

1. Introduction
Unwarranted proliferative phenotype of VSMCs is an essential feature of a number of vascular pathologies and occlusive diseases such as atherosclerosis, hypertension and arterial and in-stent restenosis. Improved understanding of the vascular proliferative diseases implicate that the inhibition of VSMC proliferation by antiproliferative therapy may be an appropriate strategy to interrupt vascular proliferative diseases [1-3]. Recent studies indicate that inhibition of cell proliferation by targeting cell-cycle regulation is a rational strategy for preventing arterial restenosis and in-stent restenosis, which has resulted in use of drug-coated stents for arresting VSMC cell proliferation following clinical procedures [1, 3-6].
HDAC inhibitors (HDACIs) are a new class of anticancer agents by virtue of their ability to arrest cell proliferation and promote cell differentiation or stimulate apoptosis [7-12]. At molecular level, HDACIs cause reactivation of epigenetically silenced genes by increasing global histone acetylation by inhibiting class I and class II HDACs [7-12]. Global hyperacetylation of histone appears to alter chromatin structure and cause relaxation of chromatin structure, which exposes DNA and allows availability of promoter sites for transcriptional activation [7-12]. Furthermore, evidence suggests that the link between hyperacetylation-induced increased transcriptional activity and growth inhibitory effect of HDACIs is reflected in transcriptional regulation of several cell cycle regulators [7, 8, 10-12].
Butyrate, a dietary HDACI, is a short chain fatty acid derived from the intestinal microbial fermentation of dietary fiber [10-12]. Several epidemiological, animal and interventional studies suggest the protective effects of dietary fiber in chronic diseases such as bowel disorders and colorectal cancer, cancer of other tissues, cardiovascular disease, diabetes, obesity and hypertension [3, 12-18] is linked to bioactivity of butyrate [3, 12, 14-18]. It elicits many cytoprotective, chemopreventive and chemotherapeutic activities mainly through arrest of cell proliferation, induction of apoptosis or stimulation of cell differentiation by selectively altering gene expression but the mechanistic basis for these actions are far from clear [3, 10-12, 18, 19-26]. Butyrate and its derivatives with longer half lives have been developed and being used in animal models and in human studies to treat different cancers [8, 9], hemoglobinopathies [22, 27], cystic fibrosis [23, 24] and Huntington's disease [25, 26]. Conversely, no similar studies are performed to indicate the protective role of butyrate in cardiovascular diseases. However, our studies [3, 12, 28, 29] and studies by others [30] have established arrest of VSMC proliferation by butyrate. Moreover, our cDNA array screening studies detected altered expression of several genes in butyrate arrested VSMC proliferation [31].
In the present study, we investigate the influence of butyrate on histone H3 posttranslational modifications and its consequence on G1-specific cell cycle regulators to elucidate the mechanistic link between chromatin remodeling and antiproliferation action of butyrate in VSMCs. Outcomes of our study indicate interplay between different site-specific posttranslational modifications of histone H3 in butyrate treated VSMCs that seem to alter chromatin structure and organization causing differential expression of both negative and positive regulators of cell cycle resulting in arrest of VSMC proliferation, a possible cause of atherosclerosis and an important critical trait of postangioplasty restenosis and in-stent restenosis.
2. Materials and Methods
2.1. Materials
Antibodies to cyclin D1, cyclin D3, p15INK4B, extracellular signal-regulated kinase 1 and 2 (ERK1/2), histone H3, phospho-histone H3Serine10 (phospho-H3Ser10), acetyl-histone H3Lysine9 (acetyl-H3Lys9), di-methyl-histone H3Lysine9 (di-methyl-H3Lys9), di-methyl-histone H3Lysine4 (di-methyl-H3Lys4), phospho-Rb-Serine807/811, (pRbSer807/811) and horse radish peroxidase (HRP)-conjugated second antibodies were obtained from Cell Signaling (Beverly, MA, USA). Anti-mouse Alexa Fluor 488, anti-rabbit Alexa Fluor 546, and Hoechst were from Molecular Probes (Carlsbad, CA, USA). Chemiluminescence luminol reagent and antibodies to p21Cip1, cdk-2, cdk-4 and cdk-6 were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Antibody to Rb protein was purchased from BD Biosciences (San Jose, CA, USA). Butyrate and antibody to smooth muscle α-actin were obtained from Sigma -Aldrich (St. Louis, MO, USA). The micro BCA protein assay kit was from Pierce (Rockford, IL, USA).
2.2. Cell Culture and Treatments
Rat VSMCs were isolated from thoracic aortas [32, 33] and cultured in complete medium consisting of DMEM supplemented with 10% fetal bovine serum, 100 U/ml penicillin and 100 μg/ml streptomycin at 37°C in a humidified atmosphere of 95% air and 5% CO2. For all experiments, VSMCs were seeded at a ratio of 1:6. One day after splitting, actively growing cells were grown in the absence or presence of 5 mM butyrate or specified concentrations of butyrate for required lengths of time [29, 34]. Culture medium was replaced every other day with fresh medium containing freshly prepared butyrate in sterile phosphate buffered saline (PBS). VSMCs of third to fifteenth passages were used for all studies. All experiments were repeated at least three times unless otherwise mentioned.
2.3. Measurement of cell proliferation
The proliferation of VSMCs was measured as described previously [29, 34]. After the required period of treatment, cells were washed three times with sterile PBS and trypsinized with trypsin-EDTA. Cell numbers were counted by trypan blue exclusion method [29, 34].
2.4. Preparation of Cell Lysates and Western Analysis
Whole cell lysates were prepared as described previously [34]. Equal amounts of denatured protein samples were fractionated on: 7.5% SDS- polyacrylamide gels to immunoblot Rb and phospho-Rb (pRb); 10% SDS- polyacrylamide gels to immunoblot cyclin D1, cyclin D3, ERK1/2, p21Cip1, cdk-2, cdk-4 and cdk-6; and 12% SDS- polyacrylamide gels to immunoblot p15INK4B and unmodified and modified histone H3. Fractionated proteins were transferred to PVDF membrane and processed for immunoblotting with indicated antibodies [34]. Immunodetection was performed with the western blot luminol reagent from Santa Cruz biotechnology. For quantitative measurements, a Molecular Imager FX Pro Plus MultiImager System and Quantity One software (Bio-Rad, CA) were used. Immunoblotting of ERK1/2 was performed with the same lysates to normalize protein loading unless otherwise mentioned.
2.5. Immunofluorescence Staining
VSMCs were fixed in cold methanol and immunostained as described previously [34]. Briefly, fixed cells were blocked with 10% heat inactivated horse serum (HS) in PBS for 1 h at room temperature. Blocked cells were incubated with appropriate antibodies in 1.5% HS for 1 to 3 h followed by three washes with PBS, each for 10 min. Similar washing protocol is used for downstream procedures. Cultures were then incubated with appropriate Alexa Fluor second antibody conjugates in 1.5% HS for 1 h. Following washing, cultures were incubated with 1 μg/ml Hoechst in 1.5% HS for 30 min. After washing with PBS cultures were subjected to fluorescence microscopy using a Nikon fluorescence microscope.
