

Abstract
We have developed an effective pathway for the prediction and characterization of novel transmembrane β-barrel proteins. The Freeman-Wimley algorithm, which is a highly accurate prediction method based on the physicochemical properties of experimentally characterized transmembrane β barrel (TMBB) structures, was used to predict TMBBs in the genome of Salmonella typhimurium LT2. The previously uncharacterized product of gene yshA was tested as a model for validating the algorithm. YshA is a highly conserved 230-residue protein that is predicted to have 10 transmembrane β-strands and an N-terminal signal sequence. All of the physicochemical and spectroscopic properties exhibited by YshA are consistent with the prediction that it is a TMBB. Specifically, recombinant YshA localizes to the outer membrane when expressed in Escherichia coli; YshA has β-sheet-rich secondary structure with stable tertiary contacts in the presence of detergent micelles or when reconstituted into a lipid bilayer; when in a lipid bilayer, YshA forms a membrane-spanning pore with an effective radius of ~0.7 nm. Taken together, these data substantiate the predictions made by the Freeman-Wimley algorithm by showing that YshA is a TMBB protein.
1 INTRODUCTION
The proteins that span the outer membrane of Gram-negative bacteria fold into the structural motif known as the transmembrane β-barrel (TMBB), examples of which are also found in the outer membranes of mitochondria and chloroplasts [1]. The only other membrane-spanning structure is the α-helical bundle, which can have as few as one or more than 20 transmembrane helices, and is found in the cytoplasmic membranes of all forms of life [2]. Although 2–3% of the genes in Gram-negative bacteria encode TMBBs, they represent less than 1% of all solved protein structures from Gram-negative organisms. Because TMBBs, like most membrane proteins, are difficult to crystallize or study otherwise in vitro, many groups have turned to computational structure prediction methods. Membrane-spanning helices can be predicted using a relatively straightforward prediction algorithm that can identify the 19–25 contiguous hydrophobic residues which constitute a single membrane-spanning helix [3–5]. However, developing an accurate algorithm for TMBB prediction has been more elusive due to the cryptic nature of the TMBB structure [6].
The structures of TMBB proteins consist of a contiguous sequence of 8–24 anti-parallel β-strands which meander in and out of the membrane in a cylindrical geometry. This arrangement gives the overall structure a barrel-like appearance with the lumen of the barrel transverse to the surface of the lipid bilayer. The meandering TM β-strands are connected by short turns of about five residues at one opening of the barrel, and loops of varying length at the other opening. Each strand is laterally connected to the next by anti-parallel, inter-strand hydrogen bonds, including the first N-terminal strand and the last C-terminal strand. The 10-residue long TM β-strands typically consist of an alternating pattern of nonpolar and polar amino acids, where the nonpolar side chains are exposed to the acyl chains of the lipid bilayer and the more polar side chains are exposed to the lumen of the barrel. The low information content in the TM segments and high variability in the loops makes the identification of TMBBs by sequence alone very difficult.
The Freeman-Wimley algorithm [7] is a highly accurate TMBB prediction method based on the spatial amino acid composition of structurally characterized TMBBs. The TM domain of a TMBB has two surfaces, the pore-exposed surface (internal) and the lipid-exposed surface (external). This algorithm utilizes information derived from known protein structures to elucidate the evolutionary preference for amino acid side-chains in the native structural environment of each surface. For example, the side chains of nonpolar amino acids such as Trp and Leu are typically oriented toward the external lipid environment, while the side chains of polar amino acids such as Glu and Arg are much more frequently oriented toward the pore of the TMBB. The Freeman-Wimley algorithm uses statistical bioinformatic data to predict the likelihood that a sequence will fold into a TMBB. The prediction accuracy of the algorithm is as high as 99% [7].
The focus of this study was to establish a set of experiments to test the predictions made by the Freeman-Wimley algorithm. There is no standard set of protocols used in the literature for deciding whether a protein is a TMBB or not, but there are some characteristics common to almost all known TMBBs. A TMBB will localize to the outer membrane of a Gram-negative organism, have a β-sheet-rich secondary structure, resist SDS-PAGE-induced unfolding, and insert as a folded protein into a lipid bilayer. Most TMBBs also form a pore across a lipid bilayer.
The gene yshA, which encodes a putative outer membrane protein, was identified in the Salmonella typhimurium genome by the Freeman-Wimley algorithm as a strong candidate, and thus was used as a model protein for characterization. The only reported studies of a YshA homolog were conducted separately by Dartigalongue, et al. and Sardesai, et al. on the Escherichia coli homolog (outer membrane porin L; OmpL), which is 91% identical to YshA of S. typhimurium [8, 9]. Dartigalongue, et al. claimed that OmpL in E. coli was involved in the efflux of an unknown redox reagent processed by the disulfide-bond-modifying protein DsbB. They also showed that OmpL was found in the outer membrane, and that it could be reconstituted into liposomes to form a pore. However, a subsequent study by Sardesai, et al. (2003) was unable to reproduce any of the data supporting the function attributed to OmpL. Although the work by Sardesai, et al. disputed the reported function of OmpL, they neither challenged nor supported the characterization of OmpL as an outer membrane protein. The suspicion cast on the validity of the study conducted by Dartigalongue, et al. suggests that a more probing investigation into the structural characteristics of YshA/OmpL is needed.
In this work, an array of biochemical and biophysical techniques were used to characterize YshA, independent of its unknown biological function. We show that YshA of S. typhimurium LT2 localizes to the outer membrane when expressed in E. coli, has β-sheet-rich secondary structure, makes stable tertiary contacts in the native structure, and can be reconstituted into a lipid bilayer, where insertion depended on the protein being folded. Finally, YshA formed a pore through the bilayer with an effective pore radius consistent with other known TMBBs. These data taken together strongly suggest that YshA is a true TMBB. These findings validate the predictive value of the TMBB prediction algorithm developed in this lab, and suggest that high-throughput structural and proteomic characterization of transmembrane β barrels is feasible.
2 MATERIALS AND METHODS
2.1 Reagents
A frozen culture of Salmonella typhimurium LT2 was a generous gift from Dr. Lucia Freytag. Rabbit anti-LamB polyclonal antibody was a generous gift from Dr. Thomas Silhavy. Endonucleases (NdeI and XhoI) were obtained from New England Biolabs, Ipswich, MA. Culturing media, Teknova Brilliant Blue R-250 and most chemicals were obtained from Fisher Scientific, Hercules, CA. All polyethylene glycols, lauroyl sarcosine (LS), kanamycin, dithiothreitol (DTT), 4-chloro-1-naphthol (4CN), and goat anti-rabbit immunoglobulin G (IgG) were obtained from Sigma-Aldrich, St. Louis, MO. Rabbit anti-YshA antisera were produced by Covance, Denver, PA. Streptavidin-bound horseradish peroxidase (SHRP) was obtained from Jackson ImmunoResearch, West Grove, PA. PCR master mix with Taq polymerase and T4 Ligase were obtained from Promega, Madison, WI. QIAPrep Miniprep and QIAQuick gel extraction kits were obtained from QIAGEN, Valencia, CA. Site-directed mutagenesis Quik-Change kit was obtained from Stratagene, La Jolla, CA. All oligonucleotides were synthesized by Integrated DNA technologies, Coralville, IA. Proteinase K, all 4–12% BisTris polyacrylamide gels, nitrocellulose membranes, molecular weight standards, electrophoresis materials, buffers and apparatus were obtained from Invitrogen, Carlsbad, CA. Detergents tetraoxyethylene monooctyl ether (C8E4) and n-dodecyl-β-D-maltopyranoside (DDM) were obtained from Anatrace, Maumee, OH. Isopropyl-β-D-1-thiogalactopyranoside (IPTG) was obtained from Inalco Spa, Milano, Italy. Palmitoyl-oleoyl-phosphatidyl-choline (POPC), palmitoyl-oleoyl-phosphatidyl-glycerol (POPG), and lissamine rhodamine B-palmitoyl-oleoyl-phosphatidyl-ethanolamine (RhoPE) were obtained from Avanti Polar Lipids, Alabaster, AL.
