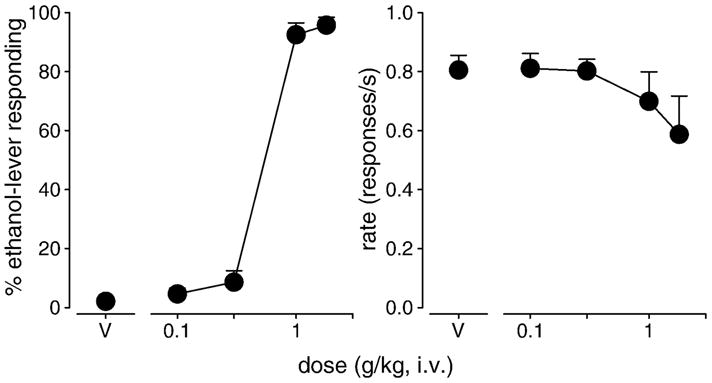
Percentage of ethanol-lever responding (left) and response rate (right) engendered by ethanol in squirrel monkeys trained to discriminate ethanol from vehicle. Points above “V” show the effects of saline administered in the first component followed by cumulative doses of ethanol. Data are means (± S.E.M.) in four subjects.
3.2. Effects of opioid receptor agonists alone
None of the opioid receptor agonists engendered ethanol-like discriminative stimulus effects (Fig. 2). The mu opioid receptor agonists fentanyl (0.0001 – 0.001 mg/kg) and buprenorphine (0.0003 – 0.03 mg/kg) produced maximums of 12 and 10% ethanol-lever responding, respectively. Compared to responding after vehicle injection, the highest doses of these compounds also reliably reduced response rates (fentanyl: F(3,12) = 4.0, P<0.05; buprenorphine: F(4,15) = 11.2, P<0.01; Bonferroni t-tests, P<0.05). Likewise, the delta opioid receptor agonists SNC 80 (0.003 – 0.03 mg/kg) and SNC 162 (0.01 – 0.3 mg/kg) produced maximums of 18 and 29% ethanol-lever responding, respectively. The highest dose of SNC 80, but not SNC 162, reliably reduced response rate compared to vehicle (F(3,12) = 3.6, P<0.05; Bonferroni t-test, P<0.05). However, SNC 162, at a dose of 0.3 mg/kg, induced a seizure in one monkey precluding the testing of higher doses.
Figure 2.

Percentage of ethanol-lever responding (top) and response rate (bottom) engendered by the mu opioid receptor agonists fentanyl and buprenorphine, and the delta opioid receptor agonists SNC 80 and SNC 162 in squirrel monkeys trained to discriminate ethanol from vehicle. Horizontal dashed lines represent mean (± S.E.M.) response rates after vehicle administration. Data are means (± S.E.M.) in four subjects. *Note that P<0.05, Bonferroni t-test.
3.3. Effects of pretreatment with the opioid receptor agonists
Pretreatment with fentanyl altered the discriminative stimulus effects of ethanol in different ways depending on the specific dose of ethanol (Fig. 3, top left). At a low dose (0.1 g/kg), fentanyl reliably enhanced the discriminative stimulus effects of ethanol such that a larger percentage of responses were made on the ethanol-paired lever after fentanyl pretreatment compared to responding after vehicle pretreatment. At the training dose (1 g/kg), fentanyl reliably attenuated the discriminative stimulus effects of ethanol such that a reduced percentage of responses were made on the ethanol-paired lever after fentanyl compared to responding after vehicle. These findings are confirmed statistically by a significant ethanol X fentanyl interaction (F(2,6) = 24.8, P<0.01; Bonferroni t-test, P<0.05). Fentanyl did not alter the discriminative stimulus effects engendered by 0.3 g/kg ethanol. Response rates engendered by ethanol following pretreatment with fentanyl were reliably reduced compared to response rates after vehicle pretreatment (F(1,3) = 16.7, P<0.05; Fig. 3, bottom left).
Figure 3.
