

Abstract
Human valacyclovirase (hVACVase) is a prodrug-activating enzyme for amino acid prodrugs including the antiviral drugs valacyclovir and valganciclovir. In hVACVase-catalyzed reactions, the leaving group of the substrate corresponds to the drug moiety of the prodrug, making the leaving group effect essential for the rational design of new prodrugs targeting hVACVase activation. In this study, a series of valine esters, phenylalanine esters and a valine amide were characterized for the effect of the leaving group on the efficiency of hVACVase-mediated prodrug activation. Except for phenylalanine methyl and ethyl esters, all of the ester substrates exhibited a relatively high specificity constant (kcat/Km), ranging from 850 to 9490 mM-1·s-1. The valine amide Val-3-APG exhibited significantly higher Km and lower kcat values compared to the corresponding ester Val-3-HPG, indicating poor specificity for hVACVase. In conclusion, the substrate leaving group has been shown to affect both binding and specific activity of hVACVase-catalyzed activation. It is proposed that hVACVase is an ideal target for α-amino acid ester prodrugs with relatively labile leaving groups, while it is relatively inactivate towards amide prodrugs.
Keywords: hVACVase, prodrug
Introduction
The prodrug strategy has proven to be a successful approach for improving oral absorption of low-permeability drugs by various mechanisms.1-4 Following absorption, the prodrug must be activated, either chemically or enzymatically, to the active parent drug. Historically, the activation mechanism was often not investigated. A more modern approach, on the other hand, would exploit the mechanism behind the activation, enabling to target the location and control over the extent of this essential process.5-8 We have recently identified an amino acid ester prodrug-activating enzyme, human valacyclovirase, hVACVase,9 which has been shown to be one of the major enzymes for the activation of the valine ester prodrugs valacyclovir and valganciclovir. We have also shown that hVACVase effectively hydrolyzes many other amino acid esters as well.10-12 Its potential to serve as a prodrug-activating enzyme for amino acid ester and amide prodrugs depends on its leaving group (drug) specificity.
hVACVase is a serine hydrolase with a catalytic triad S122-H255-D227. The very specific preference for amino acid analogues can be attributed to the electrostatic interaction between the critical residue D123, adjacent to the active S122, and the free amino group of the substrate.11 As revealed by its crystal structure, hVACVase contains a large leaving group-accommodating groove, which explains the diversity of its substrate leaving groups (potential drugs), including nucleoside analogues acyclovir, ganciclovir, floxuridine, gemcitabine, zidovudine, 2-bromo-5,6-dichloro-1-(β-D-ribofuranosyl) benzimidazole, and other alcohols such as methanol, ethanol, benzyl alcohol and [3-(hydroxymethyl)phenyl]guanidine (3-HPG).10-12 Since the leaving group corresponds to the drug moiety of the prodrug, hVACVase, due to its expression in the intestinal epithelial cells, is an ideal target for oral delivery and activation of amino acid ester prodrugs, and perhaps amide prodrugs as well. While the phenylalanine benzyl and ethyl esters are hVACVase substrates (specific activities are 358.3 and 75.3 units/mg, respectively), phenylalanine t-butyl ester is not a substrate.11 In addition, the valine 5’-floxuridine has a higher Vmax value compared with valine 3’-floxuridine ester (148 and 33 nmol/min/μg, respectively),10 thus indicating that the alcohol leaving groups do have an effect on hVACVase catalytic activity. Additionally, hVACVase does not exhibit significant hydrolytic activity toward a series of amides, including Lys-p-NA, Leu-p-NA, Pro-p-NA, Phe-p-NA, Val-p-NA and Gly-Pro-p-NA.10 This may be due to the much more stable amide bond compared with the ester bond. The ten-fold difference in Km values of the prodrugs valacyclovir and valganciclovir (0.19 and 1.90 mM, respectively) indicates that the leaving groups also affect the binding affinity.9 To better understand the hVACVase-catalyzed prodrug activation and guide future prodrug design, an investigation of the leaving group effect is essential.
