

Abstract
The SSeCKS/Gravin/AKAP12 gene, encoding a kinase scaffolding protein with metastasis-suppressing activity, is transcriptionally downregulated in Src-transformed cells through the recruitment of HDAC1 to a Src-responsive proximal promoter site charged with Sp1, Sp3 and USF1. However, the ectopic expression of these proteins formed a suppressive complex in Src-transformed but not in parental NIH3T3 cells, suggesting the involvement of additional repressor factors. Transcription Factor TFII-I (Gtf2i) was identified by mass spectrometry as being associated with the SSeCKS promoter complex in NIH3T3/Src cells, and moreover, the Src-induced tyrosine phosphorylation of TFII-I significantly increased its binding to the SSeCKS proximal promoter. siRNA-mediated knockdown of TFII-I or the expression of TFII-IY248/249F caused the derepression of SSeCKS in NIH3T3/Src cells. Taken with previous data showing that the tyrosine phosphorylation of TFII-I facilitates its nuclear translocation, these data suggest that Src-family kinase-mediated phosphorylation converts a portion of TFII-I into a transcriptional repressor.
Keywords: Src, SSeCKS/Gravin/AKAP12, Sp1, Sp3, USF1, HDAC1, TFII-I, Gtf2i, mass spectrometry
INTRODUCTION
The Src oncogene induces oncogenic transformation by several means, including altering signaling and cytoskeletal pathways through the direct phosphorylation of substrates via its intrinsic protein-tyrosine kinase activity [1][2]. A less studied mechanism involves the transcriptional regulation of genes that either promote or suppress oncogenic transformation [3]. A small number of studies have identified gene expression signatures regulated by oncogenically-activated Src [4][5], and these have been used to predict cancer clinical outcomes and subtypes [6–9].
A handful of Src-induced or suppressed genes have been studied in detail, and the Src-dependent transcriptional mechanisms governing the differential expression of these genes often involve seemingly opposite effects by the same transcription factor. For example, Src upregulates transcription of the urokinase receptor (uPAR), matrilysin, CD11c and prostaglandin synthase-2 genes via increased binding of Sp1 and AP-1 to proximal promoter sites [10–13]. In contrast, downregulation of the SSeCKS/Gravin/AKAP12 and QR1 genes by Src requires Sp1 and AP1 promoter binding events, respectively [14–16]. Indeed, Sp1 and AP1 can be influenced to either induce or repress gene transcription by cell- or cancer-specific transcription factors and/or post-translational modifications [17][18]. Thus, it is likely that Src alters their activity by controlling the expression of co-activators or co-repressors, or by directly modifying transcription factor activity through tyrosine phosphorylation.
Transcription Factor II-I (TFII-I), also known as General Transcription factor 2i (encoded by the GTF2I gene), is a ubiquitously-expressed protein first identified as a positive regulator of mitogen-induced c-fos transcription (reviewed in [19]. Its ability to function in transcriptional regulation was subsequently shown to require tyrosine phosphorylation in response to growth factor signaling [20][21] or to B or T cell receptor activation [22][23]. The direct phosphorylation of TFII-I by mitogen-activated Src on Y248 facilitates its nuclear translocation, thereby facilitating its association with c-fos promoter sites and the subsequent induction of c-fos transcriptional activity [24]. Several TFII-I splice variant products exist that exhibit non-overlapping cellular distribution, suggesting varying functions [25]. For example, TFII-Iβ is found in the nucleus constitutively whereas mitogens induce the nuclear translocation of TFII-IΔ and the egress of TFII-Iβ, and thus, TFII-IΔ is mainly responsible for growth factor induced c-Fos transcription [26]. Interestingly, the TFII-IΔ isoform also binds ERK1/2 via its poY248 domain and then shuttles it to the nucleus in response to mitogens, thereby transducing growth factor-induced cell cycle progression [26]. Although the TFII-I-related protein family member, BEN, can repress transcription of TFII-I-induced genes such as goosecoid [27], TFII-I has mainly been viewed as a transcriptional activator, in part, by its ability to bind to histone deacetylases (HDAC), thereby relieving their repressive activity at promoter sites[28]. However, TFII-I can function as a transcriptional repressor [29].
In analyzing how oncogenic Src induced the transcriptional downregulation of the SSeCKS/Gravin/AKAP12 (“SSeCKS”) gene, we identified a minimal Src-responsive proximal promoter sequence (−47/−106) requiring the binding of USF1, Sp1, Sp3 and HDAC1 [14]. Paradoxically, the binding of Sp1 and Sp3 to this promoter domain was increased in Src-transformed compared to NIH3T3 cells, although the forced overexpression of either or both of these proteins in NIH3T3 cells induced, rather than suppressed SSeCKS expression. This suggests that Src converts Sp1/Sp3 into repressive factors through modification or through the expression of co-repressors. Here, we identify TFII-I by mass spectrometry as a factor with increased binding to the Src-responsive SSeCKS proximal promoter/protein complex, and demonstrate that the downregulation of TFII-I or the expression of TFII-IY248F is sufficient to derepress SSeCKS expression in Src-transformed NIH3T3. Our data provide the first example of TFII-I repressor activity mediated by Src-induced phosphorylation, and they elucidate the mechanism by which Src promotes oncogenic progression through the transcriptional downregulation of the SSeCKS tumor suppressor.
