

Abstract
In vivo peptide inhibition of tumor suppressor p53 binding to the protein MDM2 is hampered by inefficient delivery of the peptide. Our approach to couple a hydrophobic lipid-like tail on the inhibitory peptide p5314–29 allowed its intracellular delivery in vitro, in a panel of different cell lines. The constructed chimeric molecules, termed peptide amphiphiles, further self-assembled into supramolecular structures, identified as elongated worm-like micelles. Internalization of peptides occured following micelle disassembly, partly via clathrin-mediated endocytosis of monomers. Incubation of SJSA-1 cells in hypertonic culture media, aimed to disrupt endocytic vesicles, resulted in peptide amphiphile-mediated cell death. Our results provide the basis for the construction of novel therapeutic supramolecular nanoparticles and suggest hydrophobic modification of peptides as a promising strategy for enhancing delivery of impermeable peptides.
Keywords: peptide inhibitors, intracellular delivery, p53-MDM2 interaction, cancer therapy, lipopeptides
Introduction
Inhibition of the interaction between tumor suppressor p53 and oncoproteins MDM2 and MDM4 (also known as MDMX) is a promising strategy in anticancer therapy for the subset of solid tumors that retain wild-type p53 and have amplified MDM2 and/or MDM41. To this end, small-molecules, peptides and peptidomimetics have all been considered as candidates for liberating p53 from its MDM2- and MDM4-bound state, allowing subsequent induction of cell death2, 3. Demonstrated in vitro and in vivo successes have led to preclinical and clinical trials of two types of small molecule inhibitors, Nutlins and spiro-oxindole compounds4. Despite adequate bioavailability and pharmacokinetic profiles, these small inhibitors suffer from lack of targeting specificity and inefficient simultaneous inhibition of both p53-MDM2 and p53-MDM4 interactions.
Peptide inhibitors could overcome such limitations but face impeding delivery challenges both at the whole body and cellular level: the intact peptide has to not only reach cancer cells following systemic administration but must enter into the cytoplasm and nucleus to function. As an example, the peptide corresponding to the native sequence of the MDM2-binding region of p53 (p5314–29) is cell impermeable, prone to rapid enzymatic degradation and lacks any targeting sequence to aid in its tumor tissue accumulation, even though it acts as a μM inhibitor of p53-MDM2 and p53-MDM4 in the test tube5. We pursue here an attractive strategy to tackle the obstacles in the delivery of p5314–29, and potentially a generalized strategy for peptide delivery, namely, the modification of the peptide via covalent attachment of lipid tails to construct chimeric molecules termed peptide amphiphiles (PAs)6.
Peptide amphiphiles self-assemble to form modular, nanoscale delivery structures that can be designed to actively target malignant tissues. Recently, we have shown that mixed micelles of DSPE-PEG(2000) PAs incorporating targeting peptide ligands were able to reach their corresponding targets in vivo and transport a non-targeting amphiphile to those tissue targets7, 8. This demonstrated that the stability of self-assembled peptide amphiphile micelles was sufficient for active targeting despite their inherent dynamic character9. The ability of self-assembled nanoparticles to disassemble may be an advantage but we need to know more about their fate. Once at the target tissue, it is not clear how these micelles interact with cells and how their stability influences their internalization. More specifically, the questions that arise are whether these self-assembled structures are internalized intact or following their disassembly into monomers and what internalization pathway they follow.
The presence of tethered hydrophobic tails in PAs promotes lipid membrane anchoring of monomers10, 11 and their subsequent cell internalization12–14. This is an alternative method to the use of cell-penetrating peptides (CPPs)15 or peptide stapling16, 17 for achieving peptide entry into cells. It is non-specific but nevertheless highly efficient even in cell lines that are not receptive to CPPs18. The elucidation of lipid-modified peptide internalization mechanisms, which are further linked to the fate of internalized cargo, are still under debate19, 20. Nevertheless, studies performed using live cell imaging point to energy-dependent endocytic uptake, which traps PAs into endosomal compartments12, 21. Consequently, peptides that are functional in the vicinity of the plasma or inner compartmental membranes benefit from their hydrophobic modification and exhibit increased potency due to their increased membrane affinity21–23.
Among plasma membrane constituents, glycosyl-phosphatidylinositol (GPI)-anchored proteins (GPI-APs) share structural similarities to PAs. GPI-APs possess saturated or unsaturated hydrocarbon tails, which insert in one leaflet of the plasma membrane, and a hydrophilic headgroup onto which proteins are covalently attached. GPI-APs have been shown to enter through several distinct clathrin- and caveolae-independent mechanisms19, 20, 24–26. Among them, a cdc42- and Arf1-regulated, lipid-raft mediated and dynamin- and Arf-6-independent pathway proceeds through a distinct class of tubular invaginations termed GPI-AP-enriched early endosomal compartments (GEECs)24, 27–29. This constitutive pathway is apparently also used by artificial lipidated proteins delivering internalized cargo to the recycling endosomal compartment through distinct vesicles budding from the cell surface24. The process is independent of the nature of the lipid tail anchor, even though the latter determines to a certain degree GPI-AP membrane localization13, 28, 30. Therefore, sorting at the plasma membrane cannot explain the mechanism by which GPI-APs target this pathway. Recently, Bhagatji et al. proposed a mechanism that relies on headgroup size: large headgroups exclude lipid-anchored proteins from ‘crowded’ clathrin-coated pits, whereas a fraction of amphiphiles possessing smaller headgroups still partition in them and use clathrin-mediated endocytosis (CME) as a mechanism of internalization31.
Based on the structural similarities between PAs and GPI-APs, we aimed at determining whether a double-tailed p5314–29 PA would use the same endosomal pathway as GPI-APs. Previously, we reported on palmitoylation of p5314–29 and its internalization by SJSA-1 osteosarcoma cells via an energy-dependent, endocytotic mechanism of monomeric lipopeptides following rapid micelle disassembly12. Here, we sought to increase stability by substituting the single palmitoyl tail with a synthetic analog composed of two palmitoyl tails linked through a glutamate linker. An exchange of the hydrophobic part of this PA was expected to alter its membrane affinity and localization in an analogous manner to how acylation machinery in cells regulates protein localization and function32. This report supplies the key background understanding of double tailed PA-cell interactions and provides the basis towards our overarching goal of designing efficient peptide delivery systems based on PA technology.
Experimental Section
Peptide Amphiphiles
Peptide, PA and PA− (Figure 1) were synthesized using standard solid-phase synthesis protocols. The details can be found in the supplementary data section. Synthesis of the single-tail peptide amphiphile palmitoyl-p5314–29 was previously described12.
Figure 1. Physicochemical characterization of micelles composed of double-tailed peptide amphiphiles.

Peptide p5314–29 was fluorescently labeled with rhodamine (Peptide) and a synthetic di-palmitic tail (PA). A non-fluorescent peptide amphiphile (PA−) was synthesized to vary micelle fluorescent intensity (A). When dissolved in an aqueous phosphate buffer, PA self-assembled into elongated micelles. Cryogenic (D) and negative stain (E) transmission electron microscopy were used to image the high aspect ratio micelles and dynamic (B) and static (C) light scattering were used to extract information on their size and persistence length.