The purpose of this study was to investigate the effect of leaving groups on the hVACVase-catalysed prodrug activation in a systematic fashion. We have determined the kinetic parameters of a series of valine esters (Val-3-HPG, valacyclovir, valine benzyl ester, valine p-nitrobenzyl ester), phenylalanine esters (Phe-3-HPG, phenylalanine benzyl ester, phenylalanine ethyl ester, phenylalanine methyl ester) and valine amide of [3-(aminomethyl)phenyl]guanidine, Val-3-APG (Scheme 1). This approach allows us to determine the rate-limiting step of hVACVase-catalyzed hydrolysis and the effect of leaving groups on the binding affinity and specific activity. This will allow a more rational design of successful amino acid ester and amide prodrugs.
Scheme 1.

Structures of valine and phenylalanine analogues.
Experimental Section
Materials
Boc-L-valine and N,N,N′,N′-tetramethyl-O-(1H-benzotriazol-1-yl)uronium hexafluorophosphate (HBTU) were obtained from Calbiochem-Novabiochem (San Diego, CA). Valacyclovir was a gift from GlaxoSmithKline, Inc. (Research Triangle Park, NC). L-Valine p-nitrobenzyl ester hydrobromide was obtained from Chem-Impex International, Inc. (Wood Dale, IL). High-performance liquid chromatography (HPLC) grade acetonitrile was obtained from Fisher Scientific (St. Louis, MO). 3-Aminobenzonitrile, 1,3-bis(tert-butoxycarbonyl)-2-methyl-2-thiopseudourea, mercury(II) chloride, Pd/C, 2M NH3/EtOH, trifluoroacetic acid (TFA), N,N-diisopropylethylamine, L-valine benzyl ester hydrochloride, L-phenylalanine benzyl ester hydrochloride, L-phenylalanine methyl ester hydrochloride, L-phenylalanine ethyl ester hydrochloride, and all other reagents and solvents were purchased from Sigma-Aldrich Co. (St. Louis, MO). Cell culture reagents were obtained from Invitrogen (Carlsbad, CA), and cell culture supplies were obtained from Corning (Corning, NY) and Falcon (Lincoln Park, NJ). All chemicals were either analytical or HPLC grade.
Synthesis
NMR spectra were obtained on a Bruker AVANCE DRX500 NMR spectrometer. Electronspray ionization mass spectra were obtained on a Micromass LCT time-of-flight mass spectrometer. The purity of all synthesized test compounds was at least 95% as determined by HPLC.
3-[[Bis[[(1,1-dimethylethoxy)carbonyl]amino]methylene]amino]benzonitrile (2)
To a stirred suspension of 3-aminobenzonitrile (1, 118 mg, 1.0 mmol), 1,3-bis(tert-butoxycarbonyl)-2-methyl-2-thiopseudourea (290 mg, 1 mmol) and mercury(II) chloride (353 mg, 1.3 mmol) in 5 mL of dry tetrahydrofuran (THF), 405 mg (4.0 mmol) of triethylamine was added dropwise under argon. After 2 hours, the reaction was stopped and reaction mixture was diluted with ethyl acetate and filtered through Celite®. Solvents were removed and the residue was dissolved in 40 mL of ethyl acetate and washed with 10% w/v citric acid, saturated NaHCO3 and brine. The organic phase was dried over anhydrous MgSO4 and concentrated in vacuo. Product 2 was purified by column chromatography (hexanes : ethyl acetate, 20 : 1) to give 327.9 mg white powder. Yield: 91.0%. 1H NMR (CDCl3) δ 11.65 (1H, br), 10.55 (1H, br), 8.11 (1H, s), 7.77 (1H, d, J = 7.8 Hz), 7.42 (2H, m), 1.56 (9H, s), 1.54 (9H, s); ESI-MS: 383.1 (M+Na)+.
3-[[Bis[[(1,1-dimethylethoxy)carbonyl]amino]methylene]amino]benzylamine (3)
A mixture of 2 (150 mg, 0.416 mmol) and Pd/C (10 wt % palladium on activated carbon, 150 mg) in 100 mL of 2M NH3/EtOH was hydrogenated in a Parr apparatus at 45 psi and room temperature for 9 h. The reaction mixture was filtered through Celite®. The solvents were removed and product was purified by column chromatography (dichloromethane : methanol, 15 : 1) to give 80.0 mg light yellow gum. Yield: 52.7%. 1H NMR (CDCl3) δ 11.65 (1H, br), 10.37 (1H, br), 7.56 (1H, s), 7.52 (1H, d, J = 7.8 Hz), 7.32 (1H, dd, J = 7.8 Hz, 7.8 Hz), 7.10 (1H, d, J = 7.8 Hz), 3.90 (2H, s), 1.56 (9H, s), 1.53 (9H, s); ESI-MS: 365.2 (M+H)+.