MATERIALS and METHODS
Cell culture, plasmids and nuclear lysate preparation
The growth of NIH3T3 and Src/3T3 cells, and the production of nuclear lysates was described previously [14]. The mouse TFII-I-specific siRNA (sense: 5′-UCAGCUCCAUGAGGAGGAUCU-3′; antisense: 5′-AUCCUCCUCAUGGAGCUGAUU-3′), and scrambled siRNA control (sense: 5′-UACCACUGAAGUUACUCAGCC-3′) described previously [26], were synthesized by IDT (Coralville, IA), and the silencing of transfected cells was for 10 days. Plasmids expressing GST-tagged WT- or Y248/249F-TFII-IΔ were gifts of Ananda Roy (Tufts University School of Medicine) and were described previously [24]. Stable cell lines were produced by co-transfecting the TFII-I plasmids along with pEGFP/neo (Clontech/Takara, Mountainview, CA) and then selecting for growth in media supplemented with 500 μg/mL G418 (Invitrogen, Carlsbad, CA).
Oligonucleotide pulldown and mass spectrometry
Pulldown of proteins associated with biotinylated double-stranded oligonucleotides representing the αSSeCKS proximal promoter (Fig. 1A) was described previously [14]. Proteins that were associated with the SSeCKS promoter oligonucleotides after three washes were separated by SDS-PAGE, and after the gels were stained with Deep Purple (GE Healthcare; http://www.gehealthcare.com), protein bands were scanned using a Typhoon 9410 Imager (GE Healthcare). Protein bands of interest were excised either manually or with an Ettan SpotPicker (GE Healthcare) and placed in microtubes, (0.6 mL, Axygen, pre-washed with 18 MΩ·cm water (Mili-Q) and MeOH). In-gel digestion with trypsin was performed according to standard procedures routinely used in the RPCI Proteomics Facility. Briefly, the gel pieces were destained with 50% acetonitrile (ACN)/100 mM ammonium bicarbonate (NH4HCO3) (200 μL) for 30 min with constant mixing (MixMate, Eppendorf, Westbury, NY). After removal of the solution, the gel pieces were dehydrated with ACN (100 μL) for 15 min at room temperature (RT) and dried in a Speedvac concentrator (Eppendorf). The dried gel pieces were reduced with 10 mM DTT (Sigma)/100 mM NH4HCO3 at RT for 45 min. The solution was removed, and the samples were alkylated with 50 mM iodoacetamide (Sigma, St. Louis, MO)/100 mM NH4HCO3 (200 μL) at RT in the dark for 30 min. After removal of the solution, the gel pieces were washed with of 100 mM NH4HCO3 (200 μL) and incubated with 50% ACN/100 mM NH4HCO3 (200 μL) at RT for 10 min. Gel pieces were dehydrated with ACN (100 μL), dried in a Speedvac concentrator and digested with trypsin (Promega, Madison, WI; 10 ng/μL in 10% ACN/40 mM NH4HCO3, 30–50 μL) at 37°C for 16 hrs. The digests were extracted twice with 50% ACN/0.1% TFA (100 μL) at RT for 60 min with constant mixing. The extracts were pooled and dried in a Speedvac concentrator (Thermo Scientific, Waltham, MA), and each sample was then reconstituted with 8 μL of 2% formic acid (FA).
Figure 1. Identification of proteins binding to the Src-responsive domain in the αSSeCKS proximal promoter.
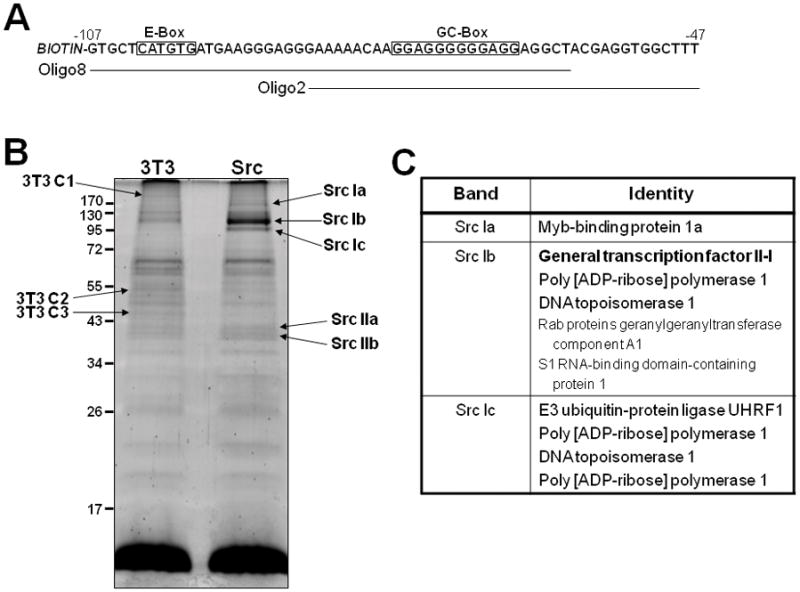
A. The minimal αSSeCKS proximal promoter region required for Src-responsiveness (−47/−107), with two biotin-labeled double-stranded oligonucleotides used for protein binding identification. Note the E- and GC-boxes previously shown to bind USF1 and Sp1/Sp3, respectively [14]. B. Proteins from nuclear lysates of NIH3T3 (“3T3”) and Src/3T (“Src”) cells (1 mg total protein each) that bind to biotin labeled, double-stranded oligonucleotides spanning −60 to −106 of the αSSeCKS proximal promoter (“oligo 8”, Fig. 1C), stained with Deep Purple according to the manufacturer’s description. Protein bands uniquely expressed in either 3T3 or Src cells (3T3 C1-3 and Src Ia–c and IIa,b) as identified by arrows. C. Identity of the proteins in bands Src Ia–c, with the top protein in each box being the most abundantly identified by MS.
