

Abstract
Background
Attention-deficit/hyperactivity disorder (ADHD) may result from delayed establishment of corticolimbic circuitry or perturbed dopamine (DA) neurotransmission. Despite the widespread use of stimulants to treat ADHD, little is known regarding their long-term effects on neurotransmitter levels and metabolism. Cyclin-dependent kinase 5 (Cdk5) regulates DA signaling through control of synthesis, postsynaptic responses, and vesicle release. Mice lacking the Cdk5-activating cofactor p35 are deficient in cortical lamination, suggesting altered motor/reward circuitry.
Methods
We employed mice lacking p35 to study the effect of altered circuitry in vivo. Positron emission tomography measured glucose metabolism in the cerebral cortex using 2-deoxy-2-[18F]fluoro-D-glucose as the radiotracer. Retrograde dye tracing and tyrosine hydroxylase immunostains assessed the effect of p35 knockout on the medial prefrontal cortex (PFC), especially in relation to mesolimbic circuit formation. We defined the influence of Cdk5/p35 activity on catecholaminergic neurotransmission and motor activity via examination of locomotor responses to psychostimulants, monoamine neurotransmitter levels, and DA signal transduction.
Results
Here, we report that mice deficient in p35 display increased glucose uptake in the cerebral cortex, basal hyperactivity, and paradoxical decreased locomotion in response to chronic injection of cocaine or methylphenidate. Knockout mice also exhibited an increased susceptibility to changes in PFC neurotransmitter content after chronic methylphenidate exposure, and altered basal DAergic activity in acute striatal and PFC slices.
Conclusions
Our findings suggest that dysregulation of Cdk5/p35 activity during development may contribute to ADHD pathology, as indicated by the behavioral phenotype, improperly established mesolimbic circuitry, and aberrations in striatal and PFC catecholaminergic signaling in p35 knockout mice.
Keywords: Cdk5, p35, dopamine, prefrontal cortex, methylphenidate, ADHD
Introduction
Attention-deficit/hyperactivity disorder (ADHD) is a heterogeneous developmental syndrome characterized by inattention, impulsivity, and hyperactivity. It is among the most common adolescent psychiatric diagnoses with an estimated prevalence of 4% (1). Motor control, vigilance, and attention are mediated through the mesocorticolimbic circuitry of the brain. In this pathway, glutamatergic pyramidal neurons of the medial prefrontal cortex (mPFC) synapse onto (γ-aminobutyric acid) GABA-ergic medium spiny neurons of the striatum. These neurons project via the direct and indirect pathways to dopaminergic neurons in the substantia nigra (SN)/ventral tegmental area (VTA). To complete the loop, SN/VTA neurons innervate the cortex either directly or via the thalamus. The transition from stimulus perception to overt action depends upon coordinated responses between this pathway and higher-association cortical circuits, which may be deficient in ADHD. Aberrations of mesocorticolimbic structure and function have been repeatedly implicated in ADHD patient imaging studies (2-7). Furthermore, high-resolution magnetic-resonance imaging links ADHD onset to a 3-4 year delay in corticogenesis (8). Thus, the size or developmental trajectory of cortical or basal ganglia structures may significantly affect attentional or motor inhibitory abilities later in life.
Dopaminergic neurotransmission may also play a central role in ADHD pathophysiology (9). Stimulants, such as methylphenidate (MPH), are the most common drug treatment for this disorder. These medications are thought to act by raising synaptic concentrations of dopamine (DA), and other catecholamines, through inhibition of presynaptic reuptake transporters (10-11). Several lines of evidence suggest that both hypo- and hyper-stimulation of DA receptors, in the PFC specifically, deprecate attention and working memory (12-14). In mice, psychostimulants increase catecholaminergic neurotransmission in the PFC selectively at therapeutic doses, which do not promote locomotion (15). Furthermore, the locomotor-suppressing effect of low-dose stimulants is observed in control and ADHD patients (16). Nuclear imaging of DA transporter binding (17-18) and studies of the DA receptor D4 tandem repeat polymorphism (19-23) repeatedly implicate deficiencies in this neurotransmitter system in ADHD.