[3-(Aminomethyl)phenyl]guanidine (3-APG, 4)
24.8 mg of 3 was dissolved in 2.2 mL of trifluoroacetic acid (TFA):CH2Cl2 (1:1.2) and stirred at room temperature for 2 hours. Then solvents were removed and the residue was dissolved in 0.1 % TFA, filtered and lyophilized. The raw product was further purified by semi-prep HPLC to give 15.4 mg white solid. Yield: 57.7%. 1H NMR (CD3OD) δ 7.57 (1H, dd, J = 7.9 Hz, 7.9 Hz), 7.44 (2H, m), 7.36 (1H, d, J = 7.9 Hz), 4.18 (2H, s); ESI-MS: 165.1 (M+H)+.
(2S)-2-Amino-3-methyl-N-[3-[(aminoiminomethyl)amino]phenyl]methylbutanamide (Val-3-APG, 6)
To a stirred solution of 3 (12.6 mg, 0.035 mmol), Boc-L-valine (9.1 mg, 0.042 mmol) and N,N,N′,N′-tetramethyl-O-(1H-benzotriazol-1-yl)uronium hexafluorophosphate (HBTU, 16.0 mg, 0.042 mmol) in 1 mL of anhydrous CH2Cl2, 18 μl of N,N-diisopropylethylamine were added dropwise under argon. The reaction mixture was stirred at ambient temperature for 3 h and then the solvents were removed. The residue was dissolved in 30 mL of CH2Cl2 and washed with 10% w/v citric acid, saturated NaHCO3 and brine. The organic phase was dried over anhydrous MgSO4 and concentrated in vacuo. The mixture was then chromatographed on silica gel (hexanes : ethyl acetate, 5 : 1) to obtain 5 as colorless oil. 1H NMR (CDCl3) δ 11.65 (1H, br), 10.35 (1H, br), 7.52 (1H, d, J=7.8 Hz), 7.47 (1H, s), 7.28 (1H, dd, J=7.8 Hz, 7.8 Hz), 7.03 (1H, d, J=7.8 Hz), 6.37 (1H, br), 5.06 (1H, m), 4.43 (2H, d, J=5.6 Hz), 3.92 (1H, m), 2.19 (1H, m), 1.42-1.53 (27 H, m), 0.96 (3H, d, J=6.8 Hz), 0.91 (3H, d, J=6.8 Hz); ESI-MS: 564.2 (M+H)+.
5 was treated with 2.2 mL of TFA : CH2Cl2 (1:1.2) for 2 hours. Solvents were removed and residue was dissolved in 0.1 % TFA, filtered and lyophilized to give 12.86 mg of 6 as light brown syrup. Yield of two steps: 74.8%. 1H NMR (CD3OD) δ 7.48 (1H, dd, J=7.9 Hz, 7.8 Hz), 7.35 (1H, d, J=7.9 Hz), 7.28 (1H, s), 7.23 (1H, d, J=7.8 Hz), 4.49 (2H, m), 3.70 (1H, d, J=5.7 Hz), 2.21 (1H, m), 1.06 (6H, m); ESI-MS: 264.1 (M+H)+.
Hydrolysis of valine and phenylalanine esters in pH 7.4 HEPES buffer
Hydrolysis in pH 7.4 HEPES buffer was determined at 37 °C. The hydrolysis reaction was initiated by adding 0.75 μL of test compound solution (200 mM in DMSO) to a reaction tube containing 749.25 μL pH 7.4 HEPES buffer. At various time points, 100 μL of the reaction mixture was removed and added to a quenching plate containing 100 μL of 1% TFA (in water) and stored in ice. Following the collection of all samples, quenching plate was filtered (2,000 rpm, 4 °C, 10 min). The filtrate was removed and assayed by HPLC.