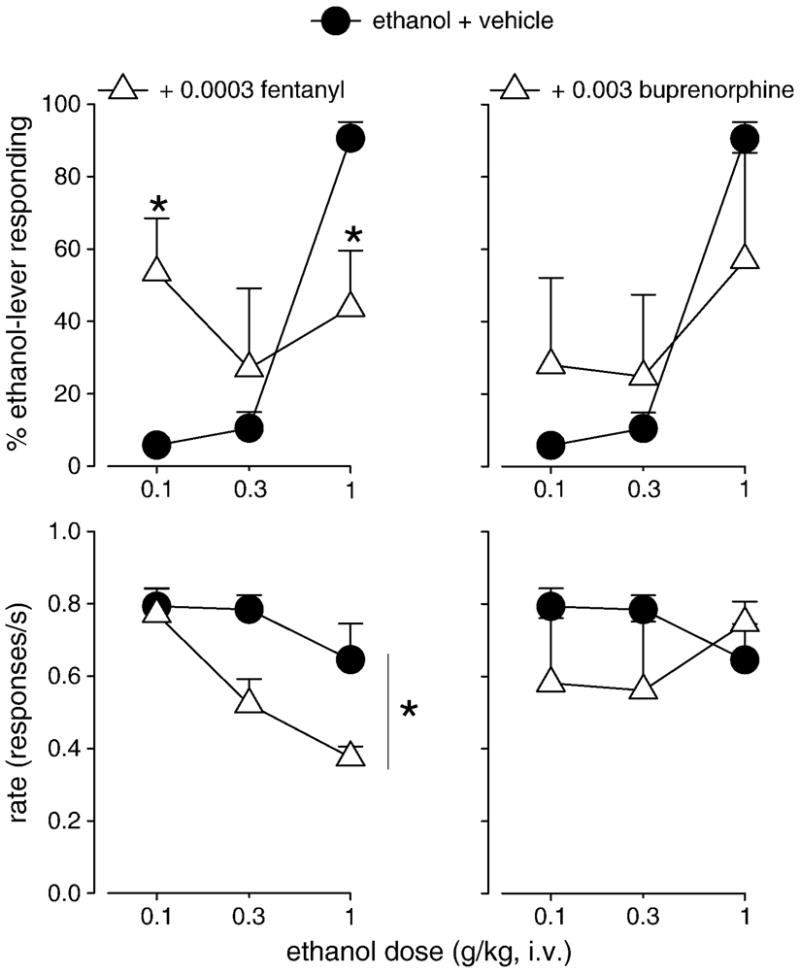
Percentage of ethanol-lever responding (top) and response rate (bottom) engendered by ethanol after vehicle pretreatment (filled circles) and after pretreatment with the mu opioid receptor agonists fentanyl (left, open triangles) and buprenorphine (right, open triangles) in squirrel monkeys trained to discriminate ethanol from saline. Data are means (± S.E.M.) in four subjects. *Note that P<0.05, Bonferroni t-test.
A similar pattern of effects was observed following pretreatment with buprenorphine in that, at low ethanol doses, a greater percentage of responses were made on the ethanol-paired lever after buprenorphine pretreatment compared to responding after vehicle pretreatment and, at the ethanol training dose, a lesser percentage of responses were made on the ethanol-paired lever after buprenorphine compared to after vehicle (Fig. 3, top right). However, none of these differences were significant. Buprenorphine did not markedly alter rates of responding at any dose of ethanol (Fig. 3, bottom right).
Like with fentanyl, pretreatment with SNC 80 altered the discriminative stimulus effects of ethanol in different ways depending upon dose (Fig. 4, top left). At an intermediate dose (0.3 g/kg), SNC 80 reliably enhanced the discriminative stimulus effects of ethanol such that a larger percentage of responses were made on the ethanol-paired lever after SNC 80 pretreatment compared to responding after vehicle pretreatment (ethanol X SNC 80 interaction: (F(2,6) = 9.4, P<0.05; Bonferroni t-test, P<0.05). Although a reduced percentage of responses were made on the ethanol-paired lever after SNC 80 compared to responding after vehicle at the training dose (1.0 g/kg), this was not significant. Response rates engendered by ethanol following pretreatment with SNC 80 were reliably reduced compared to response rates after vehicle pretreatment (F(1,3) = 13.6, P<0.05; Fig. 4, bottom left). In contrast, as shown in the right panels of Figure 4, SNC 162 neither enhanced nor attenuated the discriminative stimulus effects of ethanol. Moreover, SNC 162 did not alter response rate compared to rate after vehicle.
Figure 4.