2.6. Statistical analysis
Data is expressed as mean ± SD. Differences were assessed by Student's t-test. Statistically significant difference between data sets was determined at p < 0.01 to < 0.001.
3. Results
3.1. Link between inhibition of VSMC proliferation and temporal dynamics of histone H3 acetylation and phosphorylation by butyrate
The relationship between inhibition of VSMC proliferation and site-specific histone H3 modification by butyrate is assessed by determining the temporal effect of butyrate on VSMC proliferation, and on acetylation of histone H3-Lys9 (H3Lys9) and phosphorylation of histone H3-Ser10 (H3Ser10). The results of the study reveals that while butyrate treatment of VSMCs stimulates striking H3Lys9 acetylation all through the experimental period (Fig. 1A), it also arrests VSMC proliferation dramatically (Fig. 1B). Unlike butyrate treated VSMCs, untreated VSMCs display proliferative phenotype with very low levels of H3Lys9 acetylation. Conversely, induction of H3Ser10 phosphorylation is exclusive to untreated proliferating VSMCs, although the extent of H3Ser10 phosphorylation is significantly reduced in a time-dependent manner compared to 24 h untreated VSMCs (Fig. 1A). In contrast, butyrate treated VSMCs barely exhibit H3Ser10 phosphorylation. Enhanced acetylation of H3Lys9 in butyrate treated VSMCs and increased phosphorylation of H3Ser10 in untreated proliferating VSMCs are confirmed by intracellular immunostaining (Fig. 2). While H3Lys9 acetylation is specifically enhanced in the nuclear region of butyrate treated VSMCs, H3Ser10 phosphorylation is solely increased in the nuclei of untreated VSMCs.
Fig. 1. Link between butyrate-induced posttranslational modification of histone H3 and butyrate-inhibited VSMC proliferation.
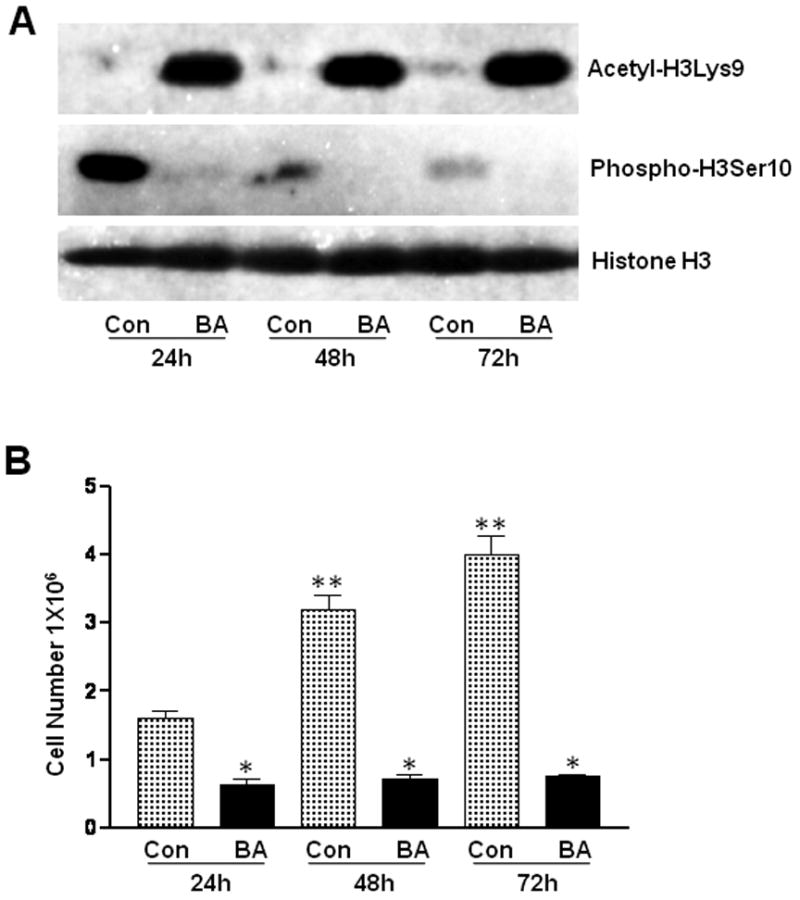
Proliferating VSMCs were untreated (Con) or treated with 5 mM butyrate (BA) for indicated periods of time. A. The levels of histone H3Lys9 acetylation (Acetyl-H3Lys9) and histone H3Ser10 phosphorylation (Phospho-H3Ser10) were determined by western analyses. Histone H3 was used for normalizing protein loading. B. Proliferation of respective VSMC cultures was assessed by counting number of VSMCs. The data is presented as mean ± S.D. *p< 0.001 compared to untreated VSMCs and **p<0.001 compared to 24 hours untreated VSMCs.
Fig. 2. Analysis of H3Lys9 acetylation and H3Ser10 phosphorylation of histone H3 in butyrate treated VSMCs by intracellular immunofluorescence staining.
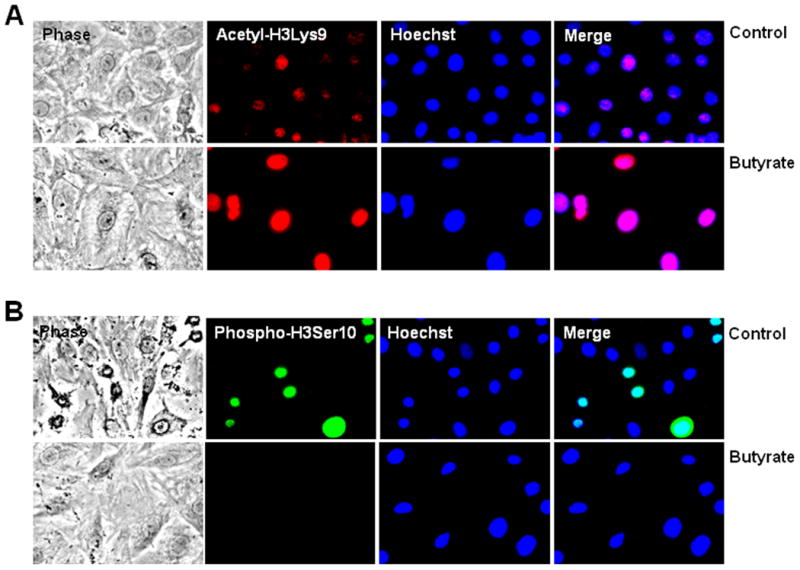
Proliferating VSMCs were untreated or treated with butyrate for 24 h and processed for immunofluorescence staining of histone H3Lys9 acetylation (A) and histone H3Ser10 phosphorylation (B) followed by nuclear staining with Hoechst. Images of stained VSMCs were captured using a Nikon fluorescence microscope with a CCD camera and 40 × objective.