LC-MS/MS Analysis
The tryptic digests (4 μL of each sample) were analyzed by LC nanoelectrospray-tandem mass spectrometry (LC-ESI-MS/MS) using a nanoACQUITY UPLC (Waters, Milford, MA) coupled through a nebulization-assisted nanospray ionization source to a Q-ToF Premier mass spectrometer (Waters/Micromass). The LC consisted of a trap column (Symmetry C18, 5μ, 180μ × 20mm, Waters), followed by separation on an analytical column (Atlantis C18, 3μ, 100μ × 10cm, Waters). Samples were loaded, trapped, and washed at a flow rate of 4 μL/min with 98% solvent A (0.1% FA)/2% solvent B (ACN containing 0.1% FA) for 5 min. Peptides were eluted with a gradient of 99% A/1% B to 50% A/50% B for 70 min at 0.4μL/min, 10% A/90% B for 20 min at 0.6 μL/min, and then 10% A/90% B at 0.6 μL/min for 10 min. Throughout the gradient, the mass spectrometer was programmed (Data Dependent Acquisition experiment, DDA) to monitor ions with m/z in the range of 320–1620, and ions with +2 to +4 charges only were selected for MS/MS experiments using the preset DDA collision energy parameters.
Database Search, Peptide and Protein Identification
MS/MS spectra were processed and transformed to the PKL file format using Proteinlynx Global Server v2.2 (Waters/Micromass) and the default parameters of MaxEnt3 (Waters/Micromass). The PKL files were used to search the Rodentia subset of the Swiss-Prot database (release 53.2, containing 21,110 sequences) using a locally installed version of MASCOT (Matrix Science, Boston, MA, v2.2.0). The search parameters were as follows: trypsin as the proteolytic enzyme with 2 possible missed cleavages, carboxyamidomethylation of cysteine as a fixed modification, oxidation of methionine and phosphorylation of STY as a variable modification, the allowable mass error was 100 ppm for peptides and 100 mDa for fragment ions, peptide charge was set to 2+ and 3+, the instrument was set to ESI-QUAD-TOF. The Mascot default significance threshold of p<0.05 for assignments was used in the searches and a minimum of two unique peptides were used as a criteria for a match.
Chromatin immunoprecipitation (ChIP)
ChIP assays were performed as described previously [14] using TFII-I antibody (Cell Signaling, Beverly, MA; cat. #4562). The primers for PCR amplification of SSeCKS α proximal promoter sequence between −270 and +33 were 5′-TGCTGCTCCTGAACCTTCTG-3′ and 5′-GATCCTGCTGAGAACACACC-3′.
Immunoblotting, immunoprecipitation and RT-PCR
Immunoblotting (IB) and immunoprecipitation (IP) were performed as described previously [14] using the following antibodies (Ab): rabbit anti-rat SSeCKS [30], TFII-I (Cell Signaling, Beverly, MA; cat. #4562, 1:1000 dilution), GAPDH (Santa Cruz Biotechnology, Santa Cruz, CA; cat. #sc-257778, 1:1000 dilution), antiphosphotyrosine MAb-4G10 (Millipore/Upstate, Billerica, MA). The semi-quantitative RT-PCR for SSeCKS α isoform and β-actin was described previously [14].
RESULTS and DISCUSSION
We previously showed that a minimal Src-responsive element in the SSeCKS promoter (−47/−106) required USF1 binding to an upstream E-box and Sp1/Sp3 binding to a downstream GC-box, plus the association of HDAC1 [14]. Moreover, whereas the level of USF1 binding did not change between NIH3T3 and Src/3T3 cells, the binding of Sp1/Sp3 increased at least 3-fold in Src/3T3 nuclear lysates compared to those of NIH3T3, corresponding to an overall transcriptional suppression of the SSeCKS gene by oncogenic Src. In some systems, Sp3 can induce transcriptional suppression by competing with Sp1 for DNA binding sites, normally a transcriptional activator [31], from promoter sites [32]. However, in our system, the ectopic expression of Sp1 and/or Sp3 led to SSeCKS upregulation. Alternatively, Sp1/Sp3 can be converted into repressors upon the recruitment of HDACs [33–35], and indeed, the forced co-expression of Sp1, Sp3 and HDAC1 induced SSeCKS transcriptional downregulation in untransformed NIH3T3 cells [14]. However, the levels of total and nuclear Sp1, Sp3 or USF1 did not vary between NIH3T3 and Src/3T3 cells, strongly suggesting that Src either modifies one or more of these factors or induces the expression of repressor proteins that complex with the Sp1/Sp3/USF1/HDAC1 complex.