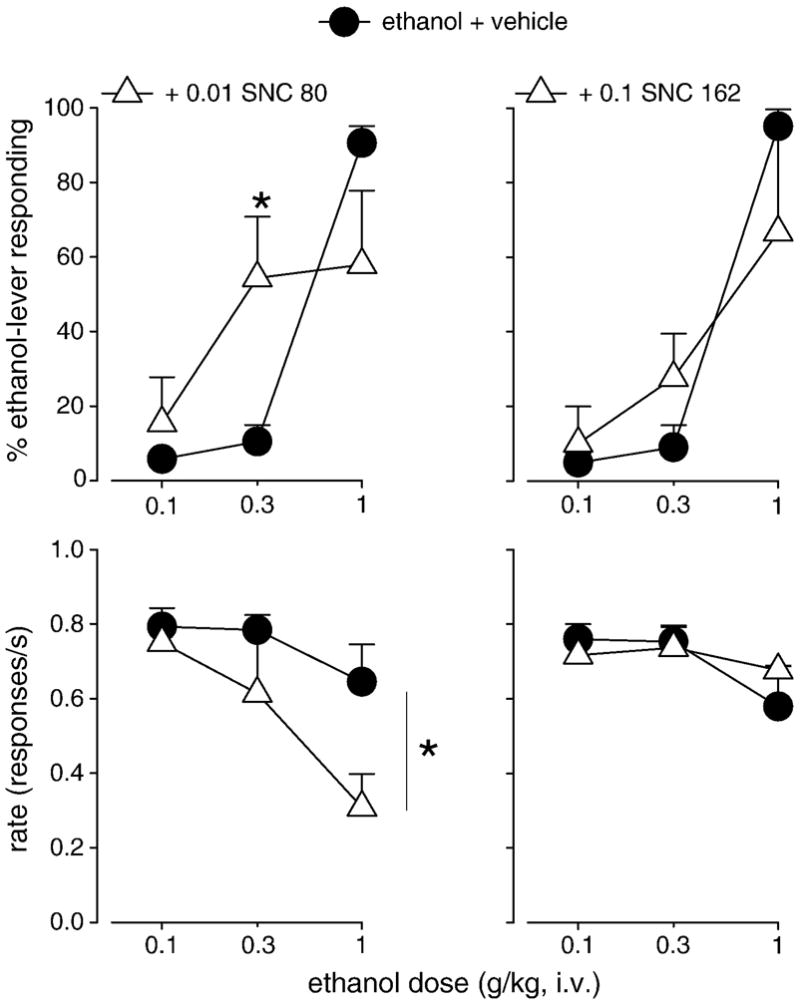
Percentage of ethanol-lever responding (top) and response rate (bottom) engendered by ethanol after vehicle pretreatment (filled circles) and after pretreatment with the delta opioid receptor agonists SNC 80 (left, open triangles) and SNC 162 (right, open triangles) in squirrel monkeys trained to discriminate ethanol from saline. Data are means (± S.E.M.) in four subjects. *Note that P<0.05, Bonferroni t-test.
3.4. Effects of the opioid receptor antagonists
When administered in the first component, ethanol (1 g/kg) engendered nearly exclusive responding on the ethanol-paired lever (Fig. 5, top panels, point over “1 ETOH”). When followed in subsequent components with saline injections, ethanol continued to engender virtually all responses on the ethanol-paired lever (data not shown). Administration of increasing doses of naltrexone (0.01 – 0.1 mg/kg) attenuated the discriminative stimulus effects of ethanol, reliably reducing the percentage of responses on the ethanol-paired lever to 24% (Fig. 5A; top, F(3,12) = 10.8, P<0.01; Bonferroni t-test, P<0.05). Alone, ethanol did not alter substantially rate of responding; nor was response rate reliably affected by increasing doses of naltrexone (Fig. 5A, bottom).
Figure 5.
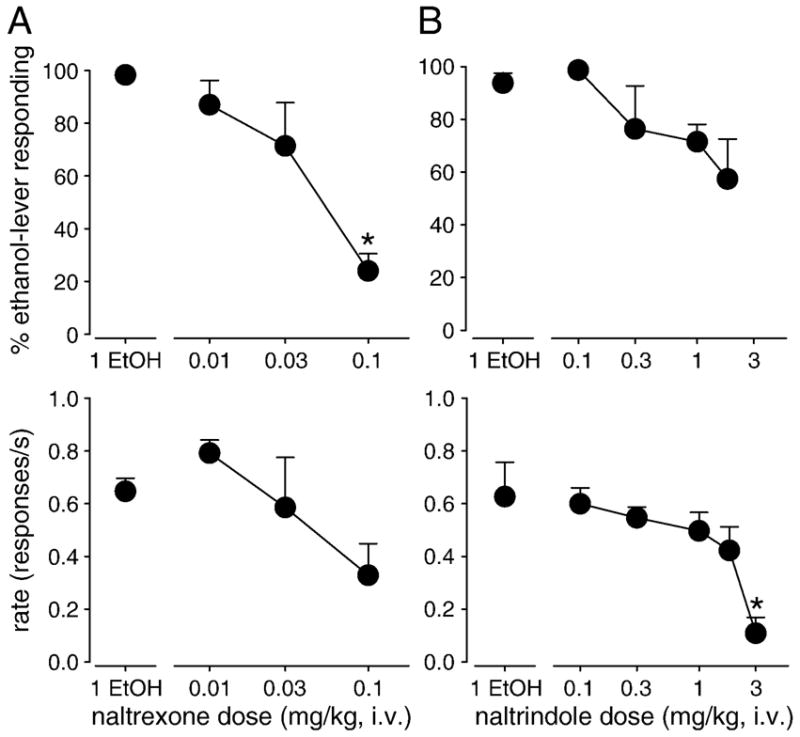
Percentage of ethanol-lever responding (top) and response rate (bottom) engendered by ethanol, alone and after pretreatment with either A) naltrexone or B) naltrindole in squirrel monkeys trained to discriminate ethanol from vehicle. Data are means (± S.E.M.) in four subjects. *Note that P<0.05, Bonferroni t-test.