3.2 Concentration and time-dependent changes in site-specific posttranslational modifications of histone H3 in butyrate-inhibited VSMC proliferation
Analysis of the patterns of histone H3 modification specific to butyrate arrested VSMC proliferation display: induction of H3Lys9 acetylation, inhibition of H3Ser10 phosphorylation, reduction of H3Lys9 di-methylation and stimulation (H3Lys4) di-methylation in a concentration dependent manner (Fig. 3A). On the other hand, the time-course studies indicate that while the changes in the levels of H3Lys9 acetylation, H3Lys9 di-methylation, and H3Lys4 di-methylation induced by 5 mM butyrate remained almost the same all through the experimental period, reduced level of H3Ser10 phosphorylation is observed in 6 h butyrate treated VSMCs, which is completely abolished with increase in time of exposure to butyrate (Fig. 3B). Conversely, 24 h (Fig. 3B) and 48 h (Fig. 3A) untreated proliferating VSMCs exhibit induction of H3Ser10 phosphorylation, inhibition of H3Lys9 acetylation, stimulation of H3Lys9 di-methylation, and reduction in H3 Lys4 di-methylation. However, except for H3Ser10 phosphorylation, no other histone H3 modifications exhibit any significant time-dependent changes in untreated proliferating cells.
Fig. 3. Concentration- and time-dependent profiles of histone H3 posttranslational modifications in butyrate treated VSMCs.
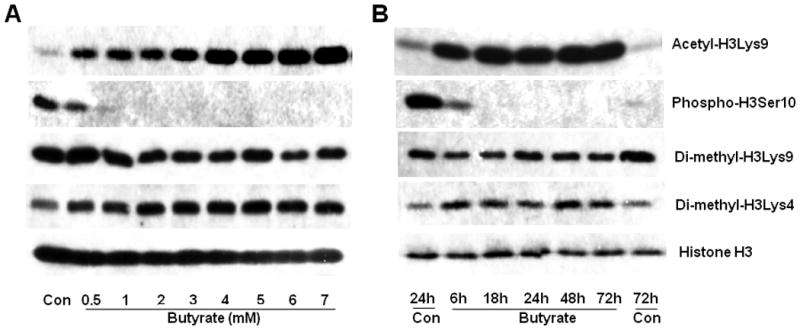
(A). For concentration-dependent study, proliferating VSMCs were treated with indicated concentrations of butyrate for 48 h. (B) For temporal effects, proliferating VSMCs were untreated (Con) or treated with 5 mM butyrate for indicated lengths of time. At the end of treatment, VSMCs were processed for western analyses to assess the levels of acetyl-H3Lys9, phospho-H3Ser10, di-methyl-H3Lys9, and di-methyl-H3Lys4. Histone H3 was immunoblotted and used as loading control.
3.3. Upregulation of cyclin D1 and cyclin D3 in butyrate-arrested VSMC proliferation
Expression of D-type cyclins that are important in driving cells through G1 phase in response to mitogenic signals are studied in butyrate treated VSMCs. Treatment of proliferating VSMCs with butyrate causes increase in cyclin D1 and D3 protein levels in a concentration (Fig. 4A) and time-dependent fashion compared to untreated proliferating VSMCs (Fig. 4B). Increase in cyclin D1 and D3 protein levels is observed at as low as 0.5mM butyrate concentration and depending on the concentration of butyrate, their protein levels further increased at the end of 48 h of treatment (Fig. 4A). The temporal study indicates about 3-fold induction of cyclin D1 after 24 h of treatment, and almost similar extent of induction is sustained all through the experimental period of 72 h (Fig. 4B). Conversely, about 3.5-fold induction of cyclin D3 is observed as early as 6 h of butyrate treatment and there onwards showed modest increase up until the end of 72 h (Fig. 4B). Moreover, intracellular immunostaining not only confirms increase in cyclin D1 (Fig. 4C) and D3 (Fig. 4D) protein levels by butyrate but also distinctly discloses their nuclear localization.
Fig. 4. Upregulation and nuclear localization of cyclin D1 and D3 proteins in butyrate treated VSMCs.
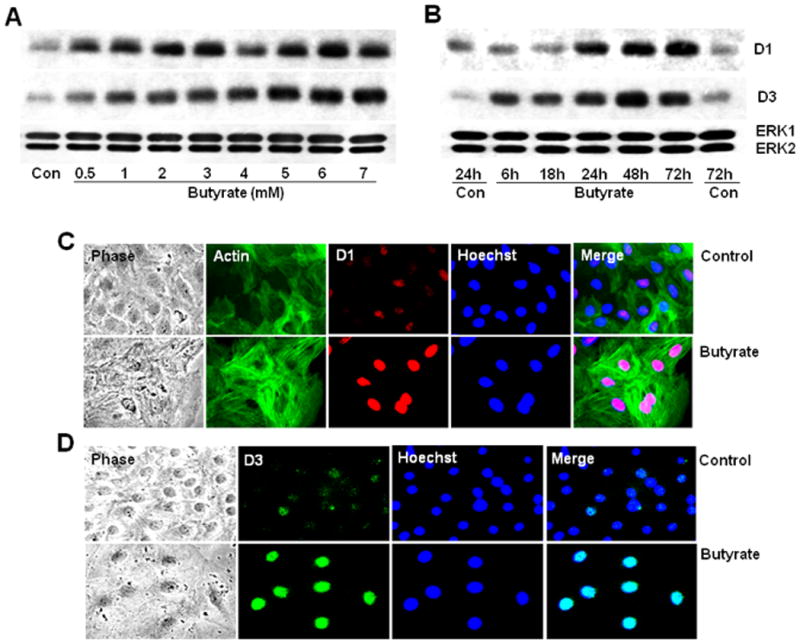
Proliferating VSMCs were treated with different concentrations of butyrate for 48 h or treated with 5 mM butyrate for indicated periods of time to determine the concentration (A) and time-dependent effects of butyrate (B). At the close of experiment, cell lysates were prepared and subjected to western analysis of cyclin D1 and D3. Intracellular localization of butyrate-induced cyclin D1 (C) and D3 (D) protein levels were determined by immunofluorescence staining of VSMCs treated with or without butyrate for 48 h. Cultures stained for cyclin D1 were also stained for smooth muscle α-actin to depict the cytoskeletal structure of VSMCs. Images of stained VSMCs were captured using a Nikon fluorescence microscope with a CCD camera and 40 × objective.
3.4. Downregulation of cyclin D- and cyclin E-specific cdks by butyrate in VSMCs
Analysis of cyclin D-specific cdk4 and cdk6 protein levels reveals that their levels are reduced in a concentration and time-dependent manner by butyrate (Fig. 5). Butyrate causes no significant effect on cdk4 and cdk6 protein levels up to 2 mM concentration but at concentrations higher than 2 mM, it causes dose-dependent reduction in their levels in comparison to untreated VSMCs (Fig. 5A). Temporal studies indicate time-dependent downregulation of cdk4 and cdk6 after 18 h of treatment with 5 mM butyrate (Fig. 5B). Moreover, butyrate displays almost similar dose- and time-dependent effects on cyclin E-dependent cdk2 protein expression that reflects downregulation profiles of cdk4 and cdk6 (Fig. 5A and 5B).
Fig. 5. Concentration- and time-dependent downregulation of cdk6, cdk4, and cdk2 in butyrate-inhibited VSMC proliferation.