In order to address the latter possibility, nuclear lysates from NIH3T3 and Src/3T3 cells were bound to a biotin-labeled, double-stranded oligonucleotide spanning the minimal Src-responsive SSeCKS promoter (Fig. 1A; “oligo 8”). The DNA proteins complexes were precipitated by binding to streptavidin-agarose beads, and after washing, the associated proteins were identified by denaturing SDS-PAGE followed by staining with Deep Purple. Fig. 1B shows five bands found more abundantly in the Src/3T3 sample, with “Src Ib” being the most prominent, as well as three bands with more abundance in NIH3T3 sample. Given the increased binding of Sp1 and Sp3 to this SSeCKS promoter region in the Src/3T3 cells [14], we analyzed the Src Ia–c bands first. Protein bands were excised from the gels, digested to completion with trypsin and then subjected to LC-MS/MS analysis. The identities of the proteins in each band are shown in Fig. 1C, with the most abundant peptides ordered from top to bottom.
We focused on general transcription factor II-I (TFII-I) because its apparent binding to the SSeCKS proximal promoter region was the most abundantly increased in the Src/3T3 cells. Moreover, TFII-I is known to interact directly with USF1 [36] via E-box promoter sequences, factors required for Src-responsiveness by the SSeCKS proximal promoter. To confirm that TFII-I binds to the SSeCKS promoter, two biotin labeled, double-stranded oligonucleotides were used to precipitate proteins from nuclear lysates of NIH3T3 or Src/3T3 cells: “oligo 8”, used in the original pulldown assay described in Fig. 1A, contains both the E- and GC-boxes and is sufficient to bind USF1 and Sp1/Sp3, respectively [14]; “oligo 2” contains only the GC-box plus several downstream bases from oligo 8. Although the levels of total TFII-I (β and Δ) isoforms) in NIH3T3 and Src/3T3 lysates are similar (see “input” lanes), there was a preferential binding of TFII-I to both oligonucleotides in the Src/3T3 lysates (Fig. 2A). The absence of detectable TFII-I binding to streptavidin beads (“no oligo”) argues against the possibility of non-specific binding in this system. The enhanced recruitment of TFII-I to the αSSeCKS proximal promoter in Src/3T3 cells was confirmed by ChIP analysis (Fig. 2B). We then investigated whether TFII-I plays a direct role in suppressing SSeCKS expression in Src-transformed cells. Treatment of Src/3T3 cells with TFII-I-specific siRNA, shown previously to knockdown mouse both TFII-Iβ and Δ isoforms [26], induced a 2.5-fold decrease in TFII-I protein levels (Fig. 2C, upper panel) and a concomitant increase in αSSeCKS RNA levels as determined by semi-quantitative RT-PCR (Fig. 2C, lower panel). Treatment with a control siRNA (scrambled) affected neither TFII-I protein nor SSeCKS RNA levels. This indicates that TFII-I plays a role in the Src-mediated downregulation of SSeCKS.
Figure 2. TFII-I binds to the αSSeCKS proximal promoter and participates in the downregulation of SSeCKS by Src.
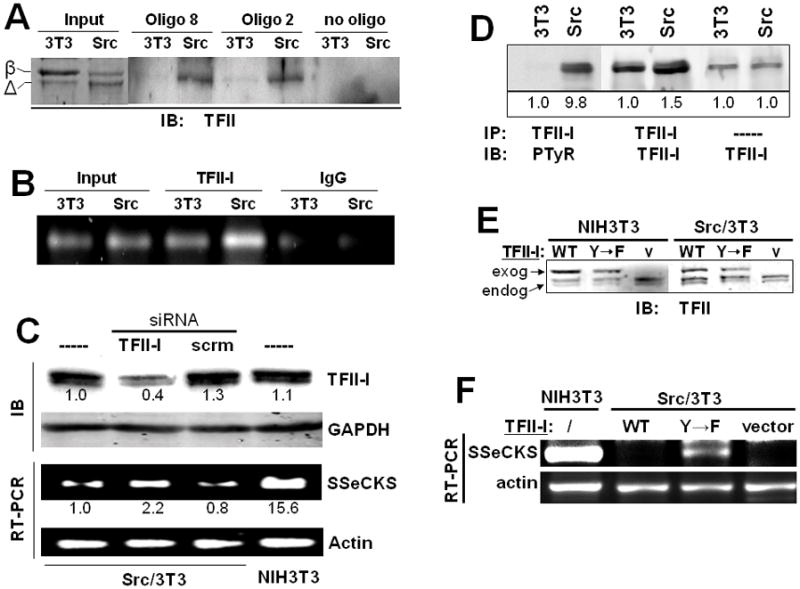