In contrast, administration of increasing doses of naltrindole (0.1 – 1.8 mg/kg) only reduced responding on the ethanol-paired lever to 57% (Fig, 5B, top). This decrease in ethanol-lever responding did not differ reliably from responding engendered by ethanol alone. When a higher dose of naltrindole (3 mg/kg) was tested for its ability to further attenuate the discriminative stimulus effects of ethanol, responding was essentially eliminated and monkeys failed to complete a single FR (F(5,18) = 4.0; P<0.05; Bonferroni t-test, P<0.05).
4. Discussion
In the present study, none of the mu or delta opioid receptor full (fentanyl, SNC 80) or partial agonists (buprenorphine, SNC 162) shared discriminative stimulus effects with ethanol. These compounds engendered primarily saline-lever responding up to doses that either significantly reduced response rates or induced untoward side effects (e.g., convulsions). We are not aware of any other studies that have administered these particular opioid receptor agonists to ethanol discriminating subjects, however, we selected these compounds based on their known selectivity and/or efficacy profiles at mu and/or delta opioid receptors (e.g., fentanyl: mu opioid receptor full agonist, ~ 100- to 250-fold selective for mu vs. delta, Emmerson et al., 1994; Jagerovic et al., 2002; buprenorphine: mu opioid receptor partial agonist, ~ 3- to 15-fold selective for mu vs. delta, and kappa opioid receptor antagonist, Rennison et al., 2007; Spagnolo et al., 2008; SNC 80: delta opioid receptor full agonist, ~ 800- to 900-fold selective for delta vs. mu, Knapp et al., 1996; Codd et al., 2009; SNC 162: partial agonist, > 1000-fold selective for delta vs. mu, Knapp et al., 1996). Our results, however, are in general concordance with other studies reporting a lack of substitution of the mu opioid receptor agonist morphine for the discriminative stimulus effects of ethanol in monkeys and rats (Winter, 1975; York and Bush, 1982; Bowen et al., 1997; Kosten et al., 1999; Platt et al., 2005). Moreover, because shared discriminative stimulus effects generally indicate common underlying receptor mechanisms, it is not surprising that mu and delta opioid receptor agonists failed to reproduce the ethanol discriminative stimulus. From ethanol self-administration studies, it is reasonably clear that mu and delta opioid receptors modulate rather than mediate the behavioral effects of ethanol.
If a receptor is hypothesized to play a role in the modulation of the discriminative stimulus effects of ethanol, one would expect an orderly progression of effects as ethanol is combined with full agonists, partial agonists and antagonists at that receptor. Namely, one would expect full agonists to enhance ethanol’s discriminative stimulus effects, partial agonists to enhance and/or attenuate ethanol’s discriminative stimulus effects depending on dose, and antagonists to only attenuate ethanol’s discriminative stimulus effects. In the present study, we observed some of these predicted effects. In the case of the mu and delta opioid receptor full agonists fentanyl and SNC 80, respectively, pretreatment with the highest doses of these compounds that did not have rate decreasing effects on their own resulted in significant enhancement of the discriminative stimulus effects of low-to-intermediate doses of ethanol. Although enhancement of the reinforcing effects of ethanol by opioid agonists is common, enhancement of the discriminative stimulus effects of ethanol by opioid agonists has rarely, if ever, been observed. The few published studies have typically investigated the interaction of mu opioid receptor agonists (e.g., morphine, hydromorphone) with ethanol and have reported either attenuation of (Kosten et al., 1999) or no effect on the discriminative stimulus or subject-rated effects of ethanol (Rush, 2001). Interestingly, though, ethanol has been shown to enhance the discriminative stimulus effects of morphine (Kosten and Bombace, 2001). Although the reasons underlying the ability of the full agonists to enhance the discriminative stimulus effects of ethanol in the present study are unclear, one cannot rule out species differences (monkey vs. rat or human), route of ethanol administration (i.v., vs. i.g. or oral), route of opioid receptor agonist administration (i.v. vs. i.p. or oral), or simply choice of test compound. These findings, nevertheless, implicate both mu and delta opioid receptors in the modulation of the discriminative stimulus effects of ethanol.