2.2 Cloning and mutagenesis of gene yshA
The cells from a 3 mL culture of S. typhimurium LT2 grown to OD600 = 1 were harvested and lysed using the QIAPrep Miniprep kit alkaline cell lysis protocol. Proteinase K was added to the cell lysate to a final concentration of 0.1 mg/ml. The cell lysate was centrifuged to pellet cellular debris and the genomic DNA was extracted from the aqueous phase by the addition of phenol: chloroform: isoamyl alcohol (25:24:1) solution and then washed with chloroform: isoamyl alcohol (24:1) solution. All washing took place in the phase-lock gel tubes supplied with the QIAPrep Miniprep kit which allowed the liquid to be removed by centrifugation and the DNA to remain in the upper compartment. After two washes, the genomic DNA was precipitated from the extract by the addition of 3 M sodium acetate and pure ethanol, and the solvent was evaporated at room temperature. The DNA was washed twice with 70% ethanol and dried at room temperature again. The DNA was subsequently re-dissolved in a standard Tris-EDTA (ethylenediamine tetraacetic acid), pH 8.0.
Gene yshA, which encodes a putative outer membrane, was cloned from the isolated genomic DNA of Salmonella typhimurium LT2 by PCR using PCR Master Mix with Taq polymerase and the following oligonucleotide primers: forward primer 5′-GCC CCT GTA GCC CCG CAT ATG AAA TCT CTG AAT C-3′ and reverse primer 5′-CCA GTC CCC ATT AGC CCT CGA GTC AGA AGA AAT ACT TCG-3′. Gene yshA was inserted into the multiple-cloning site of pET24b between the NdeI and XhoI sites using the standard techniques of digesting the amplicon and the vector with the aforementioned endonucleases for 1 h at 37 °C, subsequently purifying the products by agarose gel electrophoresis and extraction using the QIAQuick gel extraction kit, and finally ligating the purified amplicon and vector with T4 ligase overnight at 14 °C following the manufacturers’ protocols. The resulting vector was named pTF01. The plasmid was purified from the ligase-reaction mixture using the QIAPrep Miniprep kit, amplified by transformation and culturing in E. coli, followed by harvesting via Miniprep kit, and sequencing to verify successful cloning.
The sequence encoding the N-terminal export signal peptide, a domain typically absent in mature outer membrane proteins, was deleted from the gene so that the signal peptide could not interfere with protein refolding. The signal peptide was predicted using the SignalP server (http://www.cbs.dtu.dk/services/SignalP/) which uses neural networks and Hidden Markov Models to predict an export signal peptide and the position at which a signal peptidase will cleave the signal peptide from the protein precursor. The signal peptide of YshA was predicted to be cleaved C-terminal to Ala20. The plasmid vector carrying the cloned yshA gene, pTF01, was subjected to site-directed mutagenesis to delete bases 4–60. These bases encoded amino acid residues 2-20, and their deletion resulted in the plasmid pTF01Δ2-20. Site-directed mutagenesis was performed using a Quik-Change kit, following the manufacturer’s protocols, using the following oligonucleotide primers: forward primer 5′-CTC TAG AAA TAA TTT TGT TTA ACT TTA AGA AGG AGA TAT ACA TAT GGG CGC TTA TGT AGA AAA CCG TGA GGC CTA-3′ and the antisense reverse primer 5′-TAG GCC TCA CGG TTT TCT ACA TAA GCG CCC ATA TGT ATA TCT CCT TCT TAA AGT TAA ACA AAA TTA TTT CTA GAG-3′. Plasmids were transformed into competent E. coli DH5αF′ using standard techniques. The cells were plated on LB agar supplemented with 30 μg/mL of kanamycin for selective growth. Individual colonies were then selected and cultivated in LB broth supplemented with 30 μg/mL kanamycin overnight (approx. 14 h) at 37°C. The cells were pelleted by centrifugation, and the plasmids were harvested using the QIAPrep Miniprep kit.
2.3 Protein expression
The plasmids pTF01, pTF01Δ2-20, and pET24a (empty vector), were transformed into competent E. coli BL21(DE3) using standard techniques and grown in 1 L of LB broth with 30 μg/mL kanamycin at 37 °C until they reached an OD600 = 0.6. At this optical density protein expression was induced by the addition of 1 mM (final concentration) IPTG for 3 h. There were instances where IPTG was not added to cells transformed with either pTF01 or pET24a, and in these cases the cultures were grown to an OD600 = 1.0 before harvesting. The protein was expressed even in the absence of IPTG in this bacterial expression strain because it lacks control over basal levels of transcription and subsequent expression; this was desirable in order to more easily distinguish membrane-bound protein from inclusion body proteins. All cells were harvested by centrifugation at 5000 rpm for 20 min. at 4°C in a Sorvall RC-3C Plus centrifuge using a H6000A rotor. The supernatant was removed, and the pellets were resuspended in 10 mL of 50 mM sodium phosphate buffer, pH 7.6. These suspensions were then lysed via two passes through a cold French Pressure Cell. Lysates of cultures that were induced to express protein were processed for isolation of protein from the inclusion bodies, and the lysates of cultures that were not induced were processed for isolation of protein from membrane fractions.
2.4 Antibody production
To produce anti-YshA polyclonal antibodies, E. coli BL21 (DE3) transformed with pTF01 was induced to overexpress YshA, and the cells were harvested, and lysed as described above. To the protein denatured in 8 M urea was added an equal volume of 2X LDS loading dye with 0.1 M DTT, which was then boiled for 10 min. The sample was loaded onto a one-well preparative polyacrylamide gel (8×8 cm) and electrophoresed for 30 min at 200V. The protein bands were detected by Coomassie staining. The band corresponding to the approximate MW of YshA (27 kDa) was cut from the gel. The protein in the gel slice was electroeluted from the slice using the S&S Elutrap starter kit 46170 (Schleicher & Schuell) for 16 h following the manufacturer’s protocol. Approximately 2 mg of the purified protein was lyophilized and sent to Covance Research Products, Inc., where polyclonal antibodies were produced in NZW (New Zealand White) rabbits.
2.5 Subcellular fractionation
The cytosol, inner membrane, and outer membrane fractions were separated to identify where YshA localizes when expressed in vivo. The cellular lysates of the cultures transformed with pET24a and pTF01 grown as described above (not induced) were centrifuged at 10,000 × g at 4°C for 12 min to pellet the unbroken cells and inclusion bodies. The supernatant was then removed and centrifuged at 100,000 × g for 2 h at 4°C to obtain the cellular membranes. The total membrane pellet was resuspended by sonication and incubated with 25 mM LS (lauroyl sarcosine), 10 mM sodium phosphate buffer, pH 7.6 at room temperature for 45 min and then centrifuged at 100,000 × g to obtain the outer membrane pellet. The outer membrane pellet was resuspended in 150 mM C8E4, 50 mM sodium phosphate, pH 7.6 and stirred gently at room temperature for 1.5 h. The outer membrane extracts were then centrifuged at 100,000 × g for 1 h to remove detergent-insoluble material.
2.6 SDS-PAGE and western blot analysis
Sodium dodecylsulfate polyacrylamide gel electrophoresis (SDS-PAGE) was performed using either 10-well or 15-well PAGs. The samples were mixed 1:1 with 2x LDS loading dye with DTT and either boiled or not boiled (for electrophoretic mobility shift assay) before loading. Gels (8×8 cm) were electrophoresed either at 200 V (single gel) or 175 V (dual gels) for 30 to 40 min. Gels were stained with Teknova Brilliant Blue R-250 and destained with 40% methanol, 7% acetic acid. For western blots, dual gels were electrophoresed and one was transferred to nitrocellulose membranes using the Invitrogen Mini-Blot transfer module following the manufacturer’s protocol. The dried membranes were rewet with PBS with 0.5% TWEEN-20 (PBS-T) for 5 min. Next, the membranes were incubated in blocking buffer (5% powdered milk, PBS-T) for 30 min. Nitrocellulose membranes were then covered in blocking buffer containing either anti-LamB polyclonal antibodies diluted 1:750 or anti-YshA polyclonal antisera diluted 1:15,000 for 1 h followed by multiple rinses with PBS-T. Then the membranes were incubated with goat anti-rabbit IgG diluted 1:500 in blocking buffer for 1 h followed by rinsing. Next, the membranes were incubated in blocking buffer with 2 μg/ml SHRP for 1 h, and then rinsed. Finally, antibody-protein complexes were detected by immersing the membranes in 30% methanol, 0.03% hydrogen peroxide, and 0.6 mg/ml 4CN in PBS for less than 1 min, followed by rinsing with distilled deionized water.