Light Scattering
Light scattering was performed using a Brookhaven instrument that consisted of an avalanche photodiode detector to measure scattering intensity from a 632.8 nm HeNe laser (Melles Griot) as a function of delay time. Temperature was maintained at 25.0 ± 0.1°C. The first-order autocorrelation function at different angles between 40° and 120°, covering a range of scattering wave vector values, q, from 0.0090 to 0.0229 nm−1, was fit using a second-order cumulant to extract the average decay rate, Γ1. The quantity Γ1/q2 was then taken as the apparent diffusion coefficient, Dapp. Dapp at q = 0.0132 nm−1 (60°) was used to estimate the translational diffusion coefficient. The Stokes-Einstein relationship was used to calculate the size of an equivalent hydrodynamic sphere for this particle. No extrapolation was performed to q = 0 because of the uncertainty in this process for non-spherical particles.
In order to extract properties of a worm-like micelle from these data, Dapp was plotted as a function of q. Given the theoretical results of Winkler et al.33, Dapp will be independent of q for q < 1/rg, where rg is the radius of gyration of the scatterer. As q increases, two regimes may appear: an intermediate scattering vector regime (1/rg < q < p) in which Dapp increases linearly with q and a large scattering vector regime (q > p) in which internal bending modes become important causing Dapp to increase as q2/3. Here, 1/2p is the persistence length of the micelle. Thus, the scaling behavior of Dapp can provide an estimate of the radius of gyration and persistence length.
Static light scattering (SLS) measurements were performed at scattering angles ranging from 40° to 120° and the excess scattering intensity I(q) was obtained through calibration with benzene used as the standard. The q-spectrum of I(q) reveals different specific q-regions34: at low q (< 1/rg): the overall size and apparent molar mass of the scatterers are accessible, whereas at high q (> 1/2p): the length scales probed provide information on the local structure of the micelles and I(q) scales with q−4.
Transmission Electron Microscopy (TEM)
Peptide amphiphile samples (4.0 mg/ml) were imaged at 200kV with an FEI Technai G2 Sphera microscope. Images were recorded digitally with a Gatan Ultrascan 1000 CCD camera and analyzed using the Gatan Digital Micrograph software.
Negative stain samples were prepared by placing a 3 μL droplet of PA solution onto a glow discharged formvar coated copper grid for 10 minutes. The excess liquid was wicked away by filter paper. A 3 μL droplet of 2% (w/v) aqueous phosphotungstic acid was placed on the grid and left for 10 minutes before similarly being wicked away. The sample was left to dry overnight before imaging.
Cryogenic transmission electron microscopy (Cryo-TEM) samples were prepared using the environmentally controlled FEI Vitrobot Mark IV (24°C, 100% humidity). A 3.5 μL droplet of the sample was pipetted onto a glow discharged lacey carbon coated copper grid. The sample was blotted once with filter paper for 2 seconds before being plunged into liquid nitrogen cooled liquid ethane. The samples were placed in a Gatan cryo-holder and were kept below −170°C throughout imaging. During imaging, the objective lens was 2–5 μm out of focus to enhance contrast and the samples were imaged using the low-dose imaging mode.
Fluorescence Spectroscopy Studies
Fluorescence measurements were obtained using a Varian Cary Eclipse fluorescence spectrophotometer, with temperature control in a 10 mm quartz cuvette. Intensity at 585 nm (excitation at 560 nm) was monitored as a function of time following addition of a PA solution (20 μl) to a PBS solution (120 μl) to achieve a final concentration of i) 40 μM PA− ii) 0.6 mM bovine serum albumin (BSA) and iii) 300 μM egg PC in the form of small unilamellar vesicles. According to Kastantin et al. monomer desorption rates indicative of micelle stability were determined using equation (1) where k is the desorption rate constant9.
| (1) |
Imaging
For fluorescence microscopy imaging, cells were seeded in Lab-Tek chambered coverglass slides (Nalge Nunc) and incubated in the presence of our formulations for fixed periods of time. Next, medium was removed and adherent cells were washed three times with sterile filtered PBS (10 mM, pH 7.4). Cells were visualized in supplemented cell culture medium (cells retained their shape for a longer time when compared to visualization in PBS). Hoechst 33342 (Invitrogen) was added 10 minutes prior to washing in order to stain cell nuclei. A Nikon Eclipse TE-200 microscope equipped with a 10x and 100x objective and a 100W mercury arc lamp was used for fluorescence imaging. The acquired images were processed and false color was added using ImageJ software.
Flow cytometry
Flow cytometry was performed in order to quantify internalization in different cell culture conditions and/or presence of inhibitors. Following incubation for the desired time period, cells were washed twice with PBS, trypsinized, transferred to polycarbonate centrifuge tubes and centrifuged (5 min; 1500rpm). After the supernatant was discarded, cells were re-suspended in PBS and the centrifugation cycle was repeated once. The final cell suspensions were kept under ice until analyzed using a FACS Aria cytometer (BD Biosciences) equipped with a 488 nm laser and 576 nm emission filter. Live cells were gated on forward and side scatter and a total of 10,000 events in the gated population were analyzed per sample. In order to deplete cells of adenosine-5′-triphosphate (ATP): normal culture medium was replaced with medium containing 50 uM 2-D-deoxyglucose and 0.1% w/v sodium azide 30 minutes before addition of the formulations. Similarly, preincubation with culture medium containing 1–5 mM methyl-β-cyclodextrin (MβCD) was used to deplete membranes of cholesterol. Amiloride (1–3 mM) was used to inhibit Na+/H+ exchange.
Results
Double-tail PAs form worm-like micelles in water
Fluorescent labeling of p5314–29 peptide was achieved through incorporation of a lysine at the N-terminus and subsequent reaction at the ε-amine with rhodamine (Peptide). Peptide amphiphiles with two palmitic (C16) tails were synthesized with (PA) or without (PA−) the fluorophore (Figure 1A). Double-tailed PAs self-assembled in 10 mM PBS at sub-micro molar concentrations due to hydrophobic interaction of the twin palmitic tails (data not shown). Dynamic light scattering (DLS) of micelle suspensions provided an estimate for the apparent translational diffusion coefficient (Dapp=Γ1/q2=1.53×10−8 cm2/s at θ =60°): which corresponds to a hydrodynamic diameter of 319 nm, thus excluding the formation of small spherical micelles. Indeed, cryo-TEM revealed the existence of elongated micelles with a diameter of 10 nm and a few hundred nm long (Figure 1D). Negative stain TEM additionally confirmed the formation of elongated micelles (Figure 1E). The slightly higher calculated diameter of 12 nm stems from the flattening of the micelles during drying and the presence of the negative stain on the micelle surface. To obtain information on worm-like micelle properties we performed light scattering experiments. Analysis of the scaling behavior of DLS data revealed no region in which Dapp was independent of q, with Dapp ~ q2/3 and no intermediate scaling region (Figure 1B). Thus, the radius of gyration for the micelles is larger than ≈ 100 nm and they appear to be fairly stiff as their persistence length may approach rg/2 exceeding 50 nm. Static light scattering (Figure 1C) revealed a q−4 dependence of scattering intensity at q greater than ≈0.012 nm−1 indicative of Porod behavior. At this q-region it is not possible to extract information on the overall dimensions of the micelles; nevertheless we can conclude that the persistence length of the micelles is high (p > 1/2q > 40 nm) confirming the result obtained by DLS.