The apparent first-order degradation rate constants were determined by plotting the natural logarithm of test compound remaining as a function of time. The slope of this plot equals to negative rate constant (k). The degradation half-life was then estimated by the equation:
Hydrolysis of Val-3-APG in pH 7.4 HEPES buffer
The hydrolysis reaction was initiated by adding 20 μL of Val-3-APG solution (200 mM in DMSO) to a reaction tube containing 380 μL pH 7.4 HEPES buffer and incubated at 37 °C. At various time points (up to 800 h), 85 μL of the reaction mixture was removed and added to a quenching tube containing 85 μL of 10% TFA (in water), filtered (2,000 rpm, 4 °C, 10 min), and the filtrate was immediately assayed by HPLC.
The apparent first-order degradation rate constants were determined by plotting the natural logarithm of Val-3-APG percentage (1-[3-APG]/([3-HPG]+[Val-3-APG])) as a function of time. The slope equals to negative rate constant (k). The degradation half-lives were then estimated by the equation:
hVACVase-mediated hydrolysis
hVACVase was overexpressed and purified from Escherichia coli as described previously11 and used for all the test compounds except Val-3-APG. 6×His-tagged hVACVase (M - S tag - PDLGTLVPRGSMGM - hVACVase - AAALE - His tag) was produced in bacteria, purified by affinity chromatography and used for Val-3-APG. The results from the two proteins are considered to be comparable due to their very similar specific activity toward valacyclovir activation. The protein concentration was determined by Bio-Rad DC assay (Hercules, CA) with bovine serum albumin as a standard. The kinetic parameters of hVACVase-catalyzed hydrolysis were determined as follows. Kinetic measurements were carried out in 50 mM HEPES (pH 7.4) buffer at 37°C. After preincubation of the buffer for 5 min, hVACVase was added, and then the reaction was initiated by the addition of substrate. Aliquots were taken at different time points, and quenched by adding to same volume of 10% (v/v) trifluoroacetic acid. Initial velocities were calculated from the linear time course for the product formation. The kinetic parameters Km and Vmax were determined by fitting the initial velocity data to the Michaelis-Menten equation by the nonlinear least-square regression analysis in GraphPad Prism software version 4.01. The kcat value was calculated from Vmax/[enzyme]0 based on the 28.83-kDa molecular mass of hVACVase and 33.38-kDa molecular mass of 6×His-tagged hVACVase. Specific activity of valacyclovir was routinely monitored to normalize active protein concentration.
HPLC analysis
The concentrations of test compounds were determined on a Waters HPLC system (Waters Inc., Milford, MA) consisting of two Waters pumps (Model 515), a Waters auto-sampler (WISP model 712) and a Waters UV detector (996 Photodiode Array Detector). The system was controlled by Waters Millennium 32 software (Version 3.0.1). Samples were resolved in an Agilent ZORBAX Eclipse XDB-C18 column (3.5 μm, 4.6 × 150 mm) equipped with a guard column and the flow rate was 1.0 mL/min. For all the test compounds except for valine p-nitrobenzyl ester, the mobile phase consisted of 0.1% (v/v) TFA in milli-Q water (solvent A) and 0.1% (v/v) TFA in acetonitrile (solvent B) with the solvent B gradient changing from 2-30% at a rate of 2%/min during a 25 min run. The retention times for 3-HPG, Val-3-HPG, Phe-3-HPG, acyclovir, valacyclovir, 3-APG, Val-3-APG, benzyl alcohol, Val-OBn, phenylalanine, Phe-OMe, Phe-OEt, Phe-OBn were 6.7, 9.7, 12.1, 4.9, 7.9, 4.2, 8.4, 11.3, 15.7, 8.2, 10.7, 13.0, 18.9 minutes, respectively. For valine p-nitrobenzyl ester, the mobile phase consisted of 70:30 (v/v) 0.1% trifluoroacetic acid in milli-Q water: 0.1% trifluoroacetic acid in methanol. The retention times were 8.5 minutes for p-nitrobenzyl alcohol and 16.4 minutes for valine p-nitrobenzyl ester. The detection wavelength was 235 nm for 3-HPG, 3-APG, Val-3-HPG, Phe-3-HPG and Val-3-APG, 254 nm for acyclovir and valacyclovir, 275 nm for p-nitrobenzyl alcohol and L-valine p-nitrobenzyl ester and 256 nm for phenylalanine, benzyl alcohol, Val-OBn and all the phenylalanine esters except Phe-3-HPG.