2.7 Purification of YshA from inclusion bodies
The expression of the signal peptide deletion mutant, YshAΔ2-20, was induced by IPTG. Because the protein lacked a signal peptide and was so abundantly expressed, all of it was deposited into inclusion bodies. The cultures were grown, harvested, and lysed as described above. The cellular lysate was centrifuged at 12,500 × g for 30 min. at 4 °C to pellet inclusion bodies and other cellular debris. The supernatant was removed and the pellet was resuspended in 10 ml of 50 mM sodium phosphate, 25 mM LS, 1% (W/V) Triton X-100, pH 7.6 and rocked at room temperature for 1 hour. The detergent-insoluble material was pelleted and washed as above three more times. After the final detergent wash the pellet was washed twice with 50 mM sodium phosphate buffer, pH 7.6. The pellet was denatured with either 8 M urea or 250 mM SDS in 50 mM sodium phosphate buffer, pH 7.6 at room temperature by brief sonication, and then centrifuged at 100,000 × g for 1h at 4 °C to remove any remaining insoluble material.
The crude YshAΔ2-20 preparation from inclusion bodies was subjected to RP-HPLC using a Waters 600e controller and pump with Rheodyne 7725i injector. Various volumes of the crude prep (1–2 mL) were injected into a 2 ml sample loop. The proteins were resolved on either a VP 250×10 mm Nucleosil 100-7 C2 column (Macherey-Nagel) with a Trident high pressure inline filter (Restek) using a flow rate of 3 ml/min at ambient temperature or a Dynamax 250×10 mm Microsorb 300-5 C4 column (Varian) under the same conditions. The following solvents were used for the mobile phase 0.1% trifluoracetic acid (TFA) (solvent A), 0.1% TFA in acetonitrile (solvent B) and 0.1% TFA, 8 M urea (solvent C). The C2 column was equilibrated with 30% B, 70% solvent C for 5 min, followed by 70% solvent A, 30% solvent B for 10 min. Next, solvent B was linearly increased from 30% to 100% over 30 min. Then 100% solvent B flowed through the column for 5 min, followed by a linear decrease of solvent B to 30% (70% solvent A) over 5 min. The column was reequilibrated with 30% solvent B, 70% solvent C. Protein peaks were detected with a Waters 2487 Dual Absorbance Detector at 295 nm (to measure absorbance of tryptophan) and 220 nm (to measure absorbance of peptide backbone).
2.8 Protein folding
YshAΔ2-20 was refolded using a modified version of a two-step folding technique previously used to fold a mitochondrial porin [18]. HPLC-purified YshAΔ2-20 was divided into approximately 1-mg aliquots prior to dehydration under vacuum. YshAΔ2-20 was rehydrated overnight in 300 μL of 250 mM SDS, 50 mM sodium phosphate, at pH 7.6. The protein solution was diluted with phosphate buffer until the SDS concentration was 112 mM and then concentrated to a volume of 0.25 mL using a Microcon YM-10 centrifugal filter unit (Millipore; Billerica, MA). To SDS-solubilized YshAΔ2-20 was added 122.5 mg powdered DDM, followed by dilution with 0.75 mL of 50 mM sodium phosphate and rotated end-over-end overnight at room temperature. The final concentration of stock YshAΔ2-20 in SDS/DDM was 40 μM YshAΔ2-20 in 28 mM SDS, 240 mM DDM, and 50 mM phosphate buffer pH 7.6. This stock was diluted appropriately for various experiments.
2.9 Liposome and proteoliposome preparation
Liposomes and proteoliposomes containing 90% POPC and 10% POPG (unlabeled samples) and 89.5% POPC, 10% POPG and 0.5% RhoPE (fluorescent-labeled samples) were prepared as follows. Lipid films were prepared by mixing chloroform-dissolved lipids in a glass vessel and evaporating the chloroform under a stream of gaseous N2. The lipid films were further dried under vacuum overnight. Lipid films were rehydrated in 50 mM sodium phosphate, pH 7.6 for 10–15 min by intermittent vortexing. The resuspended lipids were then subjected to 10 freeze-thaw cycles (freezing in a dry ice/ethanol bath for 3–4 min. and thawing in a ~60 °C water bath) to make multi-lamellar vesicles (MLVs). The MLVs were passed through a Lipidex extruder with successive membrane pore sizes of 0.4, 0.2 and finally 0.1 μm 10–20 times to make large unilamellar vesicles (LUVs).
Proteoliposomes were prepared by mixing 5 μM refolded YshAΔ2-20 prepared as described above and LUVs containing ~2.5 mM total lipid. The detergent was removed by adsorption onto BioBeads, a hydrophobic polystyrene resin, which allowed the lipids to spontaneously aggregate and reform liposomes [22]. BioBeads were measured (1 g BioBeads/5 mL solution) and poured into a disposable 10 mL syringe with a filter needle threaded onto the Luer-Lok fitting. The beads were washed three times with full syringe volumes of methanol and five times with phosphate buffer. The protein/LUV solution was aspirated into the syringe slowly to avoid making bubbles, then rotated at room temperature for 1.5–2 h, and then ejected slowly. Proteoliposomes were then freeze-thawed as above, except thawing was performed in a water bath between 25 and 37 °C. Proteoliposomes were then extruded as above.
2.10 Circular Dichroism
Circular dichroism (CD) measurements were performed with a JASCO J-810 spectropolarimeter with a Pelltier-type thermal controller. Measurements were taken in the range of 185 nm to 250 nm for far-UV measurements and 250–350 for near-UV measurements. The samples were measured in either 0.1 or 1 cm cuvettes. The CD (ε; in mdeg) measured for each protein sample was converted to mean residue ellipticity (θ; deg*cm2*dmol−1) using the following equation:
Where NA is the number of amino acids in the protein, [S] is the molar concentration of the protein in the sample, and l is the path length of the cuvette in cm.
2.11 Liposome flotation assay
Proteoliposomes were prepared with fluorescent-labeled LUVs as described above. Total lipid concentration was 5 mM, and YshAΔ2-20 was at a final concentration of 5 μM. Liposomes were prepared to the same lipid concentrations without protein, and detergent-refolded YshAΔ2-20 was diluted to 5 μM for control experiments. The assay was performed as described by Hong and Tamm with some modifications [23]. A sucrose step gradient was prepared from bottom to top by carefully layering 30 μL of 30% sucrose, 40 μL each of 25%, 20%, and finally 10% sucrose in an Ultra Clear 8×20 mm centrifuge tube (Beckman). Sucrose solutions were made in either 50 mM sodium phosphate, pH 7.6 (non-denaturing sucrose) or 6 M urea (denaturing sucrose). Liposomes, detergent-solubilized YshAΔ2-20, and proteoliposomes were each mixed 1:1 with 60% either non-denaturing or denaturing sucrose. Samples applied to the denaturing sucrose gradients were also mixed with solid urea to a final concentration of 6 M and heated in a 95 °C water bath for 5–10 min to ensure complete protein denaturation. All samples were left on the bench to equilibrate at room temperature for at least 30 min, and then 50 μL of each sample was carefully pipetted at the bottom of a given gradient. The samples were then centrifuged for 1 h at approximately 100,000 × g in an Airfuge table-top centrifuge equipped with an A-95 rotor (Beckman) set to an air pressure of 30 psi. After centrifugation, 34 μL aliquots were taken from bottom to top and mixed 1:1 with sodium phosphate buffer. The aliquots were then transferred to a black 96-well plate (Corning) and fluorescence measurements were taken in a Synergy 2 (Biotek) fluorescent plate reader. Lissamine rhodamine fluorescence emission was measured at 590 nm after excitation at 540 nm. The tryptophan fluorescence emission of YshAΔ2-20 was measured at 360 nm after excitation at 284 nm.
2.12 Liposome swelling assay
Liposomes and proteoliposomes were prepared as above with liposome compositions of 90% POPC and 10 % POPG. For this assay 1 mM PEG 8000 was entrapped in the liposomes by adding it prior to the freeze-thaw cycles of the proteoliposome preparation. Total lipid concentration was 4 mM, and the final YshAΔ2-20 concentration was 2.5 μM. The samples were mixed in a 1 cm quartz cuvette with 1 mM of either arabinose, PEG 200, PEG 400, or PEG 600, and absorbance at 440 nm was measured in a Cary 50 UV-Vis spectrophotometer (Varian) using the kinetic measurement mode for at least 8 min. At the end of each measurement the pore-forming peptide melittin was added to a final concentration of 5 μM and the sample’s absorbance was measured again over the same time period.
3 RESULTS
3.1 Sequence analysis of YshA
The genome of Salmonella typhimurium LT2 includes 4426 genes. Nearly 100 genes (2.2%) are predicted to encode transmembrane β-barrels (TMBBs) by the Freeman-Wimley (F-W) algorithm. Approximately 70% of the predicted TMBBs are annotated as predicted, putative, or hypothetical. In order to empirically assess the prediction accuracy of the algorithm, a conserved, predicted outer membrane protein was characterized. The product of gene yshA from S. typhimurium was chosen for this study. The prediction data for YshA and other homologous sequences are listed in Table 1. The sequence is 230 residues long, and the predicted topology of YshA is a 10-stranded barrel with a very long loop between R63 and A104 as shown in Fig. 1A. The predicted transmembrane β-strands are indicated in the topology prediction plot and the protein sequence as shown in Fig. 1B.