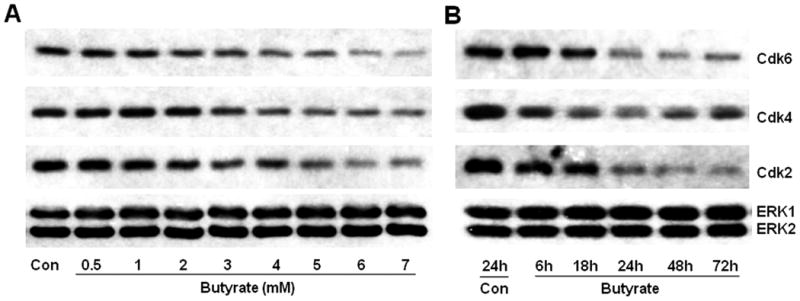
VSMCs were treated with different concentrations of butyrate for 48 h (A) or treated with 5 mM butyrate for indicated periods of time (B) to investigate concentration and temporal effects of butyrate on the expression of G1-specific cdks, respectively. At the conclusion of treatment, cell lysates were prepared and processed for western analyses of cdk6, cdk4, and cdk2.
3.5. Induction of cdk inhibitors, p15INK4b, and p21Cip1, by butyrate in VSMCs
Involvement of cdk inhibitors in butyrate arrested VSMC proliferation is evaluated by the effect of butyrate treatment on p15INK4b and p21Cip1 protein levels. Butyrate treatment causes concentration- (Fig. 6A) and time-dependent (Fig. 6B) upregulation of p15INK4b and p21Cip1 in VSMCs. Although no change in p15INK4b and p21Cip1 protein levels is observed up to 2 mM concentration, striking induction in their levels is observed between 3 mM to 7 mM concentration (Fig. 6A). Temporal studies depict p15INK4b and p21Cip1 protein levels are enhanced by about 3 to 4-fold after 18 h of treatment and most of this induction is sustained until the end of the experimental period of 72 h (Fig. 6B). Augmented expression of p15INK4b (Fig. 6C) and p21Cip1 (Fig. 6D) in butyrate treated VSMCs is further portrayed by intracellular immunostaining. Interestingly, butyrate-enhanced p15INK4b appears to be localized both in cytosolic and nuclear region of VSMCs, whereas p21Cip1 appears to be limited to nuclear region.
Fig. 6. Butyrate causes induction of p15INK4b and p21Cip1 protein expression in proliferation arrested VSMCs.
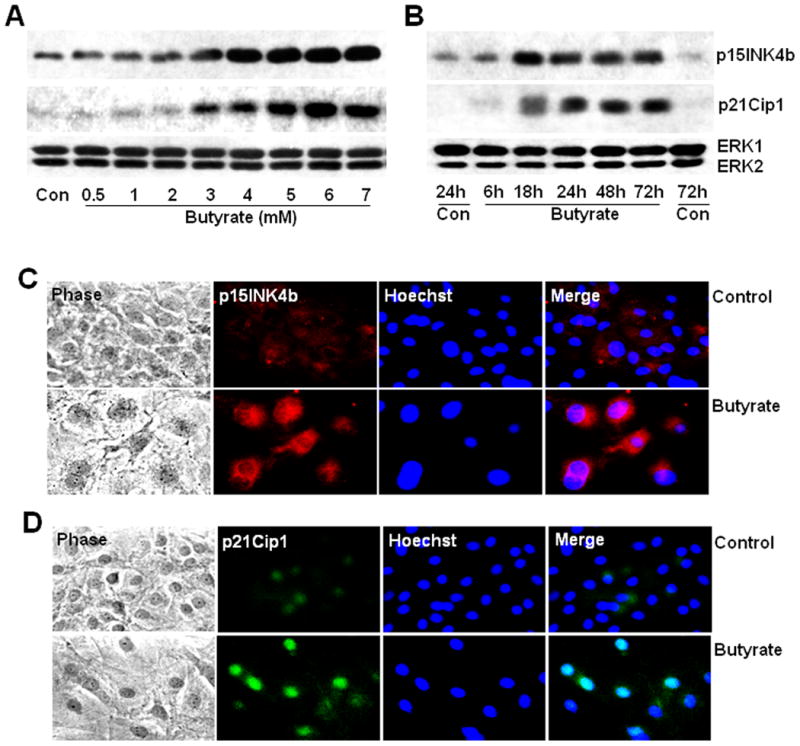
Proliferating VSMCs were treated with different concentrations of butyrate for 48 h (A) or treated with 5 mM butyrate for indicated periods of time (B) to explore concentration and temporal effects of butyrate on the expression of p15INK4b and p21Cip1 proteins in VSMCs. Cell lysates were prepared at the end of treatments and used for western analyses of p15INK4b and p21Cip1. Intracellular immunostaining of p15INK4b (C) and p21Cip1 (D) was performed to establish intracellular localization of butyrate-induced p15INK4b and p21Cip1 proteins in VSMCs.
3.6. Butyrate inhibits phosphorylation of Rb
The impact of underexpression of cyclin D-specific cdk4 and cdk6 and cyclin E-specific cdk2 and upregulation of cdk inhibitors, p15INK4b and p21Cip1, on phosphorylation state of Rb protein, is assessed by the electrophoretic migration rate upon SDS-PAGE and immunoblotting with Rb antibody (Fig. 7). Besides Rb antibody, another antibody specific to Rb protein that is phosphorylated on Ser807 and Ser811 (phospho-Rb Ser807/811) is used to determine the phosphorylation and activity state of Rb. Irrespective of the type of Rb antibodies used, highly phosphorylated Rb protein is detected in untreated proliferating VSMCs. Conversely, time-dependent inhibition of Rb phosphorylation is detected using both the antibodies in butyrate treated VSMCs. Additionally, a differently phosphorylated and slowly migrating phospho-Rb appeared along with rapidly moving unphosphorylated Rb in 6 h and 18 h butyrate treated VSMCs when Rb-specific antibody is used (Fig. 7). However, exposure to butyrate for 24 h and longer, distinctly inhibited Rb phosphorylation.
Fig. 7. Butyrate inhibits phosphorylation of Rb.
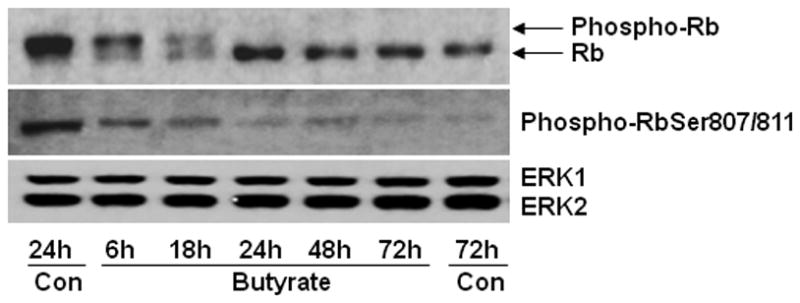
VSMCs treated with 5 mM butyrate for different lengths of time were processed for immunoblotting of phosphorylated Rb protein with two different Rb-specific antibodies: Rb antibody detects both phosphorylated and unphosphorylated Rb, which can be identified by their difference in mobility, and phospho-RbSer807/811 antibody recognizes only Rb protein that is phosphorylated at serine 807 and serine 811.