Statistical analysis
All the experiments were performed in triplicate unless stated otherwise. The data are presented as mean ± SEM. Statistical difference between groups with equal sample size was determined using the Tukey test (p < 0.01). Statistical difference between groups with unequal sample size was determined using the Tukey-Kramer test (p < 0.05). Letters designate groups with statistical significance from one another.
Results
Synthesis of 3-APG and Val-3-APG
The synthesis of 3-APG and its valine amide is summarized in Scheme 2. 3-Aminobenzonitrile was treated with 1,3-bis(tert-butoxycarbonyl)-2-methyl-2-thiopseudourea to convert the free amino group to the Boc-protected guanidino group. Then, the cyano group was reduced by hydrogenation to afford intermediate 3. Deprotection of 3 gives 3-APG. Valine-3-APG was synthesized by coupling intermediate 3 and Boc-valine followed by deprotection.
Scheme 2.

Synthesis of 3-APG and its valine amide.
Hydrolysis in buffer and cell homogenates
The half-life values of valine and phenylalanine analogues in pH 7.4 HEPES buffer are shown in Tables 1. Valacyclovir and valine benzyl ester exhibited approximately two-fold longer t1/2 than valine p-nitrobenzyl ester and Val-3-HPG. Compared with Val-3-HPG, the hydrolysis of Val-3-APG was five orders of magnitude slower. For the phenylalanine esters, the half-life values ranked in the following order: Phe-3-HPG < Phe-OBn < Phe-OMe < PheOEt.
Table 1.
Estimated half-life values of valine and phenylalanine analogues in pH 7.4 HEPES buffer (mean ± SEM; esters, n=3; Val-3-APG, n=1).
| valine analogues | t1/2 (min)a | phenylalanine analogues | t1/2 (min)a |
|---|---|---|---|
| valine p-nitrobenzyl ester | 412.3 ± 11.9 | Phe-3-HPG | 230.0 ± 6.5 |
| Val-3-HPG | 486.9 ± 6.6 | Phe-OBn | 365.0 ± 9.4 |
| valacyclovir | 742.1 ± 5.5 | Phe-OMe | 470.6 ± 12.6 |
| Val-OBn | 857.8 ± 7.6 | Phe-OEt | 1006.4 ± 26.0 |
| Val-3-APG | 6.2 × 107 |
All the groups are statistically different.
In contrast to the fast hydrolysis of Val-3-HPG (t1/2 < 5 min),12 the valine amide Val-3-APG was fairly stable in Caco-2 cell homogenates. During the 2 h incubation time no degradation of Val-3-APG was detected.
hVACVase-mediated hydrolysis
The Michaelis-Menten kinetic parameters of valine derivatives for hVACVase-mediated hydrolysis are listed in Table 2. All the valine esters showed relatively low Km values ranging from 14 to 68 μM. The kcat values of valine p-nitrobenzyl ester and Val-3-HPG were about 2-fold higher than valacyclovir and valine benzyl ester, similar to the trend shown for buffer hydrolysis rates. The valine amide Val-3-APG exhibited significantly higher Km and lower kcat values compared with the esters, indicating that it is a very poor substrate of hVACVase.
Table 2.
Michaelis-Menten kinetic parameters of valine and phenylalanine derivatives for hVACVase-mediated hydrolysis (mean ± SEM; Phe-OMe, n=2; others, n=3).