When the dose of ethanol was increased to 1 g/kg, the opioid receptor agonists, in particular fentanyl, attenuated the discriminative stimulus effects of ethanol. As mentioned above, this finding agrees with the results that Kosten and colleagues (1999) found with morphine. Morphine’s ability to block the discriminative stimulus effects of ethanol has been attributed, at least in part, to its capacity to markedly reduce plasma concentrations of ethanol and delay time to peak ethanol concentration (Kosten et al., 1999). While pharmacokinetic considerations cannot be discounted in the present study, another potential explanation is that of perceptual masking of the ethanol discriminative stimulus by the opioid receptor agonists (cf. Gauvin and Young, 1989). Perceptual masking has been inferred from demonstrations of attenuation of the discriminative stimulus effects of a drug without concomitant attenuation of its rate-altering effects. Although ethanol itself did not alter response rates systematically, response rates for ethanol after pretreatment with either fentanyl or SNC 80 were significantly reduced.
The high efficacy of fentanyl and SNC 80 at mu and delta opioid receptors, respectively, likely contributed to their ability to mask the discriminative stimulus effects of ethanol. To evaluate this possibility directly, the mu and delta opioid receptor partial agonists, buprenorphine and SNC 162, were administered as pretreatments before determining an ethanol dose-response function. It was expected that the lower efficacy compounds would not have the same masking capacity and perhaps reveal even greater enhancement of the discriminative stimulus effects of ethanol. This was not the case. The ethanol dose-response function was essentially unchanged following pretreatment with either partial agonist. These results suggest that specific efficacy conditions may have to be met at mu and/or delta opioid receptors for agonist-induced enhancement of ethanol’s discriminative stimulus effects to be observed, and they are consistent with the notion of a “threshold” efficacy requirement for the production of a particular behavioral effect (cf. Ruffolo, 1982).
It should be recalled that buprenorphine exhibits antagonist-like effects at kappa opioid receptors in addition to exhibiting partial agonist-like effects at mu opioid receptors. Because the kappa opioid system has been shown to play a role in other effects of ethanol (e.g., blockade of ethanol self-administration by kappa antagonists; Walker and Koob, 2008; Sperling et al., 2010; Walker et al., 2010), it remains possible that this receptor system also could play a role in the discriminative stimulus effects of ethanol. This prospect seems unlikely, however, given the lack of modulation of the ethanol discriminative stimulus by buprenorphine. Moreover, Spanagel (1996) failed to show modulation of the discriminative stimulus effects of ethanol in rats by the kappa opioid receptor antagonist nor-binaltorphimine. Together, these results suggest that the kappa opioid receptor system has little role in mediating the discriminative stimulus effects of ethanol.
In antagonism studies, naltrexone dose-dependently reversed the discriminative stimulus effects of ethanol at doses that did not disrupt responding, suggesting that this blockade was pharmacologically specific and not the result of a nonspecific disruption in behavior. This finding of attenuation replicates earlier findings in rats (Spanagel, 1996; Kosten et al., 1999; but see, Winter, 1975; Altshuler et al., 1981) and extends the observation to another species (squirrel monkeys) and another route of naltrexone administration (i.v. vs. s.c.). Because naltrexone has greater affinity for mu compared to delta opioid receptors (~ 14- to 60-fold, Emmerson et al., 1994; Le Bourdonnec et al., 2006) and because the dose of naltrexone found to attenuate the effects of ethanol is below that found to attenuate morphine discrimination in squirrel monkeys (cf. Platt et al., 1999), it is reasonable to assume that naltrexone is acting via mu opioid receptor blockade. Consistent with this idea, the delta opioid receptor antagonist naltrindole (~ 20- to 90-fold delta vs. mu, Emmerson et al., 1994; Schutz et al., 2002) failed to markedly reduce the discriminative stimulus effects of ethanol. At higher doses of naltrindole, significant rate disruptions were observed. Likewise, in rats, naltrindole did not block the discriminative stimulus effects of i.p. ethanol (Spanagel, 1996). Taken together, these findings imply a key role for mu, but not delta, opioid receptors in the modulation of the discriminative stimulus effects of ethanol.
In summary, our findings suggest a more prominent role for mu, rather than delta, opioid receptors in modulating the discriminative stimulus effects of ethanol in squirrel monkeys. This notion is supported by findings of enhancement of the ethanol discriminative stimulus with a mu opioid receptor selective agonist and attenuation of the ethanol discriminative stimulus with a mu opioid receptor selective antagonist. Extrapolating from ethanol self-administration studies, these results also may suggest that ethanol’s capacity to acutely impact the beta-endorphin system is an important mechanism underlying the ability of mu opioid receptor selective compounds to modulate the discriminative stimulus effects of ethanol.