4. Discussion
Here we show that butyrate-inhibited VSMC proliferation exhibits cross-talk between different site-specific posttranslation modification of histone H3 by acetylation, phosphorylation and methylation (Fig. 1-3), which has been linked to transcriptional activation, cell cycle/mitosis/meiosis and transcriptional activation/suppression, respectively, and these effects are consistent with biological effects of butyrate, antiproliferation action and differential gene expression. Butyrate's cooperative effects on histone H3Lys9 acetylation and H3Ser10 phosphorylation (Fig. 1 to 3), and contrasting effects on di-methylation of H3Lys4 and H3Lys9 (Fig. 3) appear to cause distinct chromatin-based outputs resulting in cyclin D1 and D3 induction (Fig. 4), G1-specific cdk4, cdk6 and cdk2 downregulation (Fig. 5), and cdk inhibitors, p15INK4b and p21Cip1, upregulation (Fig. 6). Although downregulation of cdk4, cdk6 and cdk2, and upregulation of p15INK4b and p21Cip1 are in accordance with antiproliferation action of butyrate, increased levels of cyclin D1 and D3 by butyrate is surprising. Irrespective of the disparities regarding butyrate's effect on D-type cyclins, downregulation of G1-specific cdks and upregulation of cdk inhibitors by butyrate appear to avert cell cycle progression by altering phosphorylation and activity state of Rb causing arrest of VSMC proliferation. Thus, butyrate exhibits potential atheroprotective role against vascular proliferative diseases due to its ability to arrest VSMC proliferation by altering expression of G1-specific cell cycle proteins by duly modifying chromatin dynamics via epigenetic modification of histones.
Chromatin structure is dynamically altered by reversible modifications of histones by the activities of HDACs and HATs, which are pivotal to transcriptional regulation, DNA replication and repair, gene silencing, and regulation of cellular differentiation and proliferation [35-39]. In our study, butyrate causes interplay between different site-specific modifications of histone H3 such as acetylation, phosphorylation and methylation in VSMCs (Fig. 1 to 3). While H3Lys9 acetylation is induced in VSMCs that are growth arrested by butyrate, it is barely noticeable in untreated proliferating VSMCs (Fig. 1 to 3). In contrast, H3Ser10 phosphorylation is inhibited in butyrate treated VSMCs whereas untreated proliferating VSMCs reveal increased H3Ser10 phosphorylation, although the extent of H3Ser10 phosphorylation is reduced with increase in time of culturing. This may be linked to reduction in proliferating cells due to contact inhibition (Fig. 1 to 3). These responses imply a cooperative cross-talk between H3Lys9 acetylation and H3Ser10 phosphorylation in butyrate treated VSMCs, which may be critical for establishing a growth-arrested phenotype. Additionally, similar interaction between H3Lys9 acetylation and H3Ser10 phosphorylation, where histone H3Ser10 phosphorylation abolishes acetylation of histone H3Lys9 is documented by some recent studies [35-37]. Histone H3 tails have been known to be susceptible for site-specific reversible modifications that are linked to discrete chromatin-based biological activities such as: H3Lys9 and H3Lys14 acetylation to transcriptional activation; H3Ser10 phosphorylation to cell cycle/mitosis/meiosis and chromosome condensation / segregation; H3Lys9 methylation to transcriptional silencing; and H3Lys4 methylation to transcriptional activation [35-39]. Moreover, modification of histone tails by acetylation that normally correlates with transcriptional activation, and phosphorylation, which is mostly seen during mitosis/meiosis/cell cycle have been linked to dynamic relaxation and condensation state of chromatin, respectively. It is possible the cooperative interplay observed between H3Lys9 acetylation and H3Ser10 phosphorylation in butyrate treated VSMCs may aptly alter chromatin structure favoring decondensation and hindering condensation of chromatin that may be crucial for establishing a growth-arrested VSMC phenotype.
Besides acetylation and phosphorylation, histone H3 amino-terminal tails are also target of methylation [35, 38-41]. In butyrate treated VSMCs, in addition to strong induction of H3Lys9 acetylation, modest contrasting effects on di-methylation state of H3Lys9 and H3Lys4 are observed compared to untreated VSMCs (Fig. 1 to 3). These responses imply two key points: First, H3Lys9 acetylation and di-methylation occur on separate sets of histone H3 molecules (Fig. 1 and 3); and second, diminished H3Lys9 di-methylation may reduce gene silencing and contribute to suppression of at least some of the genes that play a role in growth arrest of VSMC by butyrate such as G1-specific cdks (Fig. 5). Consistent with our observation, recent studies indicate that H3Lys9 acetylation and methylation do occur on independent sets of histone H3 [40- 43] but unlike in our study, some of these studies reveal increased H3Lys9 acetylation and di-methylation by butyrate in different non-vascular cell [42] implicating cell and/or species-specific disparity [44], significance of which is not clear. Contrary to H3Lys9 di-methylation, subtle increase in H3Lys4 di-methylation is detected in butyrate treated VSMCs (Fig. 3). Because H3Lys4 di-methylation is mostly restricted to active chromatin, this increase in H3Lys4 di-methylation may contribute to upregulation of certain cell cycle regulators or other proteins linked to proliferation arrest of VSMCs by butyrate (Fig. 4 to 6). All in all, acting collectively, a combination of histone H3 modifications stimulated by butyrate appears to suitably alter chromatin structural organization to form a so called “histone code” that is translated to a specific biological event exclusive to butyrate effect, namely, proliferation-arrested VSMC phenotype via altered transcriptional activation.
Many studies recognize that there is a link between HDAC inhibitors stimulated altered chromatin structure via modifications of histones and transcriptional activity of chromatin, which underlie the antiproliferative action of HDAC inhibitors including butyrate. Evaluation of butyrate's effect on the expression of G1-specific cell cycle regulatory proteins that regulate Rb protein phosphorylation status, which is critical for cell to progress through G1 to S phase, clearly strengthens the relationship between butyrate-stimulated altered chromatin structure and change in expression of G1-specific cell cycle regulatory proteins in executing their antiproliferation action. Analysis of G1-specific D-type cyclins that drive the cells through G1 phase in response to mitogenic signals [45-47] such as D1 and D3 reveals augmented levels of their proteins in butyrate treated VSMCs (Fig. 4). Based on our earlier cDNA array screening studies [31] it appears cyclin D1 increase is due to increase in synthesis whereas enhanced D3 protein levels seem to be linked to protein stability [48]. Although butyrate arrests proliferation of other cell types [12, 49] and causes variable effects on the expression of D-type cyclins [7,12,49-53], our study reveals increase in both cyclin D1 and D3 proteins in response to butyrate, which appears to be exclusive to VSMCs, significance of which is not clear (Fig. 4). However, recent evidence indicates that in addition to their role as cdk-dependent regulators of cell cycle, D-type cyclins exhibit nonredundant crucial functions in a number of cdk-independent processes [54-59]. At present it is not clear whether cyclin D1 and D3 exert any cdk-independent roles in butyrate arrested VSMC proliferation.