| valine analogues | Km (μM)b | Vmax (nmol/min/μg) | kcat (s-1)c | kcat/Km (mM-1·s-1) |
|---|---|---|---|---|
| valine p-nitrobenzyl ester | 14 ± 1 | 272 ± 3 | 130 ± 2 | 9490 ± 580 |
| Val-3-HPGa | 46 ± 5 | 321 ± 9 | 154 ± 4 | 3370 ± 460 |
| valacyclovira | 68 ± 4 | 120 ± 2 | 58 ± 1 | 850 ± 66 |
| Val-OBn | 60 ± 15 | 125 ± 7 | 60 ± 3 | 992 ± 300 |
| Val-3-APG | 1810 ± 80 | (4.12 ± 0.08) × 10-3 | (2.29 ± 0.04) × 10-3 | (1.26 ± 0.08) × 10-3 |
| phenylalanine analogues | Km (μM)d | Vmax (nmol/min/μg) | kcat (s-1)e | kcat/Km (mM-1·s-1) |
|---|---|---|---|---|
| Phe-3-HPGa | 207 ± 16 | 718 ± 16 | 345 ± 8 | 1660 ± 160 |
| Phe-OBn | 99 ± 26 | 822 ± 54 | 395 ± 26 | 3980 ± 1310 |
| Phe-OMe | 5140 ± 1120 | 474 ± 49 | 228 ± 24 | 44 ± 14 |
| Phe-OEt | 6700 ± 560 | 193 ± 8 | 93 ± 4 | 14 ± 2 |
Previously published by Sun et al.12
Statistically different groups: A. valine p-nitrobenzyl ester, Val-3-HPG, valacyclovir, Val-OBn; B. Val-3-APG
Statistically different groups: A. valine p-nitrobenzyl ester, B. Val-3-HPG, C. valacyclovir, Val-OBn; D. Val-3-APG
Statistically different groups: A. Phe-3-HPG, Phe-OBn; B. Phe-OMe, Phe-OEt
Statistically different groups: A. Phe-3-HPG, Phe-OBn, B. Phe-OMe, C. Phe-OEt
Phe-3-HPG and phenylalanine benzyl ester exhibited high specificity constants as shown in Table 2, indicating that they are good hVACVase substrates. However, the specificity constants of phenylalanine methyl and ethyl esters were much lower, which was mainly attributable to the higher Km value.
Discussion
Rational design of prodrugs includes a thorough investigation of the activation process, which releases the free active drug. hVACVase, a novel prodrug-activating enzyme, has been shown to mediate the hydrolytic activation of various amino acid ester prodrugs including the significant antiviral drugs valacyclovir and valganciclovir. In the hVACVase-mediated prodrug activation process, the leaving group corresponds to the drug moiety of the prodrug molecule; hence, investigating the leaving group effect is essential for designing new prodrugs targeting this enzyme for activation.
As a serine hydrolase, the hydrolysis process catalyzed by hVACVase is presumably a three-step reaction as shown in the following scheme, where E stands for enzyme, S stands for substrate and AA stands for amino acid:13
 |
For this kinetic scheme the Michaelis-Menten equation can be expressed as:
in which
and
It can be seen that the effect of the leaving groups depends on the rate-limiting step, which can be either acylation or deacylation. Since the leaving group (alcohol or amine) is released from the enzyme in the acylation step, it will only have an effect on the k2 value. If the acylation step is rate-limiting (k2 << k3), then the kcat value will be affected by the leaving group (kcat ≈ k2), and the Km value will approach Ks. On the other hand, if the deacylation step is rate-limiting (k2 >> k3), the kcat value will be independent of the leaving group (kcat ≈ k3), and Km value will be a function of Ks, k2 and k3. For a series of esters or amides with the same acyl group but different leaving groups with diverse lability, k2 values should be different but k3 values should be the same since the deacylation step is identical for all of the substrates. Thus, the rate-limiting step can be determined by comparing kcat (or Vmax) values for the ester and amide substrates. A classical example of this strategy is the case of chymotrypsin, where the amide exhibits a ~3 orders of magnitude lower kcat value compared with the corresponding esters, clearly indicating that the acylation step is rate-limiting for the amide substrate (Table 3).14 Further, for chymotrypsin-mediated hydrolysis of esters, while p-nitrophenol is a much better leaving group than methanol and ethanol, the very similar kcat values suggested that the deacylation step was rate-limiting (Table 3). In the present study we have applied this classical strategy to investigate hVACVase-mediated prodrug activation.
Table 3.
Kinetic parameters for N-acetyltryptophanyl substrates of chymotrypsin (adapted from Bender and Kezdy14).
| X | Km (mM) | kcat (s-1) | |
|---|---|---|---|
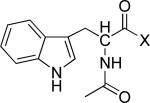
|
-OEt | 0.097 | 0.448 |
| -OMe | 0.095 | 0.462 | |
| -OPNP | 0.002 | 0.508 | |
| -NH2 | 5 | 0.0006 |
The buffer hydrolysis rate reflects the lability of the leaving groups and therefore the k2 value of the hVACVase-catalyzed hydrolysis. The buffer hydrolysis rates of valine p-nitrobenzyl ester and Val-3-HPG were two-fold faster than valine benzyl ester, consistent with the electron withdrawing effect of the nitro and guanidino groups (Table 1). The order of phenylalanine esters hydrolysis rate was also in correspondence to the leaving group lability (Table 1). The very low acidity of 3-APG makes it a poor leaving group, explaining the five orders of magnitude slower buffer hydrolysis of Val-3-APG compared with Val-3-HPG.