A. Analysis of TFII-I binding to “oligo 8” or “oligo 2” probes (see Fig. 1C), or no oligo, from 0.5 mg of 3T3 or Src nuclear lysates, probed by IB using a TFII-I-specific Ab. The two major TFII-I murine isoforms, β and Δ, are identified at left. Input, 10 μg of nuclear lysate from 3T3 or Src cells. B. ChIP analysis confirms the enhanced binding of TFII-I to αSSeCKS proximal promoter in Src/3T3 cells. C. TFII-I knockdown induces αSSeCKS transcript levels. Top panel, IB analysis of TFII-I or GAPDH protein levels in the cells from the top panel. These data are typical of three independent experiments. Bottom panel, Semi-quantitative RT-PCR of αSSeCKS or β-actin, as described previously [14], from 1 μg of total RNA isolated from Src/3T3 cells treated with TFII-I or scrambled siRNA, or from untreated Src/3T3 or NIH3T3 cells. The relative signal intensities derived from densitometry analysis are shown under the respective bands (SEM for triplicate experiments, +/− 0.2). D. Analysis of phosphotyrosyl-TFII-I in total cell 3T3 and Src lysates, based on the IP of 0.5 mg cell lysate with TFII-I-specific Ab followed by IB with either PTyr or TFII-I Ab. The two panels on the right represent a direct TFII-I-specific IB analysis of 10 μg of total 3T3 or Src cell lysate. The relative signal intensities derived from densitometry analysis are shown under the respective bands (SEM for triplicate experiments, +/− 0.2). E. TFII-I-specific IB analysis of NIH3T3 and Src/3T3 cells stably expressing GST-fused WT- or Y248/249F (“Y->F”)-TFII-I, or empty vector (“v”). Endogenous (“endog.”) and exogenous (“exog.”) TFII-I proteins are identified by arrows. F. Semi-quantitative RT-PCR analysis of αSSeCKS and β-actin transcript levels in Src/3T3 cells stably expressing GST-fused WT- or Y248/249F (“Y->F”)-TF-II-I, or empty vector (“Vector”). A parallel analysis of RNA from untreated NIH3T3 cells is shown on the left. Note that the ectopic expression of WT or the Y→F TFII-I mutant in NIH3T3 cells had no effect on αSSeCKS RNA levels (data not shown). These data are typical of three independent experiments.
In agreement with previous data [24], the relative tyrosine phosphorylation of TFII-I was increased 9.8-fold in the Src/3T3 cells compared to NIH3T3, even when normalized to the slightly higher (1.5-fold) TFII-I protein levels in the Src/3t3 cells (Fig. 2D). Because Src-induced transformation involved TFII-I tyrosine phosphorylation rather than changes in protein abundance, we addressed whether the expression of TFII-IY248/249F mutant, incapable of being phosphorylated by Src, could derepress SSeCKS. Thus, stable Src/3T3 cells expressing WT or Y248/249F TFII-I (or empty vector) were produced. Representative clones were isolated that showed similar levels of these ectopic proteins (Fig. 2E; note that the exogenous GST-TFII-I proteins are 27kDa larger than the endogenous forms). RT-PCR analysis indicates that expression of the Y->F TFII-I mutant, but not WT TFII-I, resulted in increased αSSeCKS transcript levels (Fig. 2F). Taken with the data above, this finding strengthens the notion that Src-induced phosphorylation of TFII-I followed by its recruitment to the proximal SSeCKS promoter results in the repression of SSeCKS transcription. This further implies that for some genes, Src-induced tyrosine phosphorylation converts TFII-I into a transcriptional co-repressor. Importantly, the expression of WT or Y248/249F TFII-I, or TFII-I siRNA, was not sufficient to either induce or suppress morphological transformation in NIH3T3 or Src/3T3 cells, respectively, or to suppress the loss of contact-inhibited growth in Src/3T3 cells (Fig. 3).
Figure 3. TFII-I protein or phosphotyrosyl levels are not sufficient to induce or suppress oncogenic transformation.
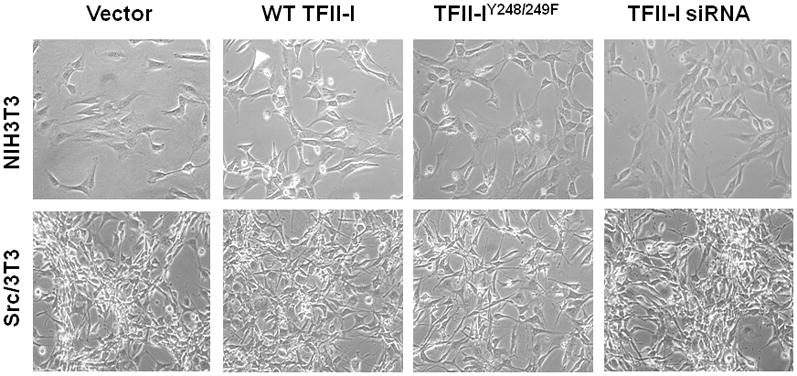
Phase contrast images of NIH3T3 and Src/3T3 stable transfectants expressing vector, GST-fused WT- or Y248/249F (“Y->F”)-TFII-I, or TFII-I siRNA.