Acknowledgments
This research was supported by USPHS grants AA16179 and RR00168.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Altshuler HL, Applebaum E, Shippenberg TS. The effects of opiate antagonists on the discriminative stimulus properties of ethanol. Pharmacol Biochem Behav. 1981;14:97–100. doi: 10.1016/0091-3057(81)90109-x. [DOI] [PubMed] [Google Scholar]
- Barson JR, Carr AJ, Soun JE, Sobhani NC, Leibowitz SF, Hoebel BG. Opioids in the nucleus accumbens stimulate ethanol intake. Physiol & Behav. 2009a;98:453–459. doi: 10.1016/j.physbeh.2009.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barson JR, Carr AJ, Soun JE, Sobhani NC, Rada P, Leibowitz SF, Hoebel BG. Opioids in the hypothalamic paraventricular nucleus stimulate ethanol intake. Alcohol Clin Exp Res. 2010;34:241–222. doi: 10.1111/j.1530-0277.2009.01084.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowen CA, Gatto GJ, Grant KA. Assessment of the multiple discriminative stimulus effects of ethanol using an ethanol-pentobarbital-water discrimination in rats. Behav Pharmacol. 1997;8:339–352. doi: 10.1097/00008877-199708000-00007. [DOI] [PubMed] [Google Scholar]
- Codd EE, Carson JR, Colburn RW, Stone DJ, Van Besien CR, Zhang SP, Wade PR, Gallantine EL, Meert TF, Molino L, Pullan S, Razler CM, Dax SL, Flores CM. JNJ-20788560 [9-(8-azabicyclo[3.2.1]oct-3-ylidene)-9H-xanthene-3-carboxylic acid diethylamine], a selective delta opioid receptor agonist, is a potent and efficacious antihyperalgesic agent that does not produce respiratory depression, pharmacologic tolerance, or physical dependence. J Pharmacol Exp Ther. 2009;329:241–251. doi: 10.1124/jpet.108.146969. [DOI] [PubMed] [Google Scholar]
- Duka T, Jackson A, Smith DC, Stephens DN. Relationship of components of an alcohol interoceptive stimulus to induction of desire for alcohol in social drinkers. Pharmacol Biochem Behav. 1999;64:301–309. doi: 10.1016/s0091-3057(99)00080-5. [DOI] [PubMed] [Google Scholar]
- De Waele JP, Gianoulakis C. Effects of single and repeated exposure to ethanol on hypothalamic β-endorphin and CRH release by the C57BL/6 and DBA/2 strains of mice. Neuroendocrinology. 1993;57:700–709. doi: 10.1159/000126428. [DOI] [PubMed] [Google Scholar]
- Emmerson PJ, Liu MR, Woods JH, Medzihradsky R. Binding affinity and selectivity of opioids at mu, delta and kappa receptors in monkey brain membranes. J Pharmacol Exp Ther. 1994;271:1630–1637. [PubMed] [Google Scholar]
- Gauvin DV, Young AM. Evidence for perceptual masking of the discriminative morphine stimulus. Psychopharmacology. 1989;98:212–221. doi: 10.1007/BF00444694. [DOI] [PubMed] [Google Scholar]
- Gianoulakis C. Characterization of the effect of acute ethanol administration on the release of β-endorphin peptides by the rat hypothalamus. Eur J Pharmacol. 1990;180:21–29. doi: 10.1016/0014-2999(90)90588-w. [DOI] [PubMed] [Google Scholar]
- Hodge CW, Niehus JS, Samson HH. Morphine induced changes in ethanol- and water-intake are attenuated by the 5-HT3/4 antagonist tropisetron (ICS 205-930) Psychopharmacology. 1995;119:186–192. doi: 10.1007/BF02246160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyytiä P. Involvement of μ-opioid receptors in alcohol drinking by alcohol-prefering AA rats. Pharmacol Biochem Behav. 1993;45:697–701. doi: 10.1016/0091-3057(93)90527-z. [DOI] [PubMed] [Google Scholar]
- Hyytiä P, Kiianmaa K. Suppression of ethanol responding by centrally administered CTOP and naltrindole in AA and Wistar rats. Alcohol Clin Exp Res. 2001;25:25–33. doi: 10.1111/j.1530-0277.2001.tb02123.x. [DOI] [PubMed] [Google Scholar]
- Jagerovic N, Cano C, Elguero J, Goya P, Callado LF, Meana JJ, Giron R, Abalo R, Ruiz D, Goicoechea C, Martin MA. Long-acting fentanyl analogues: Synthesis and pharmacology of N-(1-phenylpyrazolyl)-N-(1-phenylalkyl-4-piperidyl)propanamides. Bioorg Med Chem. 2002;10:817–827. doi: 10.1016/s0968-0896(01)00345-5. [DOI] [PubMed] [Google Scholar]