Table 1.
YshA homologs
| Organism | GIa | Name | %IDb | SPc | βBScore (MRS Passed)d | Length | C-terminuse |
|---|---|---|---|---|---|---|---|
| S. typhimurium LT2 | 16767281 | putative outer membrane protein | 100 | Y | 185 (Y) | 230 | QIRIGAKYFF |
| S. enterica serovar Paratyphi B str. SPB7 | 161617134 | putative outer membrane protein | 100 | Y | 183 (Y) | 250 | QIRIGAKYFF |
| Citrobacter koseri ATCC BAA-895 | 157147346 | outer membrane porin L | 94 | Y | 196 (Y) | 243 | QIRVGVKYFF |
| Escherichia albertii TW07627 | 170769692 | outer membrane porin | 93 | Y | 185 (Y) | 230 | QIRIGAKYFF |
| E. coli IAI39 | 218701425 | outer membrane porin L | 91 | Y | 182 (Y) | 230 | QIRIGTKYFF |
| E. coli O157:H7 str. EC4024 | 195937513 | outer membrane porin L | 90 | Y | 178 (Y) | 252 | QIRIGTKYFF |
| Citrobacter youngae ATCC 29220 | 229220568 | hypothetical protein CIT292_05286 | 88 | Y | 145 (Y) | 242 | QIRVGAKYFF |
| Shigella sp. D9 | 256021514 | outer membrane porin L | 88 | Y | 169 (Y) | 230 | QIRIGTKYFF |
| E. coli 101-1 | 194438348 | outer membrane porin | 88 | Y | 174 (Y) | 230 | QIRIGTKYFF |
| Cronobacter turicensis | 260599974 | Porin ompL | 64 | Y | 147 (Y) | 228 | RLRLGLRYSF |
| Erwinia chrysanthemi | 16075345 | KdgMf | 27 | Y | 187 (Y) | 236 | RYRVGVQYSF |
GI number assigned to each sequence in the NCBI database.
The percentage of each sequence that is identical to the YshA of S. typhimurium LT2 over the aligned region using BLAST [43]
Whether or not the protein is predicted to have a signal peptide using the HMM prediction method of the SignalP Server [11]
The β-barrel score of the sequence using the Freeman-Wimley algorithm (whether or not the sequence passed the Mean Randomized Score test) [7]
Last 10 residues of the C-terminus which signal the Omp85-like chaperones to properly orient membrane proteins in the outer membrane [44]
oligogalacturonate specific porin
Fig. 1.
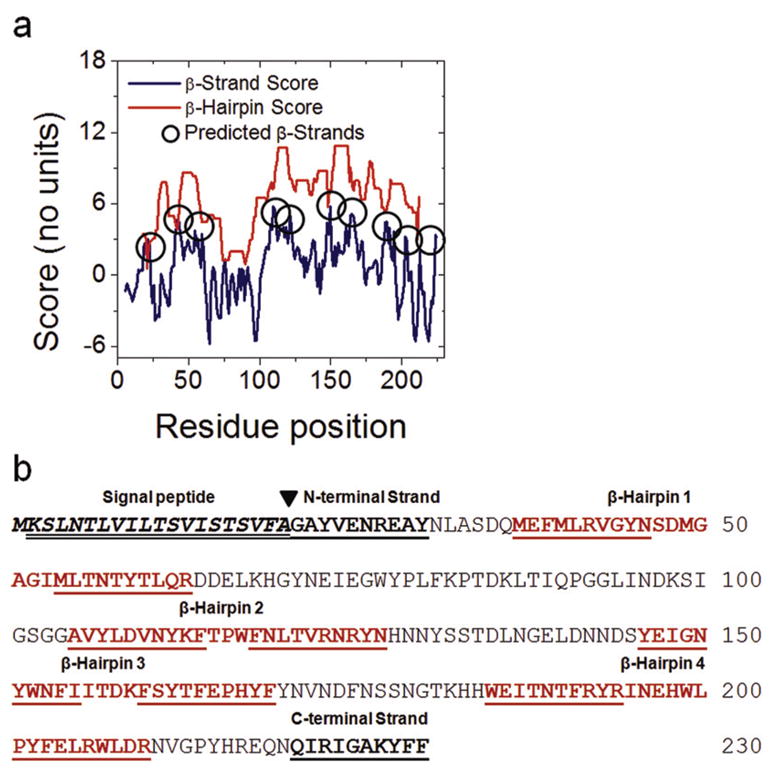
Structural prediction of YshA. A) Topology prediction of YshA was performed with the Freeman-Wimley algorithm [7]. The peaks in the β-strand score which indicate the centers of predicted β-strands are circled. B) The sequence of YshA annotated with predicted β-hairpins, predicted N-terminal export signal peptide (predicted by the SignalP server [11]) and signal peptide cleavage site (black triangle). The single underlined residues indicate predicted TM β-strands. The double underlined residues were removed to make the signal peptide deletion mutant, YshAΔ2-20.
The β-barrel score, which is a numerical estimate of the probability that a sequence folds into a TMBB, for YshA is 185; the median score of known TMBBs in the non-redundant protein database (NRPDB) compiled by this lab [7] is 168. A secondary analysis of the β-barrel score called the Mean Randomized Score (MRS) test was performed. In the MRS test the sequence is randomized 1000 times and the β-barrel scores for the random sequences are averaged together. If the overall amino acid composition of the protein is conducive to a high β-barrel score, regardless of the sequence, then the MRS is unlikely to be significantly less than the β-barrel score determined for the native sequence (p > 0.05). In a non-redundant protein database including sequences of known structures, over 80% of the known TMBBs pass the MRS test while less than 30% of non-TMBB proteins with high β-barrel scores (false positive predictions) pass the MRS test [7]. The strength of the β-barrel-score-based prediction for YshA is greatly enhanced because the MRS test p-value = 0.0001. All of the YshA homologs listed in Table 1 pass the MRS test with p-values less than 0.005.
Almost all TMBB-encoding genes have an N-terminal export signal peptide encoded within the first 30 residues of the gene product [10]. The first 20 residues of YshA were predicted with high confidence to encode an export signal peptide by the SignalP server [11] as indicated in Fig. 1B, which further supports the prediction that YshA is a TMBB (also see Fig. S1 for full genomic analysis of S. typhimurium). Since signal peptides are typically cleaved from the precursor by a signal peptidase during translocation into the periplasm, a signal peptide deletion mutant was generated based on the prediction data. Wild Type (WT) YshA protein was expressed in E. coli for the determination of subcellular localization, while the signal peptide deletion mutant, YshAΔ2-20, was expressed for in vitro characterization of the native-like protein.
3.2 Subcellular localization of YshA
The subcellular location of WT YshA was identified by expressing the protein in E. coli, followed by cellular fractionation, and immunodetection. After culturing, the cells were harvested and lysed; the soluble cytosolic proteins were separated from the insoluble material (membranes and other debris) by centrifugation. Inner membranes were selectively dissolved by the anionic detergent lauroyl sarcosine (LS); the outer membranes were then dissolved with the nonionic detergent C8E4 [12]. Each subcellular fraction was analyzed via SDS-PAGE followed by western blotting using polyclonal antibodies to detect WT YshA as well as the abundantly expressed outer membrane maltose porin, LamB (Fig. 2) [13]. The blots show that WT YshA and LamB were nearly absent from the cytosolic and inner-membrane fractions, while both proteins were enriched in the outer-membrane fractions. Moreover, YshA was only detected when the culture was transfected with a plasmid containing the cloned gene, indicating that the E. coli homolog is not being expressed. These results strongly suggest that YshA is an outer-membrane protein.
Fig. 2.