D-type cyclins grant catalytic activity to their kinase partners, cdk4, and cdk6, by forming active cyclin D-cdk4/cdk6 heterodimeric complexes [45-47]. Interestingly, analysis of cdk4 and cdk6, to evaluate whether there is any change in their levels to balance their upregulated regulatory subunits, cyclin D1 and D3, in butyrate treated VSMCs reveals a dose and time-dependent downregulation of cdk4 and cdk6 (Fig. 5). Likewise, cyclin E-dependent cdk2 expression is downregulated by butyrate (Fig. 5). Butyrate also affects cdk4, cdk6, and cdk2 of other cell types but its effects appear to be cell type specific [50, 60]. Added to the downregulation of D-type cdk4/cdk6 and cyclin E type cdk2, their catalytic activity also appears to be compromised by the induction of p15INK4b, inhibitor of cyclin D-specific cdk4/cdk6 kinases [47] and p21Cip1, inhibitor of all cyclin-cdks except cyclin D-cdk4/cdk6 [45, 61], in VSMCs growth arrested by butyrate (Fig. 6). About 4- to 5- fold induction of both p15INK4b and p21Cip1 proteins is induced by 5 mM butyrate in a time-dependent manner, which is in accordance with our earlier cDNA array data [31]. While induction of p21Cip1 in VSMCs is in-line with butyrate's almost universal transcriptional activation of p21Cip1 and similar to other HDAC inhibitors [7, 8, 10-12, 31], p15INK4b is stimulated only in certain cell types by butyrate [62]. These effects suggest that by downregulating cyclin D-cdk4/cdk6 and cyclin E-cdk2 expressions combined with upregulation of their respective catalytic inhibitors, p15INK4b and p21Cip1, butyrate causes overall net inhibition of G1-specific cdk activities. Outcome of the compromised activities of cdks are further reflected in their time-dependent failure to phosphorylate and inactivate Rb protein in butyrate treated VSMCs (Fig. 7), stalling the VSMCs in G1 phase resulting in arrest of VSMC proliferation. Taken together, downregulation of cdk4, cdk6 and cdk2, and upregulation of p15INK4b and p21Cip1 by butyrate appear to be collectively linked to VSMC proliferation arrest by failing to inactivate Rb protein. However, the significance of cyclin D1 and D3 upregulation in butyrate arrested VSMC proliferation is not clear. Considering the new cdk-independent roles of cyclin D1 and D3 that are linked gene activation, it is possible that some of these functions may be crucial for butyrate arrested VSMC proliferation. We are currently characterizing cyclin D1 promoter and its interaction with HDACs and HATs as a prelude to understanding the cdk-independent role of cyclin D1 in butyrate arrested VSMC proliferation.
In summary, butyrate, a dietary HDACI, exhibits atheroprotective antiatherogenic potential by altering G1-specific cell cycle proteins through its chromatin remodeling activity to arrest VSMCs proliferation, a critical cellular component of the blood vessel, that play a major role in the development of atherosclerosis and in the pathogenesis of clinical procedures such as arterial and in-stent restenosis. At present there is a surge towards clinical applications of HDAC inhibitors in cancer and other disease treatments but their use in preventive and therapeutic intervention of vascular diseases is very limited. Outcome of our present study and other related studies [7-12, 19] clearly indicate that butyrate and its derivatives have a potential both in cardiovascular disease prevention as a bioactive component of dietary fiber and in therapeutic intervention of arterial restenosis and in-stent-restenosis as a pharmacological agent, respectively.
Acknowledgments
This study was supported by G12RR0345 and C06RR012537-01 grants from National Center for Research Resources/National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Dzau VJ, Braun-Dullaeus RC, Sedding DG. Vascular proliferation and atherosclerosis: new perspectives and therapeutic strategies. Nat Med. 2002;8:1249–56. doi: 10.1038/nm1102-1249. [DOI] [PubMed] [Google Scholar]
- 2.Melo LG, Gnecchi M, Pachori AS, Wang K, Dzau VJ. Gene- and cell-based therapies for cardiovascular diseases: current status and future directions. Eur Heart J Suppl. 2004;6:E24–E35. [Google Scholar]
- 3.Ranganna K, Yatsu FM, Mathew OP. Insights into the pathogenesis and intervention of atherosclerosis. Vasc Dise Prev. 2006;3:375–90. [Google Scholar]
- 4.Guerin P, Sauzeau V, Rolli-Derkinderen M, Al Habbash O, Scalbert E, Crochet D, et al. Stent implantation activates RhoA in human arteries: inhibitory effect of rapamycin. J Vasc Res. 2005;42:21–8. doi: 10.1159/000082873. [DOI] [PubMed] [Google Scholar]
- 5.Morice MC, Serruys PW, Sousa JE, Fajadet J, Ban Hayashi E, Perin M, et al. A randomized comparison of a sirolimus-eluting stent with a standard stent for coronary revascularization. N Engl J Med. 2002;346:1773–80. doi: 10.1056/NEJMoa012843. [DOI] [PubMed] [Google Scholar]
- 6.Jordan MA, Toso RJ, Thrower D, Wilson L. Mechanism of mitotic block and inhibition of cell proliferation by taxol at low concentrations. Proc Natl Acad Sci USA. 1993;90:9552–6. doi: 10.1073/pnas.90.20.9552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Davis T, Kennedy C, Chiew YE, Clarke CL, DeFazio A. Histone deacetylase inhibitors decrease proliferation and modulate cell cycle gene expression in normal mammary epithelial cells. Clin Cancer Res. 2000;6:4334–42. [PubMed] [Google Scholar]
- 8.Gallinari P, Di Marco S, Jones P, Pallaoro M, Steinkühler C. HDACs, histone deacetylation and gene transcription: from molecular biology to cancer therapeutics. Cell Res. 2007;17:195–211. doi: 10.1038/sj.cr.7310149. [DOI] [PubMed] [Google Scholar]
- 9.Dokmanovic M, Clarke C, Marks PA. Histone deacetylase inhibitors: Overview and perspectives. Mol Cancer Res. 2007;5:981–9. doi: 10.1158/1541-7786.MCR-07-0324. [DOI] [PubMed] [Google Scholar]
- 10.Mehnert JM, Kelly WK. Histone deacetylase inhibitors: biology and mechanism of action. Cancer J. 2007;13:23–9. doi: 10.1097/PPO.0b013e31803c72ba. [DOI] [PubMed] [Google Scholar]