Cyclin-dependent kinase 5 (Cdk5) is a neuronal serine/threonine protein kinase critical to proper neuronal migration, corticogenesis, and regulation of postsynaptic DA signal integration. Cdk5 activity is dependent upon its association with one of two homologous cofactors, p35 (24-25) or p39 (26). Mice deficient in Cdk5 are nonviable and display severe defects in the formation of the cerebral cortex including an inversion of the typical “inside-out” lamination of cortical neurons (27-28). Mice lacking p35 (p35−/−) are viable while exhibiting reversed cortical lamination, a severe reduction in the size of the corpus callosum, and abnormal neurite outgrowth in culture. In contrast, p39−/− mice do not display these defects (29-32). Cdk5 negatively regulates DA neurotransmission through the phosphorylation of DARPP-32 (DA and cyclic AMP regulated phosphoprotein of 32 kDa) (33). Inhibition of Cdk5 through genetic or pharmacological means potentiates the behavioral effects of DA-perturbing drugs (34-36). Furthermore, Cdk5 controls the behavioral effects of caffeine (37) and sexual reproductive behavior (38), via DARPP-32-dependent mechanisms. In addition to its modulation of DARPP-32, Cdk5 may mediate these actions through auxiliary control of synaptic vesicle release and upstream synthesis of DA in the forebrain (39-41).
Given the central role of Cdk5 in both the development of the cortex and the regulation of DA neurotransmission, we hypothesized that its dysregulation could contribute to the etiology of ADHD. Here we report that loss of p35 profoundly alters glucose uptake in the cerebral cortex, mesocorticolimbic circuitry, behavioral responses to psychomotor stimulants, catecholamine levels and metabolism, and DA signal transduction and that these effects strongly reflect deficits observed in ADHD patients. Thus, developmental dysregulation of Cdk5 is implicated in ADHD pathology and p35−/− mice provide a provocative model for the study of this psychiatric disease.
Materials and Methods
PET/CT Imaging
Small animal PET/CT imaging studies were performed using a Siemens Inveon Multimodality PET/CT system (Siemens Medical Solutions Inc., Knoxville, TN, USA). Animals were anesthetized using 2% Isoflurane for the duration of the imaging. The microCT imaging was acquired at 80kV and 500mA with a focal spot of 58 μm. The total rotation of the gantry was 360° with 360 rotation steps obtained at an exposure time of 175 ms/frame. The images were obtained using CCD readout of 4096 × 3098 with a bin factor of 4 and average frame of 1. Under low magnification the effective pixel size was 102.25 μm. Total microCT scan time was approximately 6 min. CT images were reconstructed with a down sample factor of 2 using Cobra Reconstruction Software. Body temperature was maintained at 38°C using a heating pad. Injection of 100 μCi of 2-deoxy-2-[18F]fluoro-D-glucose (FDG) was followed by a 60 min dynamic scan. PET images were reconstructed using Fourier Rebinning and Ordered Subsets Expectation Maximization (FROSEM) 2D algorithm with dynamic framing every 5 min. Reconstructed images were fused and analyzed using Inveon Research Workplace (IRW) software. For quantitation, the mean ROI data from the cerebral cortex was normalized to muscle (background) and identified by visual inspection. Corresponding PET images were reconstructed using FROSEM 3D algorithm with a blur value of 0.001.
Animal Experimentation
P35 knockout mice have been previously described (29). All mice were handled in accordance with the Guide for the Care and Use of Laboratory Animals as adopted and promulgated by the U.S. National Institutes of Health. The Institutional Animal Care and Use Committee of the University of Texas Southwestern Medical Center approved the specific protocols. Before all behavioral experiments took place, 6-12 week old mice were transported to the exam room at least 1 h beforehand and left in a quiet closed room.
Locomotor activity assay
Locomotor activity was measured essentially as described (35). No differences were observed in repetitive (stereotypic) movements in any of the experiments. For experiments where drugs were injected, animals were habituated to saline injections for the first 3 days. Animals were injected and placed into the chamber for a total run time of 2 h.
Preparation and incubation of acute slices
Slices from 6-8 week old male C57BL/6 were prepared as described (33), with slight modifications. Briefly, slices were preincubated with adenosine deaminase (AD) (10 μg/mL) for 60 min, changing for fresh buffer after 30 min. Slices were then incubated with SKF-81297 (Tocris Biosciences, Bristol, UK) in Krebs buffer for the proscribed period without AD, frozen on dry ice, and stored at −80°C until further assay.