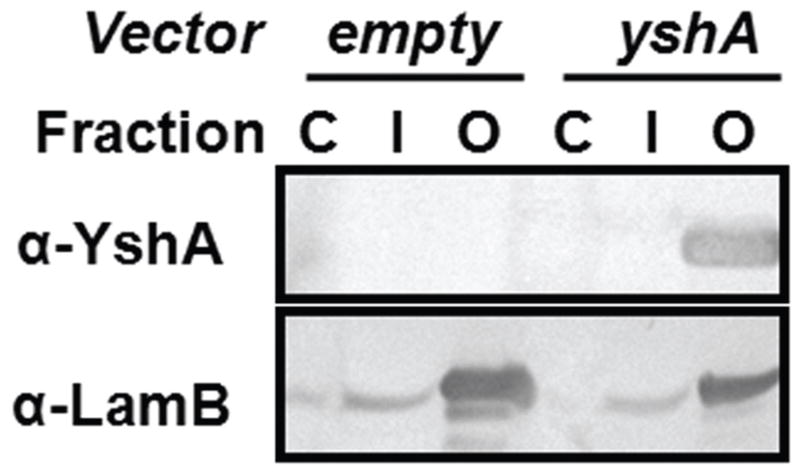
Subcellular localization of YshA. Western blots of subcellular fractions from E. coli transformed with either an empty vector (pet24a) or a vector carrying the WT yshA gene. The cytosol (C), the inner membrane (I), and the outer membrane (O). Antibodies specific to YshA showed that YshA localized to the outer membrane. The maltoporin, LamB, was also identified in the outer membrane fraction.
3.3 Folding and structural characterization of YshAΔ2-20
YshAΔ2-20 was over expressed into inclusion bodies and purified via RP-HPLC, which was expected to yield a denatured protein. The technique used to identify natively refolded YshAΔ2-20 was the electrophoretic mobility shift assay (EMSA), which is based on the observation that many TMBBs have SDS-resistant tertiary structure at room temperature, and thus migrate differently in the natively folded form compared to the unfolded form [14–17]. The SDS-PAGE mobility of WT YshA (from the outer membrane extract), was assessed with and without boiling in order to determine if there was an observable difference between native and unfolded YshA (Fig. 3A). WT YshA, found in the outer membrane, was expected to be natively folded since the protein extraction conditions are known to preserve the native structure of TMBBs [12]. The apparent molecular weight (MW) of native (not boiled) YshA was about 5 kDa smaller than denatured (boiled) YshA which had an apparent MW of 25 kDa, consistent with the theoretical mass. This showed that natively folded YshA can be detected via EMSA, and thus successful refolding of YshAΔ2-20 can be detected using this assay.
Fig. 3.

Electrophoretic mobility shift of folded YshA. A) Outer membrane fraction of E. coli that expressed WT YshA either boiled or not. The Coomassie-stained gel (C) shows total protein and the western blot (W) specifically shows YshA. B) Purified signal peptide deletion mutant YshAΔ2-20 in either SDS or SDS/DDM solution, either boiled or not.
The EMSA was used to test numerous conditions for refolding YshAΔ2-20. Ultimately, a mixed-micelle system of SDS and dodecylmaltoside (DDM) in a concentration ratio of 7:60 was found to efficiently refold YshAΔ2-20 into a native-like conformation as shown in Fig. 3B. YshAΔ2-20 in SDS alone always remained unfolded. However, when YshAΔ2-20 was in SDS/DDM and not boiled, the mobility increased substantially, indicative of native-like structure; heat-denatured YshAΔ2-20 has an apparent MW of 25 kDa regardless of sample history. Though the folding efficiency appears to be less than 100%, lowering the temperature of the apparatus (i.e. running in a cold room) reduces the intensity of the unfolded protein band, which suggests that folding efficiency could be underestimated if measured by EMSA due to heat generated during electrophoresis. Nonetheless, these results suggest that SDS/DDM folded YshAΔ2-20 into a heat-labile, native-like structure.
In order to verify that YshAΔ2-20 is making stable tertiary contacts, the tertiary order was characterized by circular dichroism (CD) spectroscopy in the near-UV range (250–350 nm). When aromatic residues, especially tyrosine and tryptophan, are ordered because of the formation of stable tertiary contacts, these residues impart ellipticity on the sample which is observed at 275 and 283 nm, respectively [18–20]. Fig. 4 shows the near-UV CD spectra of YshAΔ2-20 in either SDS or SDS/DDM. YshAΔ2-20 in SDS alone showed little ellipticity in the near-UV range of wavelengths regardless of temperature, which suggests that there was no significant tertiary order. However, YshAΔ2-20 in SDS/DDM had negative ellipticity between 275 and 285 nm which suggests a spatial ordering of tyrosine and tryptophan residues. The ellipticity of YshAΔ2-20 in SDS/DDM was lost when heated to 95 °C, and was similar to the spectrum of YshAΔ2-20 in SDS at the same temperature. Upon cooling to 20 °C, the ellipticity of YshAΔ2-20 in SDS/DDM recovered to more than half of the initial value, which suggests that at least some of the protein regains native structure after heat denaturation and rapid cooling. These data support the result from the EMSA shown in Fig. 3 and together strongly suggest that YshAΔ2-20 folds into a native-like tertiary structure in SDS/DDM.
Fig. 4.

Near-UV CD of YshA. The CD of YshAΔ2-20 in either SDS or SDS/DDM was measured at the indicated temperatures (where 95-20° C was heated and then cooled backed down) from 250 to 350 nm. YshAΔ2-20 has a distinct, heat-labile, tertiary order in SDS/DDM at 20° C.
The secondary structure of YshAΔ2-20 was characterized by CD in the far-UV range (185–265 nm). The ellipticity of YshAΔ2-20 was measured in SDS alone at 20°C and the spectrum was consistent with an α-helix-rich secondary structure, indicated by minima at 208 nm and 222 nm as shown in Fig. 5 [21]. When the sample was either heated to 95 °C, or cooled back to 20 °C, little change from the initial CD spectrum was observed. The same temperature treatment was performed on YshAΔ2-20 in SDS/DDM. The CD spectrum at 20 °C showed a single negative minimum at 215 nm and became positive at about 205 nm with a maximum at 200 nm, which indicates that the dominant secondary structure of YshAΔ2-20 in SDS/DDM is anti-parallel β-sheet [21]. Upon heating to 95 °C the ellipticity of YshAΔ2-20 in SDS/DDM closely resembled that of YshAΔ2-20 in SDS at the same temperature, i.e. minima at 208 and 222 nm, suggesting that α-helix is the dominant secondary structure of the unfolded protein in detergent. After YshAΔ2-20 in SDS/DDM was cooled back down to 20 °C the ellipticity showed that the protein reformed a β-sheet. These data suggest that natively folded YshAΔ2-20 is a β-sheet-rich protein, which is a defining structural characteristic of TMBBs.
Fig. 5.

Far-UV CD of YshA. The CD of YshAΔ2-20 in either SDS or SDS/DDM was measured at the indicated temperatures (where 95-20° C was heated and then cooled backed down) from 185 to 265 nm. YshAΔ2-20 has a β-sheet-rich secondary structure in SDS/DDM at 20° C.
3.4 Lipid insertion and structural analysis
After showing that YshAΔ2-20 folds into a native-like structure in detergent, the structure-dependent reconstitution of YshAΔ2-20 into a lipid bilayer was tested. A preparation of large unilamellar vesicles (LUVs) was mixed with detergent-refolded YshAΔ2-20 as described in methods. The detergent was in large molar excess so that it dissolved the LUVs. For reconstitution into proteoliposomes, the detergent was removed via adsorption with polystyrene BioBeads [22]. After detergent removal, the bilayers had formed and the sample was assayed for a physical association between lipids and protein using the liposome flotation assay (Fig. 6). Sucrose density gradient ultracentrifugation was used to separate protein and lipid mixtures which are not physically associated [23]. The gradient was separated into six fractions after ultracentrifugation, and each fraction was assayed for the presence of liposomes or YshAΔ2-20. The liposomes were detected by the fluorescence of the rhodamine-labeled lipids and YshAΔ2-20 was detected by tryptophan fluorescence. Denaturing and non-denaturing gradients (i.e., either containing 6M urea or not) were used to explore the importance of protein folding on lipid insertion. In the absence of lipids, YshAΔ2-20 was found exclusively in the bottom (highest density) fraction regardless of the presence of denaturant (Fig. 6A–B). In the absence of protein, liposomes always float to the top (lowest density) fraction regardless of the presence of denaturant (Fig. 6A–B). In the proteoliposome sample, under denaturing conditions, YshAΔ2-20 was found in the high-density fraction (bottom) while the liposomes were found in the low-density fraction (top) as shown in Fig. 6C. In the non-denaturing gradient, liposomes and YshAΔ2-20 were found together in the middle fractions (three and four) of the gradient (Fig. 6D). The conclusion drawn from these data is that YshAΔ2-20 inserts stably into lipid bilayers, and that insertion is dependent on the protein being folded. The folding and insertion appears to be greater than 95% efficient under these conditions as there was no protein found in the absence of lipids in the non-denatured sample.
Fig. 6.