The four to five orders of magnitude difference between the kcat values of Val-3-HPG and Val-3-APG indicates that for the amide Val-3-APG, the acylation step (k2) was rate-limiting, similar to chymotrypsin-catalyzed hydrolysis (Table 2). For the valine esters, the kcat values were also different and matched the trend of buffer hydrolysis rate. This observation suggests that the lability of the leaving groups did affect the kcat and unlike chymotrypsin, the acylation step (k2) is rate-limiting. Similar phenomenon was observed for the rat liver carboxylesterase-catalyzed hydrolysis of acetate esters, where the chain length of the alkyl alcohol leaving group significantly affects the reaction rate.15 However, because the kcat differences in the current study were relatively small (up to 2.5-fold), more compounds with a broader range of labilities will be necessary to conclusively confirm this analysis. For the phenylalanine benzyl, methyl and ethyl esters, the kcat value also matched the buffer hydrolysis rate, suggesting that the acylation step is rate-limiting. Despite the faster chemical hydrolysis of Phe-3-HPG, the kcat values of Phe-3-HPG and phenylalanine benzyl ester were similar. It may indicate that the deacylation rate becomes similar to acylation rate, but because k2 depends on multiple factors other than the leaving group lability, this hypothesis needs further evidence.
Assuming acylation as the rate-limiting step, the Km value should be close to the binding affinity Ks. As shown in Table 2, all the amino acid esters showed similarly high affinity to hVACVase except phenylalanine methyl and ethyl esters, likely because methyl and ethyl groups are too small to have favorable interactions such as hydrophobic interactions with the leaving group binding pocket. Despite the negative electrostatic potential in the leaving group-accommodating groove, Val-3-HPG and Phe-3-HPG with the positively charged leaving group exhibited similar binding affinity to valine and phenylalanine benzyl esters, respectively (Table 2). This may be due to the water molecules in the leaving group-accommodating groove interfering with expected electrostatic interaction. The relatively similar Km values of amino acid ester substrates suggest that the requirement of the alcohol leaving groups for the binding is not very stringent, in accordance with the large open leaving group-accommodating groove. Although structurally similar to Val-3-HPG, the amide Val-3-APG showed much lower affinity to hVACVase (Table 2). The difference of ester and amide could be due to the unfavorable interaction between the amide hydrogen and the hydroxyl hydrogen of S122 (Scheme 3). The high Km combined with the low kcat value explains why amides are poor substrates of hVACVase.10
Scheme 3.

Ester and amide substrates binding to hVACVase.
In summary, these results indicate that the leaving groups of substrates have significant effect on both the binding and reaction rate of hVACVase-catalysed hydrolysis. A good hVACVase substrate should have a leaving group labile enough for a reasonable activation rate, but not too labile to be chemically unstable. Alcohol leaving groups with reasonable sizes and labilities are preferred by hVACVase. On the other hand, the amino acid amide is a very poor substrate of hVACVase due to both low affinity and the much stronger amide bond. Therefore, amide prodrugs are more likely to circumvent hVACVase-catalyzed hydrolysis. This can be further exploited in rational prodrug design: if comparatively quick intra-enterocyte activation is desired, an amino acid ester prodrug would be a better choice. However, if stability of the prodrug in the enterocyte and activation at a later step with a different mechanism is desired, amino acid amide may be more suitable.
Acknowledgements
The authors want to thank Dr. Deepak Gupta for designing the synthetic method and Peter Ung for help explaining the data. This research was supported by grant 2R01GM037188-21A2 awarded by the National Institutes of Health.