- Jamensky NJ, Gianoulakis C. Comparison of the proopiomelanocortin and proenkephalin opioid peptide systems in brain regions of the alcohol-preferring C57BL/6 and alcohol-avoiding DBA/2 mice. Alcohol. 1999;18:177–187. doi: 10.1016/s0741-8329(99)00002-6. [DOI] [PubMed] [Google Scholar]
- June HL, McCane SR, Zink RW, Portoghese PS, Li TK, Froehlich JC. The δ2-opioid receptor antagonist naltriben reduces motivated responding for ethanol. Psychopharmacology. 1999;147:81–89. doi: 10.1007/s002130051145. [DOI] [PubMed] [Google Scholar]
- Knapp RJ, Santoro G, De Leon IA, Lee KB, Edsall SA, Waite S, Malatynska E, Varga E, Calderon SN, Rice KC, Rothman RB, Porreca F, Roeske WR, Yamamura HI. Structure-activity relationships for SNC80 and related compounds at cloned human delta and mu opioid receptors. J Pharmacol Exp Ther. 1996;277:1284–1291. [PubMed] [Google Scholar]
- Kosten TA, Bombace JC. Ethanol enhances naloxone sensitization and disrupts morphine discrimination – comparison to dizocilpine and pentobarbital: Explanation of enhancing acute and attenuating chronic effects. Prog Neuro-Psychopharmacol & Biol Psychiat. 2001;25:1283–1306. doi: 10.1016/s0278-5846(01)00180-4. [DOI] [PubMed] [Google Scholar]
- Kosten TA, Haile CN, Jatlow P. Naltrexone and morphine alter the discrimination and plasma levels of ethanol. Behav Pharmacol. 1999;10:1–13. doi: 10.1097/00008877-199902000-00001. [DOI] [PubMed] [Google Scholar]
- Krishnan-Sarin S, Jing SL, Kurtz DL, Zweifel M, Portoghese PS, Li TK, Froehlich JC. The delta opioid receptor antagonist naltrindole attenuates both alcohol and saccharin intake in rats selectively bred for alcohol preference. Psychopharmacology. 1995;120:177–185. doi: 10.1007/BF02246191. [DOI] [PubMed] [Google Scholar]
- Krishnan-Sarin S, Wand GS, Li XW, Portoghese PS, Froehlich JC. Effect of mu opioid receptor blockade on alcohol intake in rats bred for high alcohol drinking. Pharmacol Biochem Behav. 1998;59:627–635. doi: 10.1016/s0091-3057(97)00474-7. [DOI] [PubMed] [Google Scholar]
- Le Bourdonnec B, Goodman AJ, Graczyk TM, Belanger S, Seida PR, DeHaven RN, Dolle RE. Synthesis and pharmacological evaluation of novel octahydro-1H-pyrido[1,2-a]pyrazine as mu-opioid receptor antagonists. J Med Chem. 2006;49:7290–7306. doi: 10.1021/jm0604878. [DOI] [PubMed] [Google Scholar]
- Méndez M, Morales-Mulia M. Ethanol exposure differentially alters pro-enkephalin mRNA expression in regions of the mesocorticolimbic system. Psychopharmacology. 2006;189:117–124. doi: 10.1007/s00213-006-0503-3. [DOI] [PubMed] [Google Scholar]
- Modesto-Lowe V, Fritz EM. The opioidergic-alcohol link: Implications for treatment. CNS Drugs. 2005;19:693–707. doi: 10.2165/00023210-200519080-00005. [DOI] [PubMed] [Google Scholar]
- Nestler EJ. Is there a common molecular pathway for addiction? Nat Neurosci. 2005;8:1445–1449. doi: 10.1038/nn1578. [DOI] [PubMed] [Google Scholar]
- Oswald LM, Wand GS. Opioids and alcohol. Physiol Behav. 2004;81:339–358. doi: 10.1016/j.physbeh.2004.02.008. [DOI] [PubMed] [Google Scholar]
- Platt DM, Carey G, Spealman RD. Intravenous self-administration techniques in monkeys. In: Enna S, Williams M, Ferkany J, Kenakin T, Porsolt R, Sullivan J, editors. Current Protocols in Neuroscience. Unit 9.21. Wiley; New York, NY: 2005. [DOI] [PubMed] [Google Scholar]
- Platt DM, Duggan A, Spealman RD, Cook JM, Li X, Yin W, Rowlett JK. Contribution of α1GABAA and α5GABAA receptor subtypes to the discriminative stimulus effects of ethanol in squirrel monkeys. J Pharmacol Exp Ther. 2005;313:658–667. doi: 10.1124/jpet.104.080275. [DOI] [PubMed] [Google Scholar]
- Platt DM, Grech DM, Rowlett JK, Spealman RD. Discriminative stimulus effects of morphine in squirrel monkeys: Stimulants, opioids, and stimulant-opioid combinations. J Pharmacol Exp Ther. 1999;290:1092–1100. [PubMed] [Google Scholar]