Retrograde tracing
Mice were stereotactically injected with the retrograde tracer fluorogold (FG) (4% in 0.9% saline; Fluorochrome) in to the nucleus accumbens (NAc, shell). Mice were anesthetized with ketamine/xylazine (80/12 mg/kg, i.p.) and restrained in a stereotaxic apparatus. A glass micropipette connected to an air pressure injector system was positioned via the stereotaxic manipulator. FG was unilaterally injected into the NAc (+1.45 mm from the bregma, +0.65 mm lateral, −4.0 mm from the surface of cortex). The micropipette was removed and the incision was closed. Injection volumes were 10-50 nl, and there was no mortality. Mice were transcardially perfused 4 days later to allow for retrograde transport of FG. Injection sites were confirmed by immunostaining for FG and only mice with similar injection sites and volumes were compared.
Tissue preparation and histology
Immunohistochemistry (IHC) was performed essentially as described (41). Mice were perfused with 4% paraformaldehyde and brains were cryoprotected in 30% sucrose in PBS, and sectioned (30 μm) on a freezing microtome. Nonspecific binding was blocked with 3% donkey serum (Jackson ImmunoResearch, West Grove, PA, USA) and 0.3% Triton-X in PBS for 30–60 min. Primary antibodies included anti-Fluorogold (1:1000) (Fluorochrome, Denver, CO, USA), and anti-tyrosine hydroxylase (TH) (1:1000) (PhosphoSolutions, Aurora, CO, USA). Secondary antibodies included anti-rabbit Cy3 (Jackson ImmunoResearch). NeuroTrace Yellow Fluorescent Nissl Stain (Molecular Probes, Eugene, OR, USA) was used as instructed by manufacturer.
HPLC-EC analysis of catecholamines
Levels of striatal and mPFC DA, and its metabolites 3,4-dihydroxyphenylacetic acid (DOPAC), homovanillic acid (HVA), and 3-methoxytyramine (3-MT), as well as serotonin (5-HT) and its metabolite 5-hydroxyindoleacetic acid (5-HIAA) were quantified from striatum and PFC as described (42). Neurotransmitter monoamines and metabolites were detected using an ESA CoulArray electrochemical detector with a model 5014B cell set to a potential of +220 mV.
Immunoblot Analysis
Quantitative immunoblotting was conducted as described (43). Membranes were immunoblotted using antibodies for phospho-Ser845 GluR1 (PhosphoSolutions, Aurora, CO, USA), phospho-Thr34 DARPP-32 and total DARPP-32 (Cell-Signaling, Beverly, MA, USA), and rabbit total GluR1 (Abcam, Littleton, CO, USA).
Data analysis
Image J (NIH) was used to quantify immunoblots. Results are stated as mean ± standard error of the mean. Analysis of data involving one variable with independent observations utilized Student’s t test. For two variables, such as genotype and time, a two-way analysis of variance (ANOVA) was used. After confirmation of a significant effect of genotype, between-group comparisons were performed using Student’s t test.
Results
P35 KO results in increased rate of cortical glucose uptake
Positron emission tomography (PET) provides a noninvasive platform for the measurement of regional metabolism or neurotransmission in vivo. To assess the overall effect of p35 KO on brain function, p35−/− and wild-type (WT) littermates (10-12 weeks old) were injected with FDG and imaged by PET. A 60 min dynamic PET scan revealed a significantly greater rate of glucose uptake in the cerebral cortex of p35−/− mice (Figure 1A & 1B). Linear regression analysis of the best-fit line (R2 = 0.96 for WT, 0.97 for KO) indicated that the slope, or rate of glucose uptake, of p35−/− mice was 1.53 ± 0.11-fold higher than controls (Figure 1C). Across the time-span of the scan, statistical analysis revealed a significant effect of time and genotype (p < 0.001, two-way ANOVA, n = 5 WT, 7 KO) and no interaction (Figure 1B). Thus, p35 loss affects brain glucose metabolism, possibly through alteration of neural circuits and cytoarchitecture or neurotransmission.
Figure 1.