Abbreviations
- hVACVase
human valacyclovirase
- 3-HPG
[3-(hydroxymethyl)phenyl] guanidine
- p-NA
p-nitroanilide
- 3-APG
[3-(aminomethyl)phenyl]guanidine
- Boc
tert-butyloxycarbonyl
- TFA
trifluoroacetic acid
- HEPES
4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid
- PNP
p-nitrophenyl
References
- 1.Han HK, Amidon GL. Targeted prodrug design to optimize drug delivery. AAPS PharmSci. 2000;2(1):E6. doi: 10.1208/ps020106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ettmayer P, Amidon GL, Clement B, Testa B. Lessons learned from marketed and investigational prodrugs. J Med Chem. 2004;47(10):2393–2404. doi: 10.1021/jm0303812. [DOI] [PubMed] [Google Scholar]
- 3.De Clercq E, Field HJ. Antiviral prodrugs - the development of successful prodrug strategies for antiviral chemotherapy. Br J Pharmacol. 2006;147(1):1–11. doi: 10.1038/sj.bjp.0706446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li F, Maag H, Alfredson T. Prodrugs of nucleoside analogues for improved oral absorption and tissue targeting. J Pharm Sci. 2008;97(3):1109–1134. doi: 10.1002/jps.21047. [DOI] [PubMed] [Google Scholar]
- 5.Wu JZ, Lin CC, Hong Z. Ribavirin, viramidine and adenosine-deaminase-catalysed drug activation: implication for nucleoside prodrug design. J Antimicrob Chemother. 2003;52(4):543–546. doi: 10.1093/jac/dkg405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rooseboom M, Commandeur JN, Vermeulen NP. Enzyme-catalyzed activation of anticancer prodrugs. Pharmacol Rev. 2004;56(1):53–102. doi: 10.1124/pr.56.1.3. [DOI] [PubMed] [Google Scholar]
- 7.Landowski CP, Lorenzi PL, Song X, Amidon GL. Nucleoside ester prodrug substrate specificity of liver carboxylesterase. J Pharmacol Exp Ther. 2006;316(2):572–580. doi: 10.1124/jpet.105.092726. [DOI] [PubMed] [Google Scholar]
- 8.Shi D, Yang J, Yang D, LeCluyse EL, Black C, You L, Akhlaghi F, Yan B. Anti-influenza prodrug oseltamivir is activated by carboxylesterase human carboxylesterase 1, and the activation is inhibited by antiplatelet agent clopidogrel. J Pharmacol Exp Ther. 2006;319(3):1477–1484. doi: 10.1124/jpet.106.111807. [DOI] [PubMed] [Google Scholar]
- 9.Kim I, Chu XY, Kim S, Provoda CJ, Lee KD, Amidon GL. Identification of a human valacyclovirase: biphenyl hydrolase-like protein as valacyclovir hydrolase. J Biol Chem. 2003;278(28):25348–25356. doi: 10.1074/jbc.M302055200. [DOI] [PubMed] [Google Scholar]
- 10.Kim I, Song X, Vig BS, Mittal S, Shin HC, Lorenzi PJ, Amidon GL. A novel nucleoside prodrug-activating enzyme: substrate specificity of biphenyl hydrolase-like protein. Mol Pharm. 2004;1(2):117–127. doi: 10.1021/mp0499757. [DOI] [PubMed] [Google Scholar]
- 11.Lai L, Xu Z, Zhou J, Lee KD, Amidon GL. Molecular basis of prodrug activation by human valacyclovirase, an alpha-amino acid ester hydrolase. J Biol Chem. 2008;283(14):9318–9327. doi: 10.1074/jbc.M709530200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sun J, Dahan A, Amidon GL. Enhancing the intestinal absorption of molecules containing the polar guanidino functionality: a double-targeted prodrug approach. J Med Chem. 2010;53(2):624–632. doi: 10.1021/jm9011559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fersht A. The Basic Equations of Enzyme Kinetics. In: A. Fersht., editor. Structure and Mechanism in Protein Science: A Guide to Enzyme Catalysis and Protein Folding. 1st ed. W. H. Freeman & Co.; New York: 1999. pp. 107–108. [Google Scholar]
- 14.Bender ML, Kezdy J. Mechanism Of Action Of Proteolytic Enzymes. Annu Rev Biochem. 1965;34:49–76. doi: 10.1146/annurev.bi.34.070165.000405. [DOI] [PubMed] [Google Scholar]
- 15.Arndt R, Krisch K. Catalytic properties of an unspecific carboxylesterase (E1) from rat-liver microsomes. Eur J Biochem. 1973;36(1):129–134. doi: 10.1111/j.1432-1033.1973.tb02892.x. [DOI] [PubMed] [Google Scholar]