Liposome flotation assay. Samples of YshAΔ2-20 and/or liposomes were prepared as described in methods and subjected to ultracentrifugation in a sucrose density gradient to determine if YshAΔ2-20 could be inserted into a lipid bilayer. The control samples containing either YshAΔ2-20 in the absence of liposomes or vice-versa were centrifuged under either denaturing conditions (A), or non-denaturing conditions (B). The experimental samples where YshAΔ2-20 and liposomes were prepared together were also centrifuged under denaturing (C) or non-denaturing (D) conditions. YshAΔ2-20 and liposomes are found in the same fractions only under non-denaturing conditions when prepared together.
Next, the structure of lipid-embedded YshAΔ2-20 was characterized. The EMSA was used to determine if lipid-embedded YshAΔ2-20 had a stable tertiary structure, and far-UV CD spectroscopy was used to identify the dominant secondary structure. Fig. 7A shows a comparison of the mobility shifts for YshAΔ2-20 in either SDS/DDM or embedded in a bilayer. Both samples showed identical mobility shifts through SDS-PAGE of approximately -10 kDa. This shows that YshAΔ2-20 maintains the same native-like structure in a bilayer as in SDS/DDM. The CD spectra in Fig. 7B show that lipid-embedded YshAΔ2-20 has a virtually identical secondary structure to YshAΔ2-20 in mixed detergent micelles, which is β-sheet rich. These results confirm that the secondary and tertiary structural characteristics of YshAΔ2-20 were preserved in the transition from a mixed-detergent micelle environment to a lipid bilayer environment. The structural motif of natively folded YshAΔ2-20 is very likely a transmembrane β-sheet.
Fig. 7.

Structural analysis of lipid-embedded YshAΔ2-20. A) Coomassie-stained gel showing that bilayer-embedded YshAΔ2-20 has a similar migration shift to YshAΔ2-20 folded in SDS/DDM solution. B) The CD was measured for bilayer-embedded YshAΔ2-20 from 190 to 250 nm. The secondary structure of YshAΔ2-20 in a bilayer environment is similar to YshAΔ2-20 in SDS/DDM, but not YshAΔ2-20 in SDS alone.
3.5 Pore formation of YshAΔ2-20
Another common characteristic of most TMBBs is that they facilitate the passive diffusion of nonelectrolyte solutes across a lipid bilayer [24, 25]. The liposome swelling assay was used to measure the effective radius of the pore, if quantifiable, formed by YshA. In this assay, liposomes with a high molecular weight solute entrapped inside are mixed with an isotonic solution of a lower molecular weight solute. In the absence of a pore, there is no significant change to the liposome size. However, if there is a pore in the bilayer of the liposome large enough to accommodate the solute, then the smaller solute will diffuse into the liposome. This diffusion creates a concentration gradient, which then drives water into the liposome, thereby increasing the volume of the liposome. Increases in the liposome size are indirectly measurable by UV-Vis spectrometry because the optical density (OD) of a liposome suspension decreases with increasing liposome size.
Liposomes or proteoliposomes with YshAΔ2-20 were prepared using the freeze-thaw/extrusion method and had polyethylene glycol MW 8000 (PEG 8000) entrapped in the interior. Either liposomes or proteoliposomes were added to isotonic solutions of arabinose (MW 150.13), PEG 400, and PEG 600 with hydrodynamic radii of 0.38, 0.7 and 0.8 nm, respectively [26, 27]. The OD440 for each sample was measured over the course of 8 min. Examples are shown in Fig. 8. None of the solutes caused a significant change in OD440 when mixed with plain liposomes (Fig. 8A). At the end of each kinetic scan, the pore-forming peptide melittin (from honey bee venom) was added to the sample and then scanned over the same period of time as a positive control for leakage; melittin caused swelling in every sample (not shown). The YshAΔ2-20 proteoliposomes showed a rapid decrease in OD440 when mixed with arabinose (Fig. 8B). When YshAΔ2-20 proteoliposomes were mixed with PEG 400, the OD440 decreased slightly compared to that of plain liposomes, but was much more rapid upon the addition of melittin. There was no decrease in OD440 when PEG 600 was mixed with YshAΔ2-20 proteoliposomes, but there was a rapid decrease in OD440 upon the addition of melittin. This experiment reveals that YshAΔ2-20 forms a pore through the bilayer and the effective radius of the pore is ~0.7 nm. These results confirm that YshAΔ2-20 spans the membrane and combined with the localization and structural data supports the conclusion that the YshA proteins (Table 1) are transmembrane β-barrels as predicted by the Freeman-Wimley algorithm.
Fig. 8.

Liposome swelling assay. Liposomes (A) or proteoliposomes containing YshAΔ2-20 (B) were treated with isotonic solutions of arabinose (Ara; R(radius) = 0.38 nm), PEG 400 (R = 0.7 nm), or PEG 600 (R = 0.8 nm) while monitoring the OD440 over time.
4 DISCUSSION
The Freeman-Wimley algorithm is a statistical prediction method based strictly on the physicochemical relationship between the structure of the lipid bilayer, and the sequence and structure of known transmembrane β-barrel proteins (TMBBs) [7]. Currently, the pairwise sequence similarity to other known TMBBs is a typical criterion for identification of TMBBs in genome annotations. However, we posit that the Freeman-Wimley algorithm will be more useful in making such genomic predictions, especially in instances where no homologs have been characterized, which is often the case. The goal of this work was to establish a set of rigorous tests, which included a variety of well-established biochemical and biophysical assays, to validate the prediction that a given protein is a TMBB. The tests were used to characterize YshA, a predicted TMBB from Salmonella typhimurium.
4.1 Structural prediction of YshA reveals an interesting protein segment
The predicted topology of YshA suggests it is a 10-stranded barrel with a loop over 40 residues long between β-strands 3 and 4. This segment could be an unpredicted transmembrane β-hairpin, or it could form a plug domain, bind ligands, or participate in protein-protein interactions (PPI). On the basis of observations by Liang and coworkers, who showed that intrinsically unstable regions often correlate to transmembrane PPI domains in membrane proteins [28], we predicted that there are no PPI domains in the transmembrane region. The outer membrane phospholipase A (OmpLA) of E. coli is not predicted to have protein-protein interfaces, yet it forms dimers. Dimerization is primarily substrate-driven, and is undetectable in SDS-PAGE [28–30]. In our experiments there was no experimental evidence indicating that YshA forms any type of oligomer, however we did not specifically assay for self-association so it cannot be ruled out.
4.2 Function of YshA
The characterization of native YshA that had been assembled in cells was limited to determining the native subcellular location. Elucidating the true biological role of YshA was beyond the scope of this study. It was shown here that WT YshA cloned from S. typhimurium could be detected in an extract enriched with outer membrane proteins when expressed in E. coli. The E. coli homolog, called OmpL, is 91% identical to YshA of S. typhimurium, yet no trace of endogenous OmpL was detected in E. coli BL21 (DE3), as shown in Fig. 2, nor was YshA detected in S. typhimurium LT2 (data not shown).
YshA is hypothesized to be a homolog of the oligogalacturonate-specific porin, KdgM, from Erwinia chrysanthemi [31]. It is plausible that YshA is involved in the transport of an acidic sugar [32]. However, YshA is less than 30% identical to KdgM, and the sequences align poorly. Furthermore, there is another putative outer membrane protein in the proteome of S. typhimurium, which is more closely related to KdgM. More recent reports suggest that YshA may be a part of an operon in S. typhimurium which mediates the production of O-antigen [33, 34]. O-antigen is a polysaccharide component in the biofilm capsule formed by many enteropathogenic species of Salmonella and strains of E. coli. The strains of bacteria used in this study did not produce biofilms when cultured in liquid media, perhaps explaining why endogenous OmpL (YshA) was not detected in either E. coli or S. typhimurium. Functional studies for YshA in Salmonella are in progress.
4.3 RP-HPLC is a viable, yet underused TMBB purification technique
The overexpressed YshAΔ2-20 formed inclusion bodies and could be purified to homogeneity using reverse phase high pressure liquid chromatography (RP-HPLC). RP-HPLC has seldom been used to purify membrane proteins in large quantities. Researchers reasonably assume that RP-HPLC is ill-suited for the purification of membrane proteins, and would expect poor resolution, and very low recovery. In agreement with this assumption, we found that a C4-coated silica column developed in water/acetonitrile failed to resolve the inclusion body proteins which eluted together as a broad unresolved peak (data not shown). However, a column with C2-coated resin resolved YshAΔ2-20 much more effectively. The shorter chain length of C2 reduces the overall hydrophobicity of the column, thus weakening the interaction between protein and resin, which improved the resolution recovery dramatically. Purified YshAΔ2-20 was obtained from C2 RP-HPLC which was similar to the high purity of TMBBs purified by other means, such as ion exchange or affinity-tag chromatography [35, 36]. Moreover, refolding of C2-purified YshAΔ2-20 into a native-like structure was demonstrated in this study, thus exhibiting the viability and utility of this underused technique.