- 11.Davie JR. Inhibition of histone deacetylase activity by butyrate. J Nutr. 2003;133:2485S–93S. doi: 10.1093/jn/133.7.2485S. [DOI] [PubMed] [Google Scholar]
- 12.Ranganna K, Yatsu FM, Hayes BE. Butyrate, a small pleiotropic molecule with multiple cellular and molecular actions: Its role as an anti-atherogenic agent. Recent Res Devel Mol Cell Biochem. 2005;2:123–51. [Google Scholar]
- 13.Report of joint WHO/FAO Expert consultation. World Health Organization; Geneva: 2003. Who Technical Report Series 916. Diet, nutrition, and the Prevention of Chronic Diseases. [PubMed] [Google Scholar]
- 14.Gibson GR, Rastall RA, Roberfroid MB. Prebiotics. In: Gibson GR, Roberfroid MB, editors. Colonic Microbiota, Nutrition and Health. Dordrecht, The Netherlands: Kluwer Academic Publishers; 1999. [Google Scholar]
- 15.Kim YI. AGA technical review: impact of dietary fiber on colon cancer occurrence Gastroenterol. 2000;118:1235–57. doi: 10.1016/s0016-5085(00)70377-5. [DOI] [PubMed] [Google Scholar]
- 16.Hu FB. Plant based foods and prevention of cardiovascular disease: an overview. Am J Clin Nutr. 2003;78(Suppl):544S–551S. doi: 10.1093/ajcn/78.3.544S. [DOI] [PubMed] [Google Scholar]
- 17.Anderson JW. Whole grains protect against atherosclerotic cardiovascular disease. Proc Nutr Soc. 2003;62:135–42. doi: 10.1079/PNS2002222. [DOI] [PubMed] [Google Scholar]
- 18.Smith JG, German JB. Molecular and genetic effects of dietary derived butyric acid. Food Technol. 1995;49:87–90. [Google Scholar]
- 19.Dashwood RH, Myzak MC, Ho E. Dietary HDAC inhibitors: time to rethink weak ligands in cancer chemoprevention? Carcinogenesis. 2006;27:344–9. doi: 10.1093/carcin/bgi253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Russo GL, Della PV, Mercurio C, Palumbo R, Iacomino G, Russo M, et al. Protective effects of butyric acid in colon cancer. Adv Exp Med Biol. 1999;472:131–47. doi: 10.1007/978-1-4757-3230-6_12. [DOI] [PubMed] [Google Scholar]
- 21.Hamer HM, Jonkers D, Venema K, Vanhoutvin S, Troost FJ, Brummer RJ. The role of butyrate on colonic function. Aliment Pharmacol Ther. 2008;27:104–19. doi: 10.1111/j.1365-2036.2007.03562.x. [DOI] [PubMed] [Google Scholar]
- 22.Hoppe C, Vichinsky E, Lewis B, Foote D, Styles L. Hydroxyurea and sodium phenylbutyrate therapy in thalassemia intermedia. Am J Hematol. 1999;62:221–7. doi: 10.1002/(sici)1096-8652(199912)62:4<221::aid-ajh4>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 23.Moyer BD, Loffing-Cueni D, Loffing J, Reynolds D, Stanton BA. Butyrate increases apical membrane CFTR but reduces chloride secretion in MDCK cells. Am J Physiol. 1999;277:F271–6. doi: 10.1152/ajprenal.1999.277.2.F271. [DOI] [PubMed] [Google Scholar]
- 24.Zeitlin PL. Future pharmacological treatment of cystic fibrosis. Respiration. 2000;67:351–7. doi: 10.1159/000029528. [DOI] [PubMed] [Google Scholar]
- 25.Ferrante RJ, Kubilus JK, Lee J, Ryu H, Beesen A, Zucker B, et al. Histone deacetylase inhibition by sodium butyrate chemotherapy ameliorates the neurodegenerative phenotype in Huntington's disease mice. J Neurosci. 2003;23:9418–27. doi: 10.1523/JNEUROSCI.23-28-09418.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gardian G, Browne SE, Choi DK, Klivenyi P, Gregorio J, Kubilus JK, et al. Neuroprotective effects of phenylbutyrate in the n171-82q transgenic mouse model of Huntington's disease. J Biol Chem. 2005;280:556–63. doi: 10.1074/jbc.M410210200. [DOI] [PubMed] [Google Scholar]
- 27.Perrine SP, Ginder GD, Faller DV, Dover GH, Ikuta T, Witkowaska HE, et al. A short-term trial of butyrate to stimulate fetal-globin-gene expression in the ß-globin disorders. N Engl J Med. 1993;328:81–6. doi: 10.1056/NEJM199301143280202. [DOI] [PubMed] [Google Scholar]
- 28.Ranganna K, Joshi T, Yatsu FM. Sodium butyrate inhibits platelet-derived growth factor– induced proliferation of vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 1995;15:2273–83. doi: 10.1161/01.atv.15.12.2273. [DOI] [PubMed] [Google Scholar]
- 29.Ranganna K, Yatsu FM, Hayes BE, Milton SG, Jayakumar A. Butyrate inhibits proliferation-induced proliferating cell nuclear antigen expression (PCNA) in rat vascular smooth muscle cells. Mol Cell Biochem. 2000;205:149–61. doi: 10.1023/a:1007078200482. [DOI] [PubMed] [Google Scholar]
- 30.Feng P, Ge L, Akyhani N, Liau G. Sodium butyrate is a potent modulator of smooth muscle cell proliferation and gene expression. Cell Prolif. 1996;29:231–41. doi: 10.1046/j.1365-2184.1996.00998.x. [DOI] [PubMed] [Google Scholar]
- 31.Ranganna K, Yousefipour Z, Yatsu FM, Milton SG, Hayes BE. Gene expression profile of butyrate-inhibited vascular smooth muscle cell proliferation. Mol Cell Biochem. 2003;254:21–36. doi: 10.1023/a:1027383710582. [DOI] [PubMed] [Google Scholar]
- 32.Ramos K, Cox LR. Primary cultures of rat aortic endothelial and smooth muscle cells: I. An in vitro model to study xenobiotic-induced vascular cytotoxicity. In Vitro Cell Dev Biol. 1987;23:288–96. doi: 10.1007/BF02623712. [DOI] [PubMed] [Google Scholar]
- 33.Geisterfer AA, Peach MJ, Owens GK. Angiotensin II induces hypertrophy, not hyperplasia, of cultured rat aortic smooth muscle cells. Circ Res. 1988;62:749–56. doi: 10.1161/01.res.62.4.749. [DOI] [PubMed] [Google Scholar]
- 34.Ranganna K, Mathew OP, Yatsu FM, Yousefipour Z, Hayes BE, Milton SG. Involvement of glutathione/glutathione S-transferase antioxidant system in butyrate-inhibited vascular smooth muscle cell proliferation. FEBS J. 2007;274:5962–78. doi: 10.1111/j.1742-4658.2007.06119.x. [DOI] [PubMed] [Google Scholar]
- 35.Ito T. Role of histone modifications in chromatin dynamics. J Biochem. 2007;141:609–614. doi: 10.1093/jb/mvm091. [DOI] [PubMed] [Google Scholar]
- 36.McManus KJ, Hendzel MJ. The relationship between histone H3 phosphorylation and acetylation throughout the mammalian cell cycle. Biochem Cell Biol. 2006;84:640–57. doi: 10.1139/o06-086. [DOI] [PubMed] [Google Scholar]
- 37.Edmondson DG, Davie JK, Zhou J, Mirnikjoo B, Tatchell K, Dent SY. Site-specific loss of acetylation upon phosphorylation of histone H3. J Biol Chem. 2002;277:29496–502. doi: 10.1074/jbc.M200651200. [DOI] [PubMed] [Google Scholar]