Peptide amphiphile micelles are dynamic structures
As a result of self-assembly, rhodamine fluorescence was highly self-quenched in micelles composed solely of PA. We exploited the increase in fluorescence intensity that occurs due to de-quenching when neighboring rhodamine labeled monomers desorb from labeled micelles and partition into an excess of unlabeled (PA−) micelles, in order to probe micelle stability (Figure 2A). Monomer desorption rate constants, k−1 were in the order of hours and showed temperature-dependence, indicative of an energy-activated process.
Figure 2. Micelles are dynamic structures in equilibrium with PA monomers.

Micelle stability was studied by fluorescence dequenching experiments at 25°C and 37°C (n=2). Monomer desorption rates for each kinetic experiment were calculated from equation (1) and shown for mixing labeled micelles with an excess of unlabeled PA− micelles (A): bovine serum albumin (0.6 mM) (B) and unilamelar vesicles (100 nm in diameter) composed of egg PC (C).
Micelle stability was also tested in the presence of albumin at a concentration of 0.6 mM, which is typical of blood. Equilibration was significantly faster with desorption rates 250-fold faster than those obtained from mixing with unlabeled micelles (Figure 2B). In the presence of unilamellar egg PC vesicles of an average diameter of 150 nm, which can be taken as a rough model of the plasma membrane, fluorescence dequenching kinetics exhibited similar desorption rates when compared to micelle mixing (Figure 2C). Finally, we examined micelle breakup kinetics under the same conditions used for the in vitro cell internalization studies and verified that the presence of albumin (and perhaps more PA-binding proteins) in the serum-supplemented cell culture medium resulted in micelle breakup (Supplementary Figure S1). The slower kinetic rate (k−1=1.7 h) compared to that calculated for 0.6 mM albumin is attributed to the difference in protein concentration. Altogether our data suggest formation of dynamic micelles that exhibit enhanced kinetic stability compared to their single-tail counterparts.
Peptide amphiphiles internalize in a variety of different cell lines as monomers
PA micelles were readily internalized by a number of different cell lines as evidenced by fluorescence microscopy and flow cytometry (Figure 3). Following a 4-hour incubation in supplemented cell culture medium, we observed higher PA cellular uptake compared to the rhodamine-labeled peptide. These results demonstrate that enhanced uptake is a consequence of peptide modification with the hydrophobic tails. The above effect was observed in SJSA-1 osteosarcoma cells, HeLa cells and NIH 3T3 fibroblasts suggesting that the result is not cell-specific. The punctuate pattern observed in fluorescent micrographs implies that an endocytotic mechanism is at play, which results in peptide amphiphile entrapment in intracellular vesicles (Figure 3).
Figure 3. Covalent attachment of double tail induces peptide internalization in various cell lines.

Double-tailed PA internalizes in SJSA-1 osteosarcoma cells, HeLa cells and NIH 3T3 fibroblasts as shown by epifluorescence microscopy (top row) and flow cytometry analysis (bottom row). Cells treated for 4 h with 10 μM PA in supplemented cell culture media revealed punctuate red fluorescence inside the cell indicating vesicular localization (nuclei are stained blue by Hoechst 33342.) The mean fluorescence intensity per cell of PA-treated cells was much higher than that of peptide-treated cells. Untreated cells were used to normalize mean fluorescence per cell (fluorescence intensity=1).
As discussed above, PAs are in equilibrium between micelles and as free monomers in solution. To distinguish in which of these states peptide amphiphiles enter cells we incubated SJSA-1 cells with mixed micelles composed of 10 % PA (labeled) and 90 % PA− (unlabeled) that exhibit approximately 8-fold higher fluorescence intensity compared to PA micelles (figure 4B). Incubation with these brighter micelles resulted in lower levels of fluorescence internalized per cell. More specifically, fluorescence intensity was proportional to the amount of labeled PA, indicating that monomers instead of micelles are taken up (Figure 4A). This result is analogous to our previous observations that micelles composed of single-tailed PAs entered SJSA-1 cells as monomers12. We compared uptake of single- and double-tailed PAs by SJSA-1 cells at a fixed concentration of 10 μM and observed two-fold enhancement of double-tailed PA internalization over its single-tailed counterpart (Supplementary Figure S2).
Figure 4. Dissecting double-tail PA internalization mechanism.

(A) Fluorescence intensity per cell was proportional to the amount of labeled monomer (PA) instead of the micelle fluorescence intensity (B): as was shown for a sample prepared with 1/10th of rhodamine content, but 8-fold higher emission intensity. Inhibition of uptake was observed following ATP depletion with a mixture of sodium azide and 2-deoxy-D-glucose (C) and with increasing concentrations of amiloride (D). Methyl-β-cyclodextrin (MβCD) treatment enhanced PA uptake (E). Cell fluorescence per cell was not altered after 4 hours incubation with PA followed by a 4-hour incubation in PA-free medium, indicating lack of considerable exocytosis (F). Insets in (D) and (E) show cell viability in the presence of the corresponding inhibitor. Incubations with PA were performed for 4 h at 37°C in serum supplemented cell culture medium. Data points in (D): (E) and (F) represent the mean +/− standard deviation (n=3).
Inhibition studies of PA internalization
We sought a better understanding of the internalization pathways since they largely determine the intracellular trafficking and fate of PAs. We selected SJSA-1 cells to perform all inhibition experiments, as this cell-line is responsive to p53-MDM2 inhibition. Depletion of intracellular ATP following treatment with sodium azide and 2-deoxyglucose abolished PA uptake, indicating an active internalization process that requires energy consumption (Figure 4C). Amiloride, an inhibitor of Na+/H+ exchange35, caused a dose-dependent inhibition of internalization in a concentration range that did not affect cell viability (Figure 4D). Depletion of cell surface cholesterol with MβCD had the opposite effect. A significant increase in PA internalization was noted for up to 5 mM concentrations; higher concentrations were not tested since cell viability was compromised (Figure 4E). Following internalization, PA-containing intracellular vesicles can potentially recycle to the plasma membrane or traffic inside the cell. We examined exocytosis of PA by monitoring the amount of fluorescence retained inside SJSA-1 cells at different times following an initial 4-hour incubation step. Fluorescence levels remained the same after a 4-hour chase while 75% of fluorescence was retained after 24 hours of incubation devoid of PA in the culture medium (Figure 4F). It is not clear from our study whether the reduction in fluorescence is a result of exocytosis, whether it reflects fluorophore degradation inside the cell or is simply a consequence of cell division. Co-localization studies with fluorescein-labeled transferrin at steady state (4 h incubation) showed that the majority of transferrin positive vesicles were also positive for PA; however some PA positive-only and transferrin positive-only vesicles were observed (Figure 5).
Figure 5. PA co-localizes partially with transferrin.