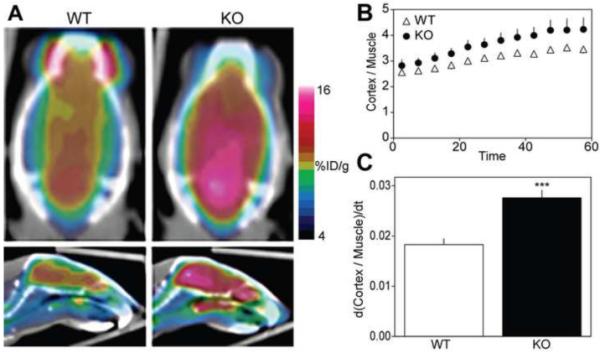
P35 knockout results in increased rate of cortical glucose uptake. (A)Representative coronal and sagittal positron emission tomography images of global brain glucose metabolism in control and p35−/− mice. Images represent the average values of a 60 min dynamic scan conducted after administration of 2-deoxy-2-[18F]fluoro-D-glucose (FDG). The color scale for each image is proportional to the percent Injected dose per gram (%ID/g) within the animal: white represents the highest, and red, yellow, green, and blue represent sequentially lower concentrations. (B) Time course of FDG uptake in the cerebral cortex. FDG uptake values (%ID/g) in the cerebral cortex are normalized to FDG uptake in muscle. Graph depicts mean values ± standard deviation of the cortex:muscle ratio in 5 min increments. (C) Rate of glucose uptake over the time-span of the 60 min scan. Values represent the mean slopes ± standard error of the best-fit lines derived from B (***p < 0.001, analysis of covariance, n = 5-7).
Upregulation of mesolimbic connectivity after p35 KO
P35 deficiency during development reverses the normal lamination of the cerebral cortex, and severely reduces the size of the corpus callosum (Figure 2A) (29-30,42). To assess how constitutive lack of p35 impacts mesocorticolimbic circuitry, we conducted immunohistochemical analysis of a retrograde tracer (44), FG, and the rate-limiting enzyme in DA biosynthesis, TH. FG was injected into the NAc in order to label neurons that synapse onto the NAc. Both p35−/− and WT mice displayed labeled neurons in the VTA, SNc, subiculum, and dorsal raphe nuclei with apparent equal cell number and distribution (data not shown). However, overt differences were found in the mPFC. In controls, afferents of the prelimbic and infralimbic cortices appeared to label discretely in layer 3, as approximated by visual inspection in conjunction with a Nissl costain. In contrast, p35−/− mice exhibited a diffuse labeling pattern (Figure 2B), with FG-tagged neurons apparently traversing layers 2-6. These results indicate that p35 KO induces a loss in the layer-specific afferentiation of descending mPFC outputs projecting to the NAc.
Figure 2.
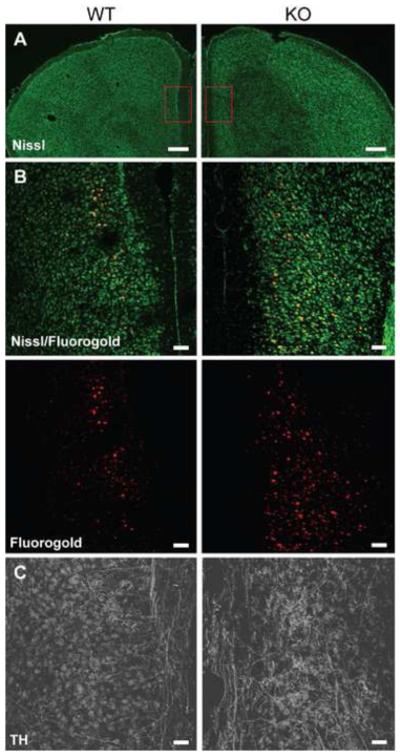
Upregulation of mesolimbic connectivity after p35 knockout. (A) Representative fluorescent Nissl stain of the prefrontal cortex in p35−/− and control mice at low magnification. Boxes indicate anatomical region of higher magnification shown in B and C. (B) Representative fluorescent stain of the medial prefrontal cortex (mPFC) for Nissl (green) and the retrograde neuronal tracer, Fluorogold (FG) (red), after injection of FG into the nucleus accumbens shell of control and p35−/− mice at high magnification. (C) Representative fluorescent stain of the mPFC for tyrosine hydroxylase (TH) at high magnification. Scale bars: 500 μm, A; 100 μm, B and C.