Although we demonstrated that TFII-I is involved in the transcriptional repression of SSeCKS by Src, SSeCKS downregulation most likely requires additional factors (e.g.- HDAC1, as we described previously [14]), since antagonism of TFII-I expression or phosphorylation only led to partial restoration of the SSeCKS expression levels found in untransformed NIH3T3 cells (Figs. 2C & F). Indeed, several of the minor binding proteins we identified in the mass spectrometry analysis in Fig. 1A could contribute to SSeCKS downregulation by Src. For example, the Myb binding protein 1a (in the “Src 1a” band) can function as a transcriptional co-repressor with NF-κB [37], and the UHRF1 E3 ubiquitin ligase (in the “Src 1c” band) is known to repress the transcription of the Tip60 gene through an interaction of HDAC1 [38]. It is also possible that the partial restoration of TFII-I was due to limited siRNA effects (Fig. 2C) or expression levels of exogenous TFII-IY248/249F (Fig. 2F).
Model
We showed previously that the overexpression of USF1, Sp1 and Sp3 resulted in the induction of the αSSeCKS promoter, driven by cognate E- and GC-box cis-acting motifs in the −106/−47 proximal promoter [14] (Fig. 4). EMSA and ChIP analyses indicated that the binding activity of Sp1/Sp3 for this proximal promoter increased in Src-transformed cells, and given that the downregulation of SSeCKS in these cells was sensitive to histone deacetylase inhibitors, we were able to show that the recruitment of HDAC1 helped convert USF1/Sp1/Sp3 to a repressor complex in the Src/3T3 cells. However, the fact that overexpression of all these proteins in untransformed cells only led to marginal downregulation of αSSeCKS promoter activity caused us to believe that additional repressive factors were critical for the greater (15-fold) SSeCKS downregulation by Src. The current data strongly suggest that recruitment of a Src-phosphorylated TFII-I enhances the transcriptional repression of SSeCKS (Fig. 4). Thus, the transcriptional downregulation of so-called Type II tumor suppressors such as SSeCKS/Gravin/AKAP12 can be facilitated by multiple, additive mechanisms during the process of oncogenic progression.
Figure 4. Model for transcriptional repression of αSSeCKS by Src.
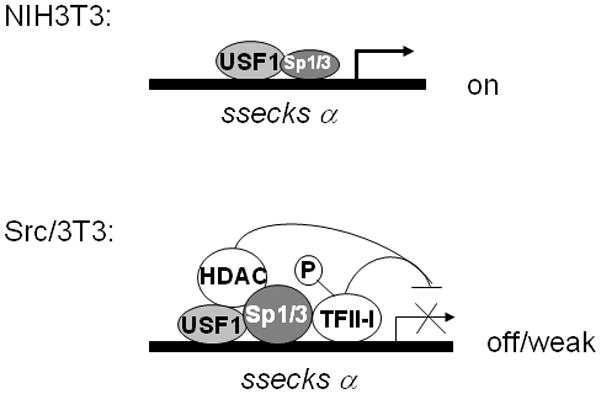
In NIH3T3 cells, the transcription factors USF1 and Sp1/Sp3 bind to their cognate αSSeCKS proximal promoter sites and activate SSeCKS transcription. In Src/3T3 cells, tyrosine phosphorylated TFII-I (induced by Src) binds to the αSSeCKS proximal promoter complex, which also contains the co-repressor HDAC1 (which is recruited via elevated Sp1/Sp3 binding), resulting in the transcriptional repression of αSSeCKS.
CONCLUSIONS
Our data suggest that the tyrosine phosphorylation of TFII-I by v-Src induces its association with a Sp1/Sp3-USF1-HDAC1 complex on the SSeCKS proximal promoter, resulting in the transcriptional repression of the SSeCKS/Gravin/AKAP12 metastasis suppressor gene (Fig. 4).
Acknowledgments
This study is supported by NIH grant CA94108 and DOD grants PC040256 and PC061246 (I.H.G.), and in part by NIH/NCI Cancer Center Support Grant 2P30 CA016056.