4.4 In vitro refolding and structural characterization of YshA
Several commonly used refolding conditions were tested with mixed success. YshAΔ2-20 was first denatured in urea, and then rapidly diluted into a battery of detergents with some refolding exhibited with n-lauroyl sarcosine (LS), and DDM, while β-octyl-glucoside (OG), lauryldimethylamine-oxide (LDAO) and a host of others did not refold YshAΔ2-20. Further, because LS and urea caused substantial background interference with circular dichroism measurements, an alternate denaturant (SDS) and the nonionic detergent DDM were used for refolding as described by Bay, et al [18]. The ratio of protein:SDS:DDM was 1:700:6000. Thus, for most experiments, 5 μM YshAΔ2-20 was refolded in 3.5 mM SDS, 30 mM DDM, in 10 mM phosphate buffer, pH 7.6. Conditions with lower concentrations of DDM, and pH below 6 or above 9 did not refold YshAΔ2-20.
Mixed anionic and nonionic detergent systems have been used to successfully refold other TMBBs including OmpA of E. coli [20], DsbB of E. coli [37], P5 outer membrane protein of Haemophilus influenza [38], and mitochondrial porin of Neurospora crassa [18]. It has been shown that mild solubilization conditions are more conducive to folding proteins from inclusion bodies than the harsh conditions in urea or guanidine-chloride solutions [39]. While folding TMBBs into a native-like structure is sensitive to multiple factors, such as protein concentration, solution ionic strength, and pH, it is likely that mixed detergent micelles can be used to fold many TMBBs and will be a valuable tool in characterizing TMBBs in the future.
Reconstitution into a lipid bilayer is routinely performed by rapidly diluting a urea-denatured TMBB, such as OmpA into a suspension of liposomes [40, 41]. However, urea-denatured YshAΔ2-20 did not refold and insert into liposomes when urea was diluted either rapidly or by dialysis. Instead, detergent-refolded protein was mixed with liposomes, which were solubilized by the detergent micelles. The detergents were removed by adsorption with BioBeads, thus allowing the phospholipids to reassemble into a lipid bilayer around the hydrophobic surface of the refolded protein. This technique has been used similarly to reconstitute a wide variety of membrane proteins into membrane Nanodiscs [42], and appears to be a promising technology in the study of TMBBs.
The liposome flotation assay results show conclusively that YshAΔ2-20 inserts into a lipid bilayer in a structure-dependent manner. This finding helped rule out the possibility that YshA is an outer membrane lipoprotein because the lipid moiety would allow the protein to bind to the membrane regardless of the presence of urea. Furthermore, the reconstitution results show that YshA can interact with the membrane independent of other proteins, which suggests it is not a periplasmic protein that binds an outer membrane protein complex. Lastly, lipid bilayer-embedded YshAΔ2-20 was structurally similar to folded YshAΔ2-20 in mixed-detergent micelles, showing that YshA is a β-sheet-rich, transmembrane protein.
The strongest evidence that YshA is a TMBB comes from the liposome-swelling assay. YshA formed a small pore which allowed the diffusion of nonelectrolyte solutes. For comparison some other porin radii are listed in Table 2, which shows that the approximate pore radius of YshAΔ2-20 (~0.7 nm) is consistent with known TMBBs. The facts that YshA adopts a transmembrane β-sheet conformation, and forms a pore when reconstituted into a lipid bilayer provide very compelling evidence that it is a TMBB.
Table 2.
Pore radii of some TMBBs
| Protein | Source Organism | Effective pore radius (nm) |
|---|---|---|
| YshA | S. typhimurium | ~0.7a |
| OmpL [8] | E. coli | <0.7a |
| OmpF [45] | E. coli | 0.7b |
| α-hemolysin [45] | S. aureus | 1.0b |
| MOMP [46] | C. trachomatis | 1.0a |
| OmpC [47] | S. typhi | 1.1a |
The pore radius was determined either by
liposome swelling or
single-channel conductance experiments.
References are indicated next to each protein.
4.5 Conclusion
In this study we used a statistical bioinformatics approach to predict the unknown outer membrane proteins of Salmonella typhimurium. We chose YshA, a highly conserved but uncharacterized putative outer membrane β-barrel protein, as an example protein to establish a set of experimental tools for the characterization of putative TMBBs. Our work clearly established that YshA is a TMBB. Guided by this information, functional and structural studies of YshA are now underway. This work establishes a complete “genome to structure” pathway for the prediction, conformation and characterization of novel TMBBs in the genomes of Gram-negative bacteria.
Supplementary Material
Acknowledgments
We would like acknowledge that this work was funded in part by NSF grant number MCB-0349578 awarded to Samuel Landry, and by NIH grant GM060000 and Louisiana Board of Regents RC/EEP-05(2007-10) awarded to William Wimley.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Wimley WC. The versatile β-barrel membrane protein. Current opinion in structural biology. 2003;13:404–411. doi: 10.1016/s0959-440x(03)00099-x. [DOI] [PubMed] [Google Scholar]
- 2.White SH, Wimley WC. Hydrophobic interactions of peptides with membrane interfaces. Biochimica et biophysica acta. 1998;1376:339–352. doi: 10.1016/s0304-4157(98)00021-5. [DOI] [PubMed] [Google Scholar]
- 3.Jayasinghe S, Hristova K, White SH. Energetics, stability, and prediction of transmembrane helices. Journal of molecular biology. 2001;312:927–934. doi: 10.1006/jmbi.2001.5008. [DOI] [PubMed] [Google Scholar]
- 4.Snider C, Jayasinghe S, Hristova K, White SH. MPEx: A tool for exploring membrane proteins. Protein Sci. 2009 doi: 10.1002/pro.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.White SH, Wimley WC. Membrane protein folding and stability: physical principles. Annual review of biophysics and biomolecular structure. 1999;28:319–365. doi: 10.1146/annurev.biophys.28.1.319. [DOI] [PubMed] [Google Scholar]
- 6.Wimley WC. Toward genomic identification of β-barrel membrane proteins: composition and architecture of known structures. Protein Sci. 2002;11:301–312. doi: 10.1110/ps.29402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Freeman TC, Jr, Wimley WC. A highly accurate statistical approach for the prediction of transmembrane β-barrels. Bioinformatics. 2010 doi: 10.1093/bioinformatics/btq308. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dartigalongue C, Nikaido H, Raina S. Protein folding in the periplasm in the absence of primary oxidant DsbA: modulation of redox potential in periplasmic space via OmpL porin. The EMBO journal. 2000;19:5980–5988. doi: 10.1093/emboj/19.22.5980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sardesai AA, Genevaux P, Schwager F, Ang D, Georgopoulos C. The OmpL porin does not modulate redox potential in the periplasmic space of Escherichia coli. The EMBO journal. 2003;22:1461–1466. doi: 10.1093/emboj/cdg152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pugsley AP. The complete general secretory pathway in gram-negative bacteria. Microbiological reviews. 1993;57:50–108. doi: 10.1128/mr.57.1.50-108.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bendtsen JD, Nielsen H, von Heijne G, Brunak S. Improved prediction of signal peptides: SignalP 3.0. Journal of molecular biology. 2004;340:783–795. doi: 10.1016/j.jmb.2004.05.028. [DOI] [PubMed] [Google Scholar]
- 12.Chimento DP, Mohanty AK, Kadner RJ, Wiener MC. Substrate-induced transmembrane signaling in the cobalamin transporter BtuB. Nature structural biology. 2003;10:394–401. doi: 10.1038/nsb914. [DOI] [PubMed] [Google Scholar]
- 13.Szmelcman S, Hofnung M. Maltose transport in Escherichia coli K-12: involvement of the bacteriophage lambda receptor. Journal of bacteriology. 1975;124:112–118. doi: 10.1128/jb.124.1.112-118.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Burgess NK, Dao TP, Stanley AM, Fleming KG. β-barrel proteins that reside in the Escherichia coli outer membrane in vivo demonstrate varied folding behavior in vitro. The Journal of biological chemistry. 2008;283:26748–26758. doi: 10.1074/jbc.M802754200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dornmair K, Kiefer H, Jahnig F. Refolding of an integral membrane protein. OmpA of Escherichia coli. The Journal of biological chemistry. 1990;265:18907–18911. [PubMed] [Google Scholar]