- 38.Eberlin A, Grauffel C, Oulad-Abdelghani M, Robert F, Torres-Padilla ME, Lambrot R, et al. Histone H3 tails containing dimethylated lysine and adjacent phosphorylated serine modifications adopt a specific conformation during mitosis and meiosis. Mol Cell Biol. 2008;28:1739–54. doi: 10.1128/MCB.01180-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ducasse M, Brown MA. Epigenetic aberrations and cancer. BMC Mol Cancer. 2006;5:60. doi: 10.1186/1476-4598-5-60. http://www.molecular-cancer.com/content/5/I/60. [DOI] [PMC free article] [PubMed]
- 40.Fischle W, Wang Y, Allis CD. Histone and chromatin cross-talk. Curr opin Cell Biol. 2003;15:172–83. doi: 10.1016/s0955-0674(03)00013-9. [DOI] [PubMed] [Google Scholar]
- 41.Fischle W, Wang Y, Allis CD. Binary switches and modification cassettes in histone biology and beyond. Nature. 2003;425:475–9. doi: 10.1038/nature02017. [DOI] [PubMed] [Google Scholar]
- 42.Bártová E, Pacherník J, Harnicarová A, Kovarík A, Kovaríková M, Hofmanová J, et al. Nuclear levels and patterns of histone H3 modification and HP1 proteins after inhibition of histone deacetylases. J Cell Sci. 2005;118:5035–46. doi: 10.1242/jcs.02621. [DOI] [PubMed] [Google Scholar]
- 43.Maison C, Bailly D, Peters AH, Quivy JP, Roche D, Taddei A, et al. Higher-order structure in pericentric heterochromatin involves a distinct pattern of histone modification and an RNA component. Nat Genet. 2002;30:329–34. doi: 10.1038/ng843. [DOI] [PubMed] [Google Scholar]
- 44.Garcia BA, Hake SB, Diaz RL, Kauer M, Morris SA, Recht J, et al. Organismal differences in post-translational modifications in histones H3 and H4. J Biol Chem. 2007;282:7641–55. doi: 10.1074/jbc.M607900200. [DOI] [PubMed] [Google Scholar]
- 45.Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 1999;13:1501–12. doi: 10.1101/gad.13.12.1501. [DOI] [PubMed] [Google Scholar]
- 46.Ortega S, Malumbres M, Barbacid M. Cyclin D-dependent kinases, INK4 inhibitors and cancer. Biochim Biophys Acta. 2002;1602:73–87. doi: 10.1016/s0304-419x(02)00037-9. [DOI] [PubMed] [Google Scholar]
- 47.Lundberg AS, Weinberg RA. Functional inactivation of the retinoblastoma protein requires sequential modification by at least two distinct cyclin-cdk complexes. Mol Cell Biol. 1998;18:753–61. doi: 10.1128/mcb.18.2.753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Santa FD, Albini S, Mezzaroma E, Baron L, Felsani A, Caruso M. pRb-dependent cyclin D3 protein stabilization is required for myogenic differentiation. Mol Cell Biol. 2007;27:7248–65. doi: 10.1128/MCB.02199-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kruh J. Effects of sodium butyrate, a pharmacological agent, on cells in culture. Mol Cell Biochem. 1982;42:65–82. doi: 10.1007/BF00222695. [DOI] [PubMed] [Google Scholar]
- 50.Siavoshian S, Segain JP, Kornprobst M, Bonnet C, Cherbut C, Galmiche JP, et al. Butyrate and trichostatin A effects on the proliferation/differentiation of human intestinal epithelial cells: induction of cyclin D3 and p21 expression. Gut. 2000;46:507–14. doi: 10.1136/gut.46.4.507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vaziri C, Stice L, Faller DV. Butyrate-induced G1 arrest results from p21-independent disruption of retinoblastoma protein-mediated signals. Cell Growth Differ. 1998;9:465–74. [PubMed] [Google Scholar]
- 52.Li C, Liu W, Meng F, Huang W, Zhou J, Sun H, et al. Effect and comparison of sodium butyrate and trichostatin A on the proliferation/differentiation of K562. J Huazhong Univ Sci Technolog Med Sci. 2003;23:249–53. doi: 10.1007/BF02829505. [DOI] [PubMed] [Google Scholar]
- 53.Lallemand F, Courilleau D, Sabbah M, Redeuilh G, Mester J. Direct inhibition of the expression of cyclin D1 gene by sodium butyrate. Biochem Biophys Res Commun. 1996;229:163–69. doi: 10.1006/bbrc.1996.1774. [DOI] [PubMed] [Google Scholar]
- 54.Han EK, Ng SC, Arber N, Begemann M, Weinstein IB. Roles of cyclin D1 and related genes in growth inhibition, senescence and apoptosis. Apoptosis. 1999;4:213–9. doi: 10.1023/a:1009618824145. [DOI] [PubMed] [Google Scholar]
- 55.Fu M, Wang C, Li Z, Sakamaki T, Pestell RG. Cyclin D1: Normal and abnormal functions. Endocrinol. 2004;145:5439–47. doi: 10.1210/en.2004-0959. [DOI] [PubMed] [Google Scholar]
- 56.Marampon F, Casimiro MC, Fu M, Powell MJ, Popov VM, Lindsay J, et al. Nerve growth factor regulation of cyclin D1 in PC12 cells through a p21RAS extracellular signal-regulated kinase pathway requires cooperative interactions between Sp1 and nuclear factor-kB. Mol Biol Cell. 2008;19:2566–78. doi: 10.1091/mbc.E06-12-1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kiess M, Gill RM, Hamel PA. Expression of the positive regulator of cell cycle progression, cyclin D3, is induced during differentiation of myoblasts into quiescent myotubes. Oncogene. 1995;10:159–66. [PubMed] [Google Scholar]
- 58.Sarruf DA, Iankova I, Abella A, Assou S, Miard S, Fajas L. Cyclin D3 promotes adipogenesis through activation of peroxisome proliferator-activated receptorγ. Mol Cell Biol. 2005;25:9985–95. doi: 10.1128/MCB.25.22.9985-9995.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fu M, Wang C, Rao M, Wu X, Bouras T, Zhang X, et al. Cyclin D1 represses p300 transactivation through a cyclin-dependent kinase-independent mechanism. J Biol Chem. 2005;280:29728–42. doi: 10.1074/jbc.M503188200. [DOI] [PubMed] [Google Scholar]
- 60.Kim J, Park H, Im JY, Choi WS, Kim HS. Sodium butyrate regulates androgen receptor expression and cell cycle arrest in human prostate cancer cells. Anticancer Res. 2007;27:3285–92. [PubMed] [Google Scholar]
- 61.Coqueret O. New roles for p21 and p27 cell-cycle inhibitors: a function for each cell compartment? Trends Cell Biol. 2003;13:65–70. doi: 10.1016/s0962-8924(02)00043-0. [DOI] [PubMed] [Google Scholar]
- 62.Hitomi T, Matsuzaki Y, Yokota T, Takaoka Y, Sakai T. p15 (INK4b) in HDAC inhibitor-induced growth arrest. FEBS Lett. 2003;554:347–50. doi: 10.1016/s0014-5793(03)01186-4. [DOI] [PubMed] [Google Scholar]