References
- 1.Frame MC. Newest findings on the oldest oncogene; how activated src does it. J Cell Sci. 2004;117:989–98. doi: 10.1242/jcs.01111. [DOI] [PubMed] [Google Scholar]
- 2.Jove R, Hanafusa H. Cell Transformation by the Viral src Oncogene. Ann Rev Cell Biol. 1987;3:31–56. doi: 10.1146/annurev.cb.03.110187.000335. [DOI] [PubMed] [Google Scholar]
- 3.Dehbi M, Bedard P-A. Regulation of gene expression in oncogenically transformed cells. Biochem Cell Biol. 1993;70:980–97. doi: 10.1139/o92-142. [DOI] [PubMed] [Google Scholar]
- 4.Bild AH, Yao G, Chang JT, Wang Q, Potti A, Chasse D, Joshi MB, Harpole D, Lancaster JM, Berchuck A, Olson JA, Jr, Marks JR, et al. Oncogenic pathway signatures in human cancers as a guide to targeted therapies. Nature. 2006;439:353–7. doi: 10.1038/nature04296. [DOI] [PubMed] [Google Scholar]
- 5.Liu Y, Gao L, Gelman IH. SSeCKS/Gravin/AKAP12 attenuates expression of proliferative and angiogenic genes during suppression of v-Src-induced oncogenesis. BMC Cancer. 2006;6:105–17. doi: 10.1186/1471-2407-6-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Acharya CR, Hsu DS, Anders CK, Anguiano A, Salter KH, Walters KS, Redman RC, Tuchman SA, Moylan CA, Mukherjee S, Barry WT, Dressman HK, et al. Gene expression signatures, clinicopathological features, and individualized therapy in breast cancer. JAMA. 2008;299:1574–87. doi: 10.1001/jama.299.13.1574. [DOI] [PubMed] [Google Scholar]
- 7.Creighton CJ. Multiple oncogenic pathway signatures show coordinate expression patterns in human prostate tumors. PLoS One. 2008;3:e1816. doi: 10.1371/journal.pone.0001816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sabbah M, Emami S, Redeuilh G, Julien S, Prevost G, Zimber A, Ouelaa R, Bracke M, De Wever O, Gespach C. Molecular signature and therapeutic perspective of the epithelial-to-mesenchymal transitions in epithelial cancers. Drug Resist Updat. 2008;11:123–51. doi: 10.1016/j.drup.2008.07.001. [DOI] [PubMed] [Google Scholar]
- 9.Zhang XH, Wang Q, Gerald W, Hudis CA, Norton L, Smid M, Foekens JA, Massague J. Latent bone metastasis in breast cancer tied to Src-dependent survival signals. Cancer Cell. 2009;16:67–78. doi: 10.1016/j.ccr.2009.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leupold JH, Asangani I, Maurer GD, Lengyel E, Post S, Allgayer H. Src induces urokinase receptor gene expression and invasion/intravasation via activator protein-1/p-c-Jun in colorectal cancer. Mol Cancer Res. 2007;5:485–96. doi: 10.1158/1541-7786.MCR-06-0211. [DOI] [PubMed] [Google Scholar]
- 11.Noti JD, Reinemann BC, Petrus MN. Sp1 binds two sites in the CD11c promoter in vivo specifically in myeloid cells and cooperates with AP1 to activate transcription. Mol Cell Biol. 1996;16:2940–50. doi: 10.1128/mcb.16.6.2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rivat C, Le Floch N, Sabbah M, Teyrol I, Redeuilh G, Bruyneel E, Mareel M, Matrisian LM, Crawford HC, Gespach C, Attoub S. Synergistic cooperation between the AP-1 and LEF-1 transcription factors in the activation of the matrilysin promoter by the src oncogene: implications in cellular invasion. The FASEB Journal. 2003;17:1721–3. doi: 10.1096/fj.03-0132fje. [DOI] [PubMed] [Google Scholar]
- 13.Xie W, Herschman HR. v-src Induces Prostaglandin Synthase 2 Gene Expression by Activation of the c-Jun N-terminal Kinase and the c-Jun Transcription Factor. J Biol Chem. 1995;270:27622–8. doi: 10.1074/jbc.270.46.27622. [DOI] [PubMed] [Google Scholar]
- 14.Bu Y, Gelman IH. v-Src-mediated Down-regulation of SSeCKS Metastasis Suppressor Gene Promoter by the Recruitment of HDAC1 into a USF1-Sp1-Sp3 Complex. J Biol Chem. 2007;282:26725–39. doi: 10.1074/jbc.M702885200. [DOI] [PubMed] [Google Scholar]
- 15.Pierani A, Pouponnot C, Calothy G. Transciptional downregulation of the retina-specific QR1 gene by pp60v-src and identification of a novel v-src responsive unit. Mol Cell Biol. 1993;13:3401–14. doi: 10.1128/mcb.13.6.3401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Provot S, Pouponnot C, Lecoq O, Calothy G, Felder-Schmittbuhl MP. Characterization of a novel quiescence responsive element downregulated by v-Src in the promoter of the neuroretina specific QR1 gene. Oncogene. 2000;19:4736–45. doi: 10.1038/sj.onc.1203837. [DOI] [PubMed] [Google Scholar]
- 17.Verde P, Casalino L, Talotta F, Yaniv M, Weitzman JB. Deciphering AP-1 function in tumorigenesis: fra-ternizing on target promoters. Cell Cycle. 2007;6:2633–9. doi: 10.4161/cc.6.21.4850. [DOI] [PubMed] [Google Scholar]
- 18.Wierstra I. Sp1: emerging roles--beyond constitutive activation of TATA-less housekeeping genes. Biochem Biophys Res Commun. 2008;372:1–13. doi: 10.1016/j.bbrc.2008.03.074. [DOI] [PubMed] [Google Scholar]
- 19.Roy AL. Transcription factor TFII-I conducts a cytoplasmic orchestra. ACS Chem Biol. 2006;1:619–22. doi: 10.1021/cb6004323. [DOI] [PubMed] [Google Scholar]