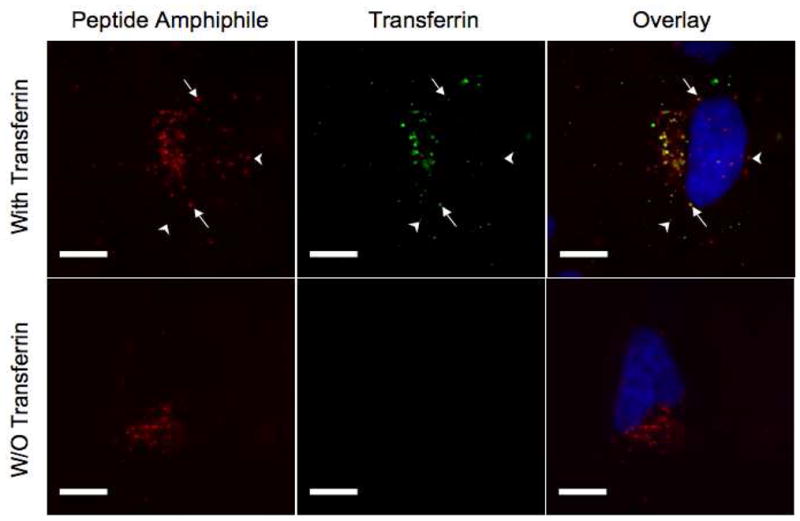
PA (25 μM) was incubated along with fluorescein-labeled transferrin (0.77 mg/ml) for 4 hours in the presence of SJSA-1 cells. Arrows denote vesicles containing both PA and transferrin and arrowheads point to vesicles containing only one of the labeled entities. The images in the lower row confirm the absence of light leakage from different filters (the microscope settings are identical for the two rows).
PAs decrease cell proliferation in cells that contain wild type p53 following endosomal disruption
The observed intracellular distribution of PAs was not expected to allow interactions with MDM2, which is mainly located in the cytoplasm and nucleus. Assuming that PAs were entrapped in endolysosomal vesicles, we sought a method to disrupt them and thus release their contents inside the cells. Addition of sucrose in the culture medium has been previously employed to burst endolysosomes due to osmotic shock36, 37. Indeed, 0.25 M sucrose resulted in swelling of fluorescent intracellular vesicles after 4 hours; however, diffuse fluorescence that would indicate release into the cytoplasm was not readily observed (Figure 6). PA escaping the endosomal compartments would be diluted in a much larger volume making its detection challenging with epifluorescence microscopy. A reduction in fluorescence per cell in the presence of sucrose was initially noted by visual inspection; quantification of uptake using flow cytometry revealed a 50% inhibition of uptake in the presence of 0.3 M sucrose at 4 hours; in this time range, no significant changes in cell viability were recorded (Supplementary Figure S3).
Figure 6. Sucrose treatment causes swelling of endocytic vesicles.

Fluorescence micrographs of rhodamine-labeled PA (25 μM) incubated for 4 hours with SJSA-1 cells in absence (left) and presence of 0.25 M sucrose (right). Sucrose induces endosome swelling. Diffuse cytoplasmic fluorescence was not observed with these settings and at this time point. Scale bar is 5 μm.
Sucrose-mediated disruption of intracellular vesicles was expected to release a fraction of PA in the cytoplasm of the cells. We monitored cell proliferation after a 4-hour pulse and 20-hour chase in the presence or absence of sucrose, and/or 100 μM PA, in two different cell lines: SJSA-1 and MDA-MB-435. SJSA-1 cells possess wild type tumor suppressor p53 (wt p53): whereas p53 is mutated in MDA-MB-435 cells (mut p53). We also confirmed that PA is internalized in MDA-MB-435 cells (Supplementary Figure S4). Incubation of both cell lines with sucrose was well tolerated at concentrations up to 0.2 M but resulted in approximately 40% decrease in cell proliferation at 0.3 M sucrose (Figure 7). The presence of PA did not affect viability in MDA-MB-435 cells. In contrast, cell viability was compromised in SJSA-1 cells in response to PA treatment. Cell proliferation was inhibited from a modest 10% in the absence of sucrose to 70% at 0.3 M sucrose. These results show that presence of PA induced cell death exlusively in the cell line with wt p53 and therefore suggest specific inhibition of the p53-MDM2 interaction.
Figure 7. p5314–29 peptide amphiphiles induce cell death in cells containing wild type p53.
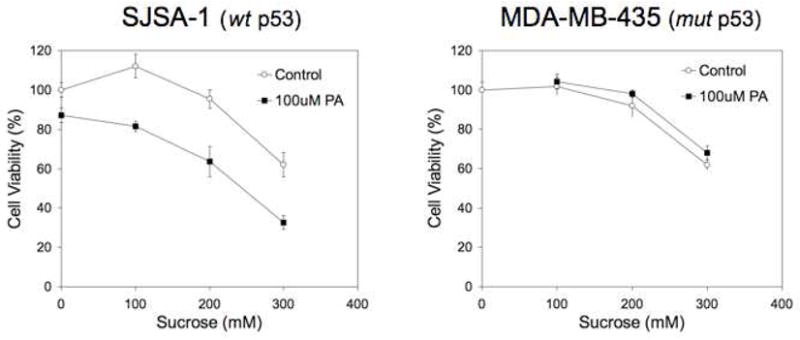
Cell viability of SJSA-1 (left) and MDA-MB-435 (right) cells shows reduced viability in the presence of PA only in cells expressing wild type p53 (SJSA-1 cells). Cells were incubated for 4 hours with 100 μM PA and varying concentrations of sucrose, washed and incubated an additional 20 hours prior to the cell viability assay. Each data point represents the mean +/− standard deviation of 5 wells.
Discussion
Modification of p5314–29 peptide with a synthetic hydrophobic tail, composed of two saturated C16 alkane chains, was carried out with two goals in mind. The first goal was to permit peptide amphiphile inclusion into modular, self-assembled structures destined to serve as drug delivery vehicles. The second goal was to promote cell internalization of PAs following initial interactions with the plasma membrane. Effective control over kinetic stability can lead to the desired scenario where self-assembled integrity is maintained during in vivo targeting followed by micelle disassembly, peptide amphiphile insertion into the cell membrane and intracellular localization. The present study provides fundamental insight into the latter process in an in vitro setting.
The physical nature of attractive interactions between PAs in micelles is an attractive feature because of the anticipated straightforward long-term bioelimination of the low molecular weight synthetic building blocks. However, micelle stability constitutes a challenge concerning the integrity of the structure during delivery and a crucial factor in determining pharmacokinetics and biodistribution. Recently, we have shown that a substantial fraction of intact, actively targeted micelles based on PEG-lipids reached their target site in vivo, despite their inherent instability upon dilution in blood7. Moreover, accumulation in RES organs like the liver and spleen was low. At the target tissue, intracellular localization was suggested but not investigated in detail.