DAergic innervation was assessed by immunostaining for TH. The VTA, SNc, and striatum, of 6-8 week old p35−/− mice displayed indistinguishable staining intensity and distribution compared to WT littermates. However, in the infralimbic, prelimbic, and cingulate cortices of KO mice, an increased density of DA fiber innervation was observed (Figure 2C). This finding suggests that p35 deficiency results in an increase in DAergic afferents projecting from the VTA to the mPFC. Taken together, these results corroborate previous reports showing abnormal neurite outgrowth when Cdk5 activity is ablated during development, but go further in implicating the Cdk5/p35 complex as a key promoter of proper mesolimbic circuit formation.
p35 KO induces a locomotor profile reminiscent of ADHD
Because p35 deficiency alters the formation of circuitry governing motor control, we measured spontaneous activity after injection of saline, chronic MPH, or chronic cocaine in a novel environment. P35−/− mice exhibited significant hyperactivity (Figure 3A); recording 1.65 ± 0.07-fold more locomotor counts over a 60 min testing period compared to controls. This effect was specific to p35, as mice lacking p39 did not display hyperactivity (data not shown).
Figure 3.
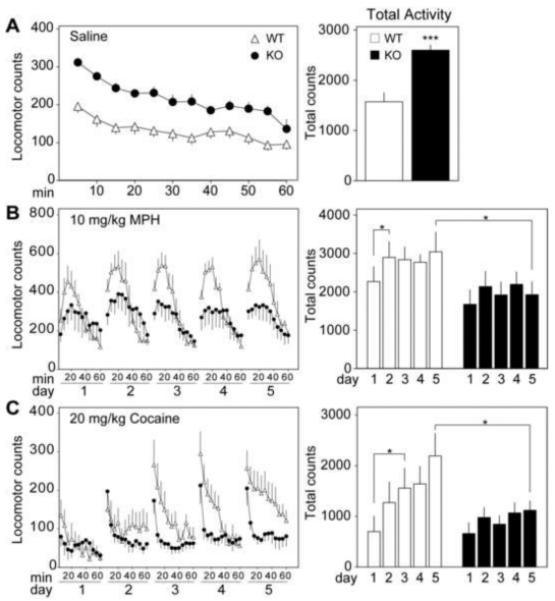
p35 knockout induces a locomotor profile reminiscent of ADHD. (A) Time course of the locomotor response to saline injection in control and p35−/− mice. Mean locomotor counts ± standard error for the 60 min following saline injection are shown (left). Each time point represents the average sum of locomotor counts from the previous five minutes, with the first point depicting 5 minutes of post-injection activity. Total locomotor counts ± standard error over the 60 min are also depicted (right) (***p < 0.001, Student’s unpaired t test, n = 5-7). (B) Time course of the locomotor response to chronic administration of methylphenidate (MPH, 10 mg/kg). Graph on the left shows mean locomotor counts ± standard error for 60 min intervals over a period of 5 days. Histogram at the right depicts total activity for each day ± standard error for the first 30 min of each session (*p < 0.05, Student’s paired t test, n = 5). (C) Time course of the locomotor response to chronic cocaine (20 mg/kg). Graph on the left shows mean locomotor counts ± standard error for 60 min intervals over a period of 5 days. Histogram at the right depicts total activity for each day ± standard error (*p < 0.05, Student’s unpaired t test, n = 5-7).
Interestingly, when dosed repeatedly with psychostimulants p35−/− mice displayed hypolocomotion. By the fifth day of MPH dosing, locomotor activity of the p35−/− group was reduced to 63 ± 11% of WT littermates. Furthermore, after administration of MPH WT mice exhibited mildly sensitized responses over the 5 experimental days while p35−/− mice were deficient in this effect. Statistical analysis of the first 30 min of cumulative activity for the MPH sensitization (Figure 3B) revealed a significant effect of genotype (p < 0.01, two-way RM ANOVA, n = 5) and no effect of treatment day or interaction.
To further assess the effect of p35 KO on behavioral responses to psychostimulants, mice were also given repeated injections of cocaine. Again, p35−/− mice exhibited a hypolocomotive response and were deficient in sensitization. On the fifth day of testing the locomotor counts of p35−/− mice were reduced to 51 ± 9% of controls over the 60 min time period. Analysis of total activity for sensitization to cocaine (Figure 3C) also revealed a significant effect of genotype (p < 0.01, two-way RM ANOVA, n = 6-7), drug (p < 0.05), and no interaction. Interestingly, a five-day treatment of cocaine, but not MPH, served to lower the locomotor counts of p35−/− mice such that there was no significant difference between MPH-treated KO animals and saline-treated WT littermates (p = 0.12, Student’s unpaired t test, n = 6-7). These data indicate that congenital KO of p35 results in basal hyperactivity and paradoxical hypolocomotion in response to chronic methylphenidate or cocaine exposure.