- 20.Kim DW, Cheriyath V, Roy AL, Cochran BH. TFII-I enhances activation of the c-fos promoter through interactions with upstream elements. Mol Cell Biol. 1998;18:3310–20. doi: 10.1128/mcb.18.6.3310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Novina CD, Cheriyath V, Roy AL. Regulation of TFII-I activity by phosphorylation. J Biol Chem. 1998;273:33443–8. doi: 10.1074/jbc.273.50.33443. [DOI] [PubMed] [Google Scholar]
- 22.Sacristan C, Schattgen SA, Berg LJ, Bunnell SC, Roy AL, Rosenstein Y. Characterization of a novel interaction between transcription factor TFII-I and the inducible tyrosine kinase in T cells. Eur J Immunol. 2009;39:2584–95. doi: 10.1002/eji.200839031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sacristan C, Tussie-Luna MI, Logan SM, Roy AL. Mechanism of Bruton’s tyrosine kinase-mediated recruitment and regulation of TFII-I. J Biol Chem. 2004;279:7147–58. doi: 10.1074/jbc.M303724200. [DOI] [PubMed] [Google Scholar]
- 24.Cheriyath V, Desgranges ZP, Roy AL. c-Src-dependent transcriptional activation of TFII-I. J Biol Chem. 2002;277:22798–805. doi: 10.1074/jbc.M202956200. [DOI] [PubMed] [Google Scholar]
- 25.Cheriyath V, Roy AL. Alternatively spliced isoforms of TFII-I. Complex formation, nuclear translocation, and differential gene regulation. J Biol Chem. 2000;275:26300–8. doi: 10.1074/jbc.M002980200. [DOI] [PubMed] [Google Scholar]
- 26.Hakre S, Tussie-Luna MI, Ashworth T, Novina CD, Settleman J, Sharp PA, Roy AL. Opposing functions of TFII-I spliced isoforms in growth factor-induced gene expression. Mol Cell. 2006;24:301–8. doi: 10.1016/j.molcel.2006.09.005. [DOI] [PubMed] [Google Scholar]
- 27.Ku M, Sokol SY, Wu J, Tussie-Luna MI, Roy AL, Hata A. Positive and negative regulation of the transforming growth factor beta/activin target gene goosecoid by the TFII-I family of transcription factors. Mol Cell Biol. 2005;25:7144–57. doi: 10.1128/MCB.25.16.7144-7157.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tussie-Luna MI, Bayarsaihan D, Seto E, Ruddle FH, Roy AL. Physical and functional interactions of histone deacetylase 3 with TFII-I family proteins and PIASxbeta. Proc Natl Acad Sci U S A. 2002;99:12807–12. doi: 10.1073/pnas.192464499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hakimi MA, Dong Y, Lane WS, Speicher DW, Shiekhattar R. A candidate X-linked mental retardation gene is a component of a new family of histone deacetylase-containing complexes. J Biol Chem. 2003;278:7234–9. doi: 10.1074/jbc.M208992200. [DOI] [PubMed] [Google Scholar]
- 30.Lin X, Tombler E, Nelson PJ, Ross M, Gelman IH. A novel src- and ras-suppressed protein kinase C substrate associated with cytoskeletal architecture. J Biol Chem. 1996;271:28430–8. doi: 10.1074/jbc.271.45.28430. [DOI] [PubMed] [Google Scholar]
- 31.Safe S, Abdelrahim M. Sp transcription factor family and its role in cancer. Eur J Cancer. 2005;41:2438–48. doi: 10.1016/j.ejca.2005.08.006. [DOI] [PubMed] [Google Scholar]
- 32.Hagen G, Muller S, Beato M, Suske G. Sp1-mediated transcriptional activation is repressed by Sp3. EMBO J. 1994;13:3843–51. doi: 10.1002/j.1460-2075.1994.tb06695.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Choi HS, Lee JH, Park JG, Lee YI. Trichostatin A, a histone deacetylase inhibitor, activates the IGFBP-3 promoter by upregulating Sp1 activity in hepatoma cells: alteration of the Sp1/Sp3/HDAC1 multiprotein complex. Biochem Biophys Res Commun. 2002;296:1005–12. doi: 10.1016/s0006-291x(02)02001-6. [DOI] [PubMed] [Google Scholar]
- 34.Doetzlhofer A, Rotheneder H, Lagger G, Koranda M, Kurtev V, Brosch G, Wintersberger E, Seiser C. Histone deacetylase 1 can repress transcription by binding to Sp1. Mol Cell Biol. 1999;19:5504–11. doi: 10.1128/mcb.19.8.5504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhao S, Venkatasubbarao K, Li S, Freeman JW. Requirement of a specific Sp1 site for histone deacetylase-mediated repression of transforming growth factor beta Type II receptor expression in human pancreatic cancer cells. Cancer Res. 2003;63:2624–30. [PubMed] [Google Scholar]
- 36.Roy AL, Du H, Gregor PD, Novina CD, Martinez E, Roeder RG. Cloning of an inr- and E-box-binding protein, TFII-I, that interacts physically and functionally with USF1. EMBO J. 1997;16:7091–104. doi: 10.1093/emboj/16.23.7091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Owen HR, Elser M, Cheung E, Gersbach M, Kraus WL, Hottiger MO. MYBBP1a is a novel repressor of NF-kappaB. J Mol Biol. 2007;366:725–36. doi: 10.1016/j.jmb.2006.11.099. [DOI] [PubMed] [Google Scholar]
- 38.Achour M, Fuhrmann G, Alhosin M, Ronde P, Chataigneau T, Mousli M, Schini-Kerth VB, Bronner C. UHRF1 recruits the histone acetyltransferase Tip60 and controls its expression and activity. Biochem Biophys Res Commun. 2009;390:523–8. doi: 10.1016/j.bbrc.2009.09.131. [DOI] [PubMed] [Google Scholar]