In contrast to the PEG-lipids mentioned above, which form spherical micelles9, the peptide amphiphiles used in this study self-assembled into elongated worm-like micelles with a high persistence length. Monomer geometry is responsible for determining micelle size and shape and is dependent on on both the nature of hydrophobic tails as well as the hydrophilicity, size, flexibility and presence of interactions between the peptide headgroups6. These properties additionally determine the rate of monomer exchange or micelle disassembly, with hydrophobic interactions having the predominant role38–40. Accordingly, monomer desorption rates calculated in this study for double-tail PAs were strikingly lower compared to those of single-tail PAs possessing the same peptide headgroup12. The desorption rates were also lower than reported values determined for PEG lipids in micelles9 or liposomes38. In this case the differences arise from dissimilar headgroup properties since PEG lipids possessed the same or similar (di-C18 versus di-C16) hydrophobic tails. The higher entropic gain associated with the polymer configurational flexibility in the monomeric form, the large headgroup size41 as well as the increased hydrophilic character of the PEG headgroup could explain the observed higher desorption rates of PEG lipids. Morever, the presence of inter-peptide interactions on the micelle corona42 and fluidity state of the micelle core9 may also contribute to the enhanced stability of PA micelles compared to PEG lipids and merits further investigation.
A dramatic increase in monomer desorption rates was recorded in the presence of albumin (kmic/kAlb ≈ 290). Albumin is known to bind hydrophobic molecules including fatty acids, lipid dyes and sterols; the higher kinetic constant for albumin reflects albumin-facilitated monomer desorption and partitioning in the binding pockets of the protein. Albumin is the most abundant protein in the blood and this affinity is expected to influence how PAs are transported throughout the body. Lipoprotein particles (LDL and HDL) are also expected to compete for peptide amphiphile binding and influence biodistribution as was shown for lipidated siRNA43. An interesting finding in the aforementioned study is that while delivery was mediated through interactions with albumin and lipoprotein particles in vivo, cellular uptake occurred independent of carrier endocytosis43. Similarly, we envision micelles for in vivo delivery that upon reaching their target would disassemble and result in cellular uptake of the PAs.
Our in vitro cell internalization findings support such a scenario. PAs were taken up as individual monomers instead of intact micelles as previously demonstrated for single-tail PAs12. Interestingly, the amount internalized was higher for double-tail PAs compared to single-tail PAs, despite the enhanced micelle stability of the former. The higher hydrophobicity of double-tail PAs enables firmer anchoring to the plasma membrane41 and therefore increases the probability of internalization. We believe this factor counterbalances the lower ‘free’ monomer concentration and results in the observed enhancement of internalization for double-tail PAs. It would be of interest to determine whether our findings are extended to previously reported single-tail modified peptides21, 23, 44 and how this would affect their activity.
Results in a panel of 3 different cell lines suggested the lack of cell-specific protein machinery in PA uptake and the generality of the process. This finding is consistent with the absence of any known membrane receptor for the peptide and its reported impermeability. We therefore attribute the initial cell-binding step to insertion of the hydrophobic tails of PA monomers in the outer leaflet of the lipid bilayer10, 45. Membrane localization of PAs, in the absence of specific peptide-mediated lateral associations, is known to depend on length and saturation of the hydrophobic tails. The two saturated C16 tails of PA are expected to sequester in ordered lipid microdomains, commonly referred to as detergent-resistant membranes (DRM) or lipid rafts10, 30. Following this first step of membrane anchoring, internalization was found to proceed in an active, energy-dependent manner, localizing PAs in intracellular vesicles. We next discuss the possible mechanism(s) that were used for genesis of these vesicles based on our data.
The observed inhibition of uptake by amiloride led us to consider macropinocytosis as a potential route of entry in cells46. However, cholesterol is also necessary for this process and here its depletion did not inhibit uptake but instead showed the opposite trend, i.e. stimulated uptake. Moreover, the size of intracellular vesicles was not reminiscent of the polydisperse and often large (up to few μm) macropinosomes46. Co-localization of PA with transferrin further argues against the involvement of this pathway since macropinosomes do not fuse with transferrin-containing endosomes47. Amiloride inhibition is not alone sufficient to assign macropinocytosis as the internalization route of the cargo46 and it has been shown to inhibit other pathways as well48. Observations of specific, amiloride-induced inhibition of macropinocytosis and not clathrin mediated endocytosis (CME) were recorded for short treatment times35, 49 compared to our experiments. Prolonged amiloride presence causes acidification of the cytosol50, which is known to inhibit CME51. Based on the above, we rule out macropinocytosis as the main pathway of internalization and suggest that amiloride inhibition could arise from impairment of CME. In order to unambiguously determine whether macropinocytosis participates to some limited degree in peptide amphiphile uptake, internalization studies following stimulation with phorbol esters should be carried out.
We next considered CME and the CLIC/GEEC pathway as potential routes of PA entry into cells. Recently, synthetic PEGylated lipids of small headgroup size, which ressemble closely our constructs, have been shown to internalize using both of these mechanisms31. Our finding that cholesterol depletion stimulated internalization instead of inhibiting it rules out the CLIC/GEEC pathway as the principal mode of internalization29. It also excludes similar lipid-raft mediated uptake mechanisms, including caveolae- and flotillin-mediated internalization19. Kalia et al. reported that amiloride did not affect the GEEC pathway in CHO cells27 even though a dose response study was not performed and concentrations used in their study were 10-fold lower, albeit with a more potent amiloride analog. Our data showed >80% inhibition with 3 mM amiloride further argued that the CLIC/GEEC pathway is not involved. Clathrin-mediated endocytosis (CME) is the major pathway transferrin receptor employs to internalize bound transferrin. The observed co-localization of PA with transferrin implies that uptake occurs, at least partly, through this pathway. An alternative scenario would be that PA-containing vesicles rapidly fuse with clathrin-coated pit vesicles24. Studies at earlier time points would shed light on this possibility. Inhibition of internalization in presence of hypertonic medium (0.3 M sucrose) is also consistent with involvement of CME52.
Further indirect proof for involvement of CME is obtained by the surprising observation that MβCD treatment stimulated internalization of PAs. Possible explanations are either an up-regulation of alternative pathways or a redistribution of PAs on the plasma membrane. In the former case, the cell would up-regulate cholesterol-independent pathways to counterbalance the impairment of those dependent on cholesterol. In the latter case, PAs could be dissociated from lipid-rafts (that require cholesterol to form) and populate plasma membrane sections that constitutively internalize, thus increasing the effective amount internalized. It is not possible from our present data to distinguish between the two cases. Enhanced uptake after cholesterol depletion has been previously documented, however, for receptors or cargo that employed CME in parallel with some other cholesterol-dependent pathway53–56. Our data are reminiscent of these reports and further suggest CME as a principal mode of internalization. We should note that some studies reported inhibition of CME following cholesterol depletion57, 58. These latter observations that contrast our hypothesis might be due to cell-type differences and/or depletion agent concentration59.
Based on the above, we argue that PAs are taken up at least partly through CME, even though additional mechanisms cannot be excluded at this point. Internalization is not mediated by specific receptor binding but occurs as a consequence of normal cell processing. According to our hypothesis, following endocytosis, PAs would be trapped in the inner leaflet of the endosomal compartment membrane. The nature of the lipid tail and geometry of the molecule, is believed to govern peptide amphiphile sorting and trafficking thereafter by determining the preference to sites of different curvature or association with different lipid phases13. In particular, the saturated C16 tails of PA are expected to direct them in regions of convex curvature and ordered phases13. The pulse-chase experiment we performed showed that PA was not recycled to the plasma membrane but was retained inside some endosomal compartment whose identity is at the present unknown.