- 16.Kleinschmidt JH, Wiener MC, Tamm LK. Outer membrane protein A of E. coli folds into detergent micelles, but not in the presence of monomeric detergent. Protein Sci. 1999;8:2065–2071. doi: 10.1110/ps.8.10.2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ohnishi S, Kameyama K, Takagi T. Characterization of a heat modifiable protein, Escherichia coli outer membrane protein OmpA in binary surfactant system of sodium dodecyl sulfate and octylglucoside. Biochimica et biophysica acta. 1998;1375:101–109. doi: 10.1016/s0005-2736(98)00145-x. [DOI] [PubMed] [Google Scholar]
- 18.Bay DC, O’Neil JD, Court DA. Two-step folding of recombinant mitochondrial porin in detergent. Biophysical journal. 2008;94:457–468. doi: 10.1529/biophysj.107.115196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bousquet JA, Duranton J, Mely Y, Bieth JG. Conformational change in elastase following complexation with alpha1-proteinase inhibitor: a CD investigation. The Biochemical journal. 2003;370:345–349. doi: 10.1042/BJ20020890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ohnishi S, Kameyama K. Escherichia coli OmpA retains a folded structure in the presence of sodium dodecyl sulfate due to a high kinetic barrier to unfolding. Biochimica et biophysica acta. 2001;1515:159–166. doi: 10.1016/s0005-2736(01)00410-2. [DOI] [PubMed] [Google Scholar]
- 21.Johnson WC., Jr Protein secondary structure and circular dichroism: a practical guide. Proteins. 1990;7:205–214. doi: 10.1002/prot.340070302. [DOI] [PubMed] [Google Scholar]
- 22.Rigaud JL, Mosser G, Lacapere JJ, Olofsson A, Levy D, Ranck JL. Bio-Beads: an efficient strategy for two-dimensional crystallization of membrane proteins. Journal of structural biology. 1997;118:226–235. doi: 10.1006/jsbi.1997.3848. [DOI] [PubMed] [Google Scholar]
- 23.Hong H, Tamm LK. Elastic coupling of integral membrane protein stability to lipid bilayer forces. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:4065–4070. doi: 10.1073/pnas.0400358101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nikaido H, Luckey M, Rosenberg EY. Nonspecific and specific diffusion channels in the outer membrane of Escherichia coli. Journal of supramolecular structure. 1980;13:305–313. doi: 10.1002/jss.400130304. [DOI] [PubMed] [Google Scholar]
- 25.Nikaido H, Nikaido K, Harayama S. Identification and characterization of porins in Pseudomonas aeruginosa. The Journal of biological chemistry. 1991;266:770–779. [PubMed] [Google Scholar]
- 26.Peyronnet O, Nieman B, Genereux F, Vachon V, Laprade R, Schwartz JL. Estimation of the radius of the pores formed by the Bacillus thuringiensis Cry1C delta-endotoxin in planar lipid bilayers. Biochimica et biophysica acta. 2002;1567:113–122. doi: 10.1016/s0005-2736(02)00605-3. [DOI] [PubMed] [Google Scholar]
- 27.Schultz SG, Solomon AK. Determination of the effective hydrodynamic radii of small molecules by viscometry. The Journal of general physiology. 1961;44:1189–1199. doi: 10.1085/jgp.44.6.1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Naveed H, Jackups R, Jr, Liang J. Predicting weakly stable regions, oligomerization state, and protein-protein interfaces in transmembrane domains of outer membrane proteins. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:12735–12740. doi: 10.1073/pnas.0902169106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stanley AM, Chuawong P, Hendrickson TL, Fleming KG. Energetics of outer membrane phospholipase A (OMPLA) dimerization. Journal of molecular biology. 2006;358:120–131. doi: 10.1016/j.jmb.2006.01.033. [DOI] [PubMed] [Google Scholar]
- 30.Stanley AM, Fleming KG. The role of a hydrogen bonding network in the transmembrane β-barrel OMPLA. Journal of molecular biology. 2007;370:912–924. doi: 10.1016/j.jmb.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 31.Blot N, Berrier C, Hugouvieux-Cotte-Pattat N, Ghazi A, Condemine G. The oligogalacturonate-specific porin KdgM of Erwinia chrysanthemi belongs to a new porin family. The Journal of biological chemistry. 2002;277:7936–7944. doi: 10.1074/jbc.M109193200. [DOI] [PubMed] [Google Scholar]
- 32.Condemine G, Berrier C, Plumbridge J, Ghazi A. Function and expression of an N-acetylneuraminic acid-inducible outer membrane channel in Escherichia coli. Journal of bacteriology. 2005;187:1959–1965. doi: 10.1128/JB.187.6.1959-1965.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Crawford RW, Gibson DL, Kay WW, Gunn JS. Identification of a bile-induced exopolysaccharide required for Salmonella biofilm formation on gallstone surfaces. Infection and immunity. 2008;76:5341–5349. doi: 10.1128/IAI.00786-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gibson DL, White AP, Snyder SD, Martin S, Heiss C, Azadi P, Surette M, Kay WW. Salmonella produces an O-antigen capsule regulated by AgfD and important for environmental persistence. Journal of bacteriology. 2006;188:7722–7730. doi: 10.1128/JB.00809-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Charbonnier F, Kohler T, Pechere JC, Ducruix A. Overexpression, refolding, and purification of the histidine-tagged outer membrane efflux protein OprM of Pseudomonas aeruginosa. Protein expression and purification. 2001;23:121–127. doi: 10.1006/prep.2001.1473. [DOI] [PubMed] [Google Scholar]
- 36.Kashino Y. Separation methods in the analysis of protein membrane complexes. Journal of chromatography. 2003;797:191–216. doi: 10.1016/s1570-0232(03)00428-8. [DOI] [PubMed] [Google Scholar]
- 37.Otzen DE. Folding of DsbB in mixed micelles: a kinetic analysis of the stability of a bacterial membrane protein. Journal of molecular biology. 2003;330:641–649. doi: 10.1016/s0022-2836(03)00624-7. [DOI] [PubMed] [Google Scholar]
- 38.Webb DC, Cripps AW. A method for the purification and refolding of a recombinant form of the nontypeable Haemophilus influenzae P5 outer membrane protein fused to polyhistidine. Protein expression and purification. 1999;15:1–7. doi: 10.1006/prep.1998.0990. [DOI] [PubMed] [Google Scholar]
- 39.Singh SM, Panda AK. Solubilization and refolding of bacterial inclusion body proteins. Journal of bioscience and bioengineering. 2005;99:303–310. doi: 10.1263/jbb.99.303. [DOI] [PubMed] [Google Scholar]
- 40.Kleinschmidt JH, Tamm LK. Secondary and tertiary structure formation of the β-barrel membrane protein OmpA is synchronized and depends on membrane thickness. Journal of molecular biology. 2002;324:319–330. doi: 10.1016/s0022-2836(02)01071-9. [DOI] [PubMed] [Google Scholar]
- 41.Surrey T, Jahnig F. Kinetics of folding and membrane insertion of a β-barrel membrane protein. The Journal of biological chemistry. 1995;270:28199–28203. doi: 10.1074/jbc.270.47.28199. [DOI] [PubMed] [Google Scholar]
- 42.Bayburt TH, Sligar SG. Membrane protein assembly into Nanodiscs. FEBS letters. 2010;584:1721–1727. doi: 10.1016/j.febslet.2009.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. Journal of molecular biology. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 44.Robert V, Volokhina EB, Senf F, Bos MP, Van Gelder P, Tommassen J. Assembly factor Omp85 recognizes its outer membrane protein substrates by a species-specific C-terminal motif. PLoS biology. 2006;4:e377. doi: 10.1371/journal.pbio.0040377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rostovtseva TK, Nestorovich EM, Bezrukov SM. Partitioning of differently sized poly (ethylene glycol)s into OmpF porin. Biophysical journal. 2002;82:160–169. doi: 10.1016/S0006-3495(02)75383-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sun G, Pal S, Sarcon AK, Kim S, Sugawara E, Nikaido H, Cocco MJ, Peterson EM, de la Maza LM. Structural and functional analyses of the major outer membrane protein of Chlamydia trachomatis. Journal of bacteriology. 2007;189:6222–6235. doi: 10.1128/JB.00552-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sundara Baalaji N, Mathew MK, Krishnaswamy S. Functional assay of Salmonella typhi OmpC using reconstituted large unilamellar vesicles: a general method for characterization of outer membrane proteins. Biochimie. 2006;88:1419–1424. doi: 10.1016/j.biochi.2006.05.010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.