Effect of p35 KO and chronic methylphenidate on catecholamine levels and metabolism in the prefrontal cortex
Psychostimulants invoke their locomotor-activating effect by raising synaptic concentrations of DA and other catecholamines in the NAc (51). To evaluate the effects of psychostimulants on catecholamine levels in WT versus p35−/− mice, absolute concentrations of neurotransmitters in the striatum and PFC were measured by high-performance liquid chromatography (HPLC). Two sets of samples were used: one group of WT and p35−/− mice was sacrificed after 20 min in a novel environment with no drug administration (Pre-MPH). Another group of WT and p35−/− mice was dosed with MPH (10 mg/kg) for 5 consecutive days and sacrificed 20 min after the final injection (Chronic-MPH).
P35 KO did not significantly affect basal (pre-MPH) DA or DOPAC (Figure 4A). However DA and DOPAC were reduced to 27 ± 3 and 39 ± 10% respectively, of their pre-MPH levels in p35−/− mice. Furthermore DA turnover, represented as the ratio of HVA over DA, was increased 1.62 ± 0.19-fold in response to repeated MPH treatment of p35−/− mice. In contrast, MPH induced no significant effects in DA (p = 0.18, Student’s unpaired t test, n = 5-6), DOPAC (p = 0.21), or DA turnover (p = 0.48), in WT mice, although a clear trend towards the reduction of DA and metabolite levels was apparent. Moreover, after chronic MPH treatment p35−/− mice exhibited significantly reduced DA turnover (51% ± 6 less) compared to controls. In other measures, small but significant elevations in basal 5-HT (1.10 ± 0.01-fold higher) and 5-HIAA (1.23 ± 0.08-fold higher) were observed in pre-MPH p35−/− mice compared to controls (Figure 4B). Furthermore, chronic MPH induced a 1.08 ± 0.03-fold increase in 5-HT, a 18 ± 5% decrease in 5-HIAA, and a 23 ± 5% decrease in 5-HT turnover (5-HIAA to 5-HT ratio) in p35−/− mice, while no effect was detected in controls. For the levels of DOPAC, 5-HT, 5-HIAA, and the ratio of HVA to DA, statistical analysis showed a significant effect of genotype and drug (p < 0.05, 2-way ANOVA, n = 5-7), and no significant interaction across the span of the drug treatment.
Figure 4.
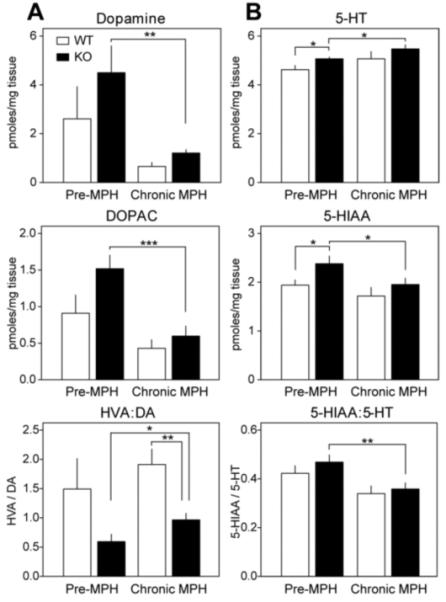
Effect of p35 knockout and chronic methylphenidate on catecholamine levels and metabolism in the prefrontal cortex. (A) Dopamine levels and metabolism in control and p35−/− mice before and after 5 consecutive injections of methylphenidate (MPH, 10 mg/kg). Graphs depict picomoles of dopamine (DA), 3,4-dihydroxyphenylacetic acid (DOPAC), and DA turnover (ratio of homovanillic acid over DA) normalized to tissue weight ± standard error (*p < 0.05, **p < 0.01, ***p < 0.001, Student’s unpaired t test, n = 5-7 per group). (B) Serotonin levels and metabolism in control and p35−/− mice before and after chronic MPH. Graphs show picomoles of serotonin (5-HT), 5-hydroxyindoleacetic acid (5-HIAA), and 5-HT turnover (ratio of 5-HIAA over 5-HT) normalized to tissue weight ± standard error (*p < 0.05, **p < 0.01, Student’s unpaired t test, n = 5-7 per group).