Escape from such an endosomal compartment is therefore the main obstacle for cytoplasmic delivery of p5314–29 peptide. To achieve this and corroborate our hypothesis, we employed sucrose, which disrupts endosomes after build up of osmotic pressure36, 37. High sucrose concentrations were associated with reduced viability and use of sucrose in vivo is not deemed feasible. The fact that cell death was enhanced following sucrose treatment in the presence of p5314–29 PAs only in the cell line with wt p53 provides indirect evidence that the cytotoxic effect was specific. At this point, however, we cannot attribute unequivocally this specificity to disruption of the p53-MDM2 since many other differences exist between the two cell lines used. More detailed localization studies to establish PA distribution in both cell lines following sucrose treatment and biochemical assays of p53 activation are now required.
Since the first description of the p53-MDM2 interaction60, several peptides with increased potency compared to p5314–29 have been reported. These inhibitors additionally target the p53-MDM4 binding, which has now been established as critical in p53 regulation5. Having acquired the basic understanding of the PA internalization process, our future efforts will employ more potent inhibiting peptides and will focus on their incorporation in structures capable of delivering the peptide amphiphiles in vivo. In parallel, having determined endosomal escape to be the barrier to cytoplasmic delivery, strategies for overcoming this obstacle, suitable for translation in vivo will be sought.
In summary, this work presents an alternative method for intracellular delivery of a pro-apoptotic peptide in cultured cells in vitro. The significance of this work lies on one hand on the generality of the approach, and on the other hand on the potential of this particular p5314–29 peptide amphiphile in constructing nano-sized structures for cancer therapy. The strategy for peptide modification presented here can be extended to peptides synthesized using solid-state phase synthesis, is simple and scaleable. The amphiphilic character of the resulting peptide amphiphiles allows them to self-assemble into dynamic structures, interact with cells and localize in intracellular compartments. Escape from these compartments therefore constitutes the main obstacle to cytoplasmic localization. The finding that uptake occurs following micelle disassembly has implications on the design of drug delivery vehicles: stability is often required for transportation but does not necessarily favor internalization. Instead, peptide amphiphiles are able to enter cells and remain in intracellular compartments for long times after extracellular peptide amphiphiles are removed. Overall, our study demonstrates that chemical modification of peptides with a lipid-like tail is a promising strategy towards efficient delivery of peptide inhibitors of the p53-MDM2 interaction in cancer cells.
Supplementary Material
Acknowledgments
This work was supported by National Heart, Lung and Blood Institute grant 5 U54 CA119335-04 and the MRSEC Program of the National Science Foundation under award DMR05-20415.
Footnotes
Supporting Information Available. Fluorescence dequenching kinetics in culture medium (Figure S1): PA uptake quantification in presence of sucrose (Figure S2) and PA uptake by MDA-MB-435 cells (Figure S3) are presented. Additional experimental procedures are also provided. This information is available free of charge via the Internet at http://pubs.acs.org.
Contributor Information
Dimitris Missirlis, Email: dimis@berkeley.edu.
Daniel V. Krogstad, Email: dkrogstad@engineering.ucsb.edu.
Matthew Tirrell, Email: mvtirrell@berkeley.edu.
References
- 1.Wade M, Wahl GM. Targeting Mdm2 and Mdmx in cancer therapy: better living through medicinal chemistry? Mol Cancer Res. 2009;7(1):1–11. doi: 10.1158/1541-7786.MCR-08-0423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chene P. Inhibiting the p53-MDM2 interaction: an important target for cancer therapy. Nat Rev Cancer. 2003;3(2):102–109. doi: 10.1038/nrc991. [DOI] [PubMed] [Google Scholar]
- 3.Murray JK, Gellman SH. Targeting protein-protein interactions: Lessons from p53/MDM2. Biopolymers. 2007;88(5):657–686. doi: 10.1002/bip.20741. [DOI] [PubMed] [Google Scholar]
- 4.Shangary S, Wang S. Small-molecule inhibitors of the MDM2-p53 protein-protein interaction to reactivate p53 function: a novel approach for cancer therapy. Annu Rev Pharmacol Toxicol. 2009;49:223–241. doi: 10.1146/annurev.pharmtox.48.113006.094723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hu B, Gilkes DM, Chen J. Efficient p53 Activation and Apoptosis by Simultaneous Disruption of Binding to MDM2 and MDMX. Cancer Res. 2007;67(18):8810–8817. doi: 10.1158/0008-5472.CAN-07-1140. [DOI] [PubMed] [Google Scholar]
- 6.Kokkoli E, Mardilovich A, Wedekind A, Rexeisen EL, Garg A, Craig JA. Self-assembly and applications of biomimetic and bioactive peptide-amphiphiles. Soft Matter. 2006;2(12):1015–1024. doi: 10.1039/b608929a. [DOI] [PubMed] [Google Scholar]
- 7.Karmali PP, Kotamraju VR, Kastantin M, Black M, Missirlis D, Tirrell M, Ruoslahti E. Targeting of albumin-embedded paclitaxel nanoparticles to tumors. Nanomedicine. 2009;5(1):73–82. doi: 10.1016/j.nano.2008.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Peters D, Kastantin M, Kotamraju VR, Karmali PP, Gujraty K, Tirrell M, Ruoslahti E. Targeting atherosclerosis by using modular, multifunctional micelles. Proc Natl Acad Sci U S A. 2009;106(24):9815–9819. doi: 10.1073/pnas.0903369106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kastantin M, Ananthanarayanan B, Karmali P, Ruoslahti E, Tirrell M. Effect of the lipid chain melting transition on the stability of DSPE-PEG(2000) micelles. Langmuir. 2009;25(13):7279–7286. doi: 10.1021/la900310k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang TY, Leventis R, Silvius JR. Artificially lipid-anchored proteins can elicit clustering-induced intracellular signaling events in Jurkat T-lymphocytes independent of lipid raft association. J Biol Chem. 2005;280(24):22839–22846. doi: 10.1074/jbc.M502920200. [DOI] [PubMed] [Google Scholar]
- 11.Peitzsch RM, McLaughlin S. Binding of acylated peptides and fatty acids to phospholipid vesicles: pertinence to myristoylated proteins. Biochemistry. 1993;32(39):10436–10443. doi: 10.1021/bi00090a020. [DOI] [PubMed] [Google Scholar]