- Poplawski MM, Boyadjieva N, Sarkar DK. Vasoactive intestinal peptide and corticotropin-releasing hormone increase beta-endorphin release and proopiomelanocortin messenger RNA levels in primary cultures of hypothalamic cells: Effects of acute and chronic ethanol treatment. Alcohol Clin Exp Res. 2005;29:648–655. doi: 10.1097/01.alc.0000158834.11252.2e. [DOI] [PubMed] [Google Scholar]
- Rasmussen DD, Bryant CA, Boldt BM, Colasurdo EA, Levin N, Wilkinson CW. Acute alcohol effects on opiomelanocortinergic regulation. Alcohol Clin Exp Res. 1998;22:789–801. [PubMed] [Google Scholar]
- Rennison D, Neal AP, Cami-Kobeci G, Aceto MD, Martinez-Bermejo F, Lewis JW, Husbands SM. Cinnamoyl derivatives of 7alpha-aminomethyl-6,14-endo-ethanotetrahydrothebaine and 7alpha-aminomethyl-6,14-endo-ethanotetrahydrooripavine and related opioid ligands. J Med Chem. 2007;50:5176–5182. doi: 10.1021/jm070255o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruffolo RR. Review: Important concepts of receptor theory. J Auton Pharmacol. 1982;2:277–295. doi: 10.1111/j.1474-8673.1982.tb00520.x. [DOI] [PubMed] [Google Scholar]
- Rush CR. Pretreatment with hydromorphone, a μ-opioid agonist, does not alter the acute behavioral and physiological effects of ethanol in humans. Alcohol Clin Exp Res. 2001;25:9–17. [PubMed] [Google Scholar]
- Sabino V, Cottone P, Steardo L, Schmidhammer H, Zorrilla EP. 14-methoxymetopon, a highly potent mu opioid agonist, biphasically affects ethanol intake in Sardinian alcohol-preferring rats. Psychopharmacology. 2007;192:537–546. doi: 10.1007/s00213-007-0746-7. [DOI] [PubMed] [Google Scholar]
- Schutz J, Dersch CM, Horel R, Spetea M, Koch M, Meditz R, Greiner E, Rothman RB, Schmidhammer H. Synthesis and biological evaluation of 14-alkoxymorphinans. 17. Highly delta opioid receptor selective 14-alkoxy-substituted indolo- and benzofuromorphinans. J Med Chem. 2002;45:5378–5383. doi: 10.1021/jm020940p. [DOI] [PubMed] [Google Scholar]
- Shelton KL, Macenski MJ, Meisch RA. Reinforcing effects of a combination of ethanol and methadone relative to each drug alone. Pharmacol Biochem Behav. 1998;61:367–374. doi: 10.1016/s0091-3057(98)00100-2. [DOI] [PubMed] [Google Scholar]
- Spagnolo B, Calo G, Polgar WE, Jiang F, Olsen CM, Berzetei-Gurske I, Khroyan TV, Husbands SM, Lewis JW, Toll L, Zaveri NT. Activities of mixed NOP and mu-opioid receptor ligands. Br J Pharmacol. 2008;153:609–619. doi: 10.1038/sj.bjp.0707598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spanagel R. The influence of opioid antagonists on the discriminative stimulus effects of ethanol. Pharmacol Biochem Behav. 1996;54:645–649. doi: 10.1016/0091-3057(95)02288-0. [DOI] [PubMed] [Google Scholar]
- Sperling RE, Gomes SM, Sypek EI, Carey AN, McLaughlin JP. Endogenous kappa-opioid mediation of stress-induced potentiation of ethanol-conditioned place preference and self-administration. Psychopharmacology. 2010;210:199–209. doi: 10.1007/s00213-010-1844-5. [DOI] [PubMed] [Google Scholar]
- Walker BM, Koob GF. Pharmacological evidence for a motivational role of kappa-opioid systems in ethanol dependence. Neuropsychopharmacology. 2008;33:643–652. doi: 10.1038/sj.npp.1301438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker BM, Zorrilla EP, Koob GF. Systemic kappa-opioid receptor antagonism by nor-binaltorphimine reduces dependence-induced excessive alcohol self-administration in rats. Addict Biol. 2010 doi: 10.1111/j.1369-1600.2010.00226.x. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter JC. The stimulus properties of morphine and ethanol. Psychopharmacologia. 1975;21:209–214. doi: 10.1007/BF00428896. [DOI] [PubMed] [Google Scholar]
- York JL, Bush R. Studies on the discriminative stimulus properties of ethanol in squirrel monkeys. Psychopharmacology. 1982;77:212–216. doi: 10.1007/BF00464568. [DOI] [PubMed] [Google Scholar]