- 12.Missirlis D, Khant H, Tirrell M. Mechanisms of peptide amphiphile internalization by SJSA-1 cells in vitro. Biochemistry. 2009;48(15):3304–3314. doi: 10.1021/bi802356k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mukherjee S, Soe TT, Maxfield FR. Endocytic sorting of lipid analogues differing solely in the chemistry of their hydrophobic tails. J Cell Biol. 1999;144(6):1271–1284. doi: 10.1083/jcb.144.6.1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carrigan CN, Imperiali B. The engineering of membrane-permeable peptides. Anal Biochem. 2005;341(2):290–298. doi: 10.1016/j.ab.2005.03.026. [DOI] [PubMed] [Google Scholar]
- 15.Foerg C, Merkle HP. On the biomedical promise of cell penetrating peptides: limits versus prospects. J Pharm Sci. 2008;97(1):144–162. doi: 10.1002/jps.21117. [DOI] [PubMed] [Google Scholar]
- 16.Bernal F, Tyler AF, Korsmeyer SJ, Walensky LD, Verdine GL. Reactivation of the p53 Tumor Suppressor Pathway by a Stapled p53 Peptide. J Am Chem Soc. 2007;129(9):2456–2457. doi: 10.1021/ja0693587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Walensky LD, Kung AL, Escher I, Malia TJ, Barbuto S, Wright RD, Wagner G, Verdine GL, Korsmeyer SJ. Activation of apoptosis in vivo by a hydrocarbon-stapled BH3 helix. Science. 2004;305(5689):1466–1470. doi: 10.1126/science.1099191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nelson AR, Borland L, Allbritton NL, Sims CE. Myristoyl-based transport of peptides into living cells. Biochemistry. 2007;46(51):14771–14781. doi: 10.1021/bi701295k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hansen CG, Nichols BJ. Molecular mechanisms of clathrin-independent endocytosis. J Cell Sci. 2009;122(Pt 11):1713–1721. doi: 10.1242/jcs.033951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mayor S, Pagano RE. Pathways of clathrin-independent endocytosis. Nat Rev Mol Cell Biol. 2007;8(8):603–612. doi: 10.1038/nrm2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rajendran L, Schneider A, Schlechtingen G, Weidlich S, Ries J, Braxmeier T, Schwille P, Schulz JB, Schroeder C, Simons M, Jennings G, Knolker HJ, Simons K. Efficient inhibition of the Alzheimer’s disease beta-secretase by membrane targeting. Science. 2008;320(5875):520–523. doi: 10.1126/science.1156609. [DOI] [PubMed] [Google Scholar]
- 22.Ingallinella P, Bianchi E, Ladwa NA, Wang YJ, Hrin R, Veneziano M, Bonelli F, Ketas TJ, Moore JP, Miller MD, Pessi A. Addition of a cholesterol group to an HIV-1 peptide fusion inhibitor dramatically increases its antiviral potency. Proc Natl Acad Sci U S A. 2009;106(14):5801–5806. doi: 10.1073/pnas.0901007106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eichholtz T, de Bont DB, de Widt J, Liskamp RM, Ploegh HL. A myristoylated pseudosubstrate peptide, a novel protein kinase C inhibitor. J Biol Chem. 1993;268(3):1982–1986. [PubMed] [Google Scholar]
- 24.Sabharanjak S, Sharma P, Parton RG, Mayor S. GPI-anchored proteins are delivered to recycling endosomes via a distinct cdc42-regulated, clathrin-independent pinocytic pathway. Dev Cell. 2002;2(4):411–423. doi: 10.1016/s1534-5807(02)00145-4. [DOI] [PubMed] [Google Scholar]
- 25.Naslavsky N, Weigert R, Donaldson JG. Characterization of a nonclathrin endocytic pathway: membrane cargo and lipid requirements. Mol Biol Cell. 2004;15(8):3542–3552. doi: 10.1091/mbc.E04-02-0151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fivaz M, Vilbois F, Thurnheer S, Pasquali C, Abrami L, Bickel PE, Parton RG, van der Goot FG. Differential sorting and fate of endocytosed GPI-anchored proteins. Embo J. 2002;21(15):3989–4000. doi: 10.1093/emboj/cdf398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kalia M, Kumari S, Chadda R, Hill MM, Parton RG, Mayor S. Arf6-independent GPI-anchored protein-enriched early endosomal compartments fuse with sorting endosomes via a Rab5/phosphatidylinositol-3′-kinase-dependent machinery. Mol Biol Cell. 2006;17(8):3689–3704. doi: 10.1091/mbc.E05-10-0980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sharma P, Varma R, Sarasij RC, Ira, Gousset K, Krishnamoorthy G, Rao M, Mayor S. Nanoscale organization of multiple GPI-anchored proteins in living cell membranes. Cell. 2004;116(4):577–589. doi: 10.1016/s0092-8674(04)00167-9. [DOI] [PubMed] [Google Scholar]
- 29.Chadda R, Howes MT, Plowman SJ, Hancock JF, Parton RG, Mayor S. Cholesterol-sensitive Cdc42 activation regulates actin polymerization for endocytosis via the GEEC pathway. Traffic. 2007;8(6):702–717. doi: 10.1111/j.1600-0854.2007.00565.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zacharias DA, Violin JD, Newton AC, Tsien RY. Partitioning of lipid-modified monomeric GFPs into membrane microdomains of live cells. Science. 2002;296(5569):913–916. doi: 10.1126/science.1068539. [DOI] [PubMed] [Google Scholar]
- 31.Bhagatji P, Leventis R, Comeau J, Refaei M, Silvius JR. Steric and not structure-specific factors dictate the endocytic mechanism of glycosylphosphatidylinositol-anchored proteins. J Cell Biol. 2009;186(4):615–628. doi: 10.1083/jcb.200903102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Resh MD. Trafficking and signaling by fatty-acylated and prenylated proteins. Nat Chem Biol. 2006;2(11):584–590. doi: 10.1038/nchembio834. [DOI] [PubMed] [Google Scholar]
- 33.Winkler R, Harnau L, Reineker P. Distribution functions and dynamical properties of stiff macromolecules. Macromol Theory Simul. 1997;6(6):1007–1035. [Google Scholar]
- 34.Dreiss CA. Wormlike micelles: where do we stand? Recent developments, linear rheology and scattering techniques. Soft Matter. 2007;3(8):956–970. doi: 10.1039/b705775j. [DOI] [PubMed] [Google Scholar]
- 35.West MA, Bretscher MS, Watts C. Distinct endocytotic pathways in epidermal growth factor-stimulated human carcinoma A431 cells. J Cell Biol. 1989;109(6 Pt 1):2731–2739. doi: 10.1083/jcb.109.6.2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kato T, Okada S, Yutaka T, Yabuuchi H. The effects of sucrose loading on lysosomal hydrolases. Mol Cell Biochem. 1984;60(1):83–98. doi: 10.1007/BF00226302. [DOI] [PubMed] [Google Scholar]
- 37.Caron NJ, Quenneville SP, Tremblay JP. Endosome disruption enhances the functional nuclear delivery of Tat-fusion proteins. Biochem Biophys Res Commun. 2004;319(1):12–20. doi: 10.1016/j.bbrc.2004.04.180. [DOI] [PubMed] [Google Scholar]
- 38.Silvius JR, Zuckermann MJ. Interbilayer transfer of phospholipid-anchored macromolecules via monomer diffusion. Biochemistry. 1993;32(12):3153–3161. doi: 10.1021/bi00063a030. [DOI] [PubMed] [Google Scholar]
- 39.Wimley WC, Thompson TE. Exchange and flip-flop of dimyristoylphosphatidylcholine in liquid-crystalline, gel, and two-component, two-phase large unilamellar vesicles. Biochemistry. 1990;29(5):1296–1303. doi: 10.1021/bi00457a027. [DOI] [PubMed] [Google Scholar]
- 40.Fenske DB, Palmer LR, Chen T, Wong KF, Cullis PR. Cationic poly(ethyleneglycol) lipids incorporated into pre-formed vesicles enhance binding and uptake to BHK cells. Biochim Biophys Acta. 2001;1512(2):259–272. doi: 10.1016/s0005-2736(01)00327-3. [DOI] [PubMed] [Google Scholar]
- 41.Shahinian S, Silvius JR. Doubly-lipid-modified protein sequence motifs exhibit long-lived anchorage to lipid bilayer membranes. Biochemistry. 1995;34(11):3813–3822. doi: 10.1021/bi00011a039. [DOI] [PubMed] [Google Scholar]
- 42.Missirlis D, Farine M, Kastantin M, Ananthanarayanan B, Neumann T, Tirrell M. Linker Chemistry Determines Secondary Structure of p53(14–29) in Peptide Amphiphile Micelles. Bioconjug Chem. 2010;21(3):465–475. doi: 10.1021/bc900383m. [DOI] [PubMed] [Google Scholar]
- 43.Wolfrum C, Shi S, Jayaprakash KN, Jayaraman M, Wang G, Pandey RK, Rajeev KG, Nakayama T, Charrise K, Ndungo EM, Zimmermann T, Koteliansky V, Manoharan M, Stoffel M. Mechanisms and optimization of in vivo delivery of lipophilic siRNAs. Nat Biotechnol. 2007;25(10):1149–1157. doi: 10.1038/nbt1339. [DOI] [PubMed] [Google Scholar]
- 44.Andrieu M, Loing E, Desoutter JF, Connan F, Choppin J, Gras-Masse H, Hanau D, Dautry-Varsat A, Guillet JG, Hosmalin A. Endocytosis of an HIV-derived lipopeptide into human dendritic cells followed by class I-restricted CD8(+) T lymphocyte activation. Eur J Immunol. 2000;30(11):3256–3265. doi: 10.1002/1521-4141(200011)30:11<3256::AID-IMMU3256>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 45.Kato K, Itoh C, Yasukouchi T, Nagamune T. Rapid protein anchoring into the membranes of Mammalian cells using oleyl chain and poly(ethylene glycol) derivatives. Biotechnol Prog. 2004;20(3):897–904. doi: 10.1021/bp0342093. [DOI] [PubMed] [Google Scholar]
- 46.Mercer J, Helenius A. Virus entry by macropinocytosis. Nat Cell Biol. 2009;11(5):510–20. doi: 10.1038/ncb0509-510. [DOI] [PubMed] [Google Scholar]
- 47.Hewlett LJ, Prescott AR, Watts C. The coated pit and macropinocytic pathways serve distinct endosome populations. J Cell Biol. 1994;124(5):689–703. doi: 10.1083/jcb.124.5.689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fretz M, Jin J, Conibere R, Penning NA, Al-Taei S, Storm G, Futaki S, Takeuchi T, Nakase I, Jones AT. Effects of Na+/H+ exchanger inhibitors on subcellular localisation of endocytic organelles and intracellular dynamics of protein transduction domains HIV-TAT peptide and octaarginine. J Control Release. 2006;116(2):247–54. doi: 10.1016/j.jconrel.2006.07.009. [DOI] [PubMed] [Google Scholar]
- 49.Racoosin EL, Swanson JA. M-CSF-induced macropinocytosis increases solute endocytosis but not receptor-mediated endocytosis in mouse macrophages. J Cell Sci. 1992;102(Pt 4):867–880. doi: 10.1242/jcs.102.4.867. [DOI] [PubMed] [Google Scholar]
- 50.Taouil K, Feray JC, Brunet J, Christen MO, Garay RP, Hannaert P. Inhibition by xipamide of amiloride-induced acidification in cultured rat cardiocytes. Eur J Pharmacol. 1997;324(2–3):289–294. doi: 10.1016/s0014-2999(97)00087-3. [DOI] [PubMed] [Google Scholar]
- 51.Sandvig K, Olsnes S, Petersen OW, van Deurs B. Acidification of the cytosol inhibits endocytosis from coated pits. J Cell Biol. 1987;105(2):679–689. doi: 10.1083/jcb.105.2.679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Heuser JE, Anderson RG. Hypertonic media inhibit receptor-mediated endocytosis by blocking clathrin-coated pit formation. J Cell Biol. 1989;108(2):389–400. doi: 10.1083/jcb.108.2.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Morris DP, Lei B, Wu YX, Michelotti GA, Schwinn DA. The alpha1a-adrenergic receptor occupies membrane rafts with its G protein effectors but internalizes via clathrin-coated pits. J Biol Chem. 2008;283(5):2973–2985. doi: 10.1074/jbc.M705795200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yumoto R, Nishikawa H, Okamoto M, Katayama H, Nagai J, Takano M. Clathrin-mediated endocytosis of FITC-albumin in alveolar type II epithelial cell line RLE-6TN. Am J Physiol Lung Cell Mol Physiol. 2006;290(5):L946–955. doi: 10.1152/ajplung.00173.2005. [DOI] [PubMed] [Google Scholar]
- 55.Tagawa M, Yumoto R, Oda K, Nagai J, Takano M. Low-affinity transport of FITC-albumin in alveolar type II epithelial cell line RLE-6TN. Drug Metab Pharmacokinet. 2008;23(5):318–327. doi: 10.2133/dmpk.23.318. [DOI] [PubMed] [Google Scholar]
- 56.Salikhova A, Wang L, Lanahan AA, Liu M, Simons M, Leenders WP, Mukhopadhyay D, Horowitz A. Vascular endothelial growth factor and semaphorin induce neuropilin-1 endocytosis via separate pathways. Circ Res. 2008;103(6):e71–79. doi: 10.1161/CIRCRESAHA.108.183327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Subtil A, Gaidarov I, Kobylarz K, Lampson MA, Keen JH, McGraw TE. Acute cholesterol depletion inhibits clathrin-coated pit budding. Proc Natl Acad Sci U S A. 1999;96(12):6775–6780. doi: 10.1073/pnas.96.12.6775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sharma DK, Brown JC, Choudhury A, Peterson TE, Holicky E, Marks DL, Simari R, Parton RG, Pagano RE. Selective stimulation of caveolar endocytosis by glycosphingolipids and cholesterol. Mol Biol Cell. 2004;15(7):3114–3122. doi: 10.1091/mbc.E04-03-0189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zidovetzki R, Levitan I. Use of cyclodextrins to manipulate plasma membrane cholesterol content: evidence, misconceptions and control strategies. Biochim Biophys Acta. 2007;1768(6):1311–1324. doi: 10.1016/j.bbamem.2007.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kussie PH, Gorina S, Marechal V, Elenbaas B, Moreau J, Levine AJ, Pavletich NP. Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science. 1996;274(5289):948–953. doi: 10.1126/science.274.5289.948. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.