Surprisingly, in the caudatoputamen, no significant differences were observed between WT and p35−/− mice before or after MPH administration (Supplement: Figure S1). Thus, p35 deficiency rendered mice responsive to the modulation of PFC catecholamine metabolism by MPH, with the stimulant profoundly lowering DA and raising 5-HT selectively in p35−/− mice.
Effect of p35 KO on dopaminergic neurotransmission in acute striatal and prefrontal cortical slices
DA neurotransmission was evaluated using the selective D1 DA receptor (D1DR) agonist, SKF-81297 (1 μmol/L, 5 min). The PKA-dependent phosphorylation states of the GluR1 subunit of the α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate (AMPA) receptor at Serine-845 (S845) (46) or DARPP-32 at Threonine-34 (T34) (47) were monitored in acute striatal and PFC slices to assess the responsiveness of the D1DR/cAMP/PKA cascade.
Activation of the D1DR in striatal slices from WT mice induced a 2.14 ± 0.20-fold increase in P-S845 GluR1 levels (Figure 5A). Consistent with previous reports (33), this response was potentiated to a 3.79 ± 0.78-fold increase in p35−/− mice. Interestingly, total levels of GluR1 in p35 KO slices were reduced to 68 ± 10% of untreated controls by SKF-81297, while no effect was observed in slices from WT littermates. Furthermore, the basal level of total GluR1 was 1.73 ± 0.17-fold higher in p35−/− versus WT mice. As a result, when P-S845 levels are normalized, a decrease in basal P-S845 to 61% ± 3 of controls was detected in KO mice. D1DR activation induced 2.36 ± 0.32- and 4.43 ± 0.99-fold increases in normalized P-S845 GluR1 in WT and p35−/− striatal slices, respectively. Likewise, 2.86 ± 0.38- and 6.98 ± 0.91-fold increases in P-T34 DARPP-32 were induced by SKF-81297 treatment of striatal slices from WT and p35−/− mice, respectively. Additionally, KO mice exhibited 44 ± 5% of WT normalized basal P-T34 DARPP-32 levels. Neither p35 KO nor drug treatment had an effect on total levels of striatal DARPP-32.
Figure 5.
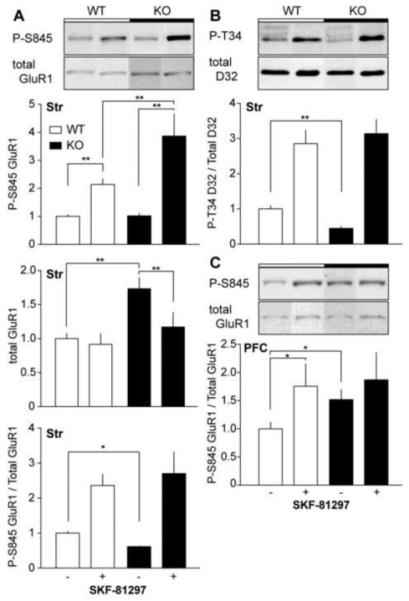
Effect of p35 knockout on dopaminergic neurotransmission in acute striatal and prefrontal cortical slices. (A) Immunoblot analysis of phospho-Ser845 GluR1 in striatal slices from control and p35−/− mice treated with SKF-81297 (1 μM, 5min). Blots were probed for phospho-Ser845, and total GluR1. Quantitation of the phosphoprotein, total protein, and the phospho/total protein ratio ± standard error normalized to wild-type controls are shown (*p < 0.05, **p < 0.01, Student’s unpaired t test, n = 4-6 per group). (B) Immunoblot analysis of phospho-Thr34 DARPP-32 in striatal slices. Blots were probed for phospho-phospho-Thr34 of DARPP-32, and total DARPP-32 (**p < 0.01, Student’s unpaired t test, n = 4-6 per group). (C) Immunoblot analysis of phospho-Ser845 GluR1 in acute prefrontal cortical slices (*p < 0.05, Student’s unpaired t test, n = 5-9 per group).
In WT PFC slices, SKF-81297 treatment induced a 1.75 ± 0.4-fold increase in P-S845 GluR1, but had no effect on slices from p35−/− mice. Statistical analysis revealed no effect of genotype, drug, or interaction (p > 0.05, two-way ANOVA, n = 5-9) across the time span of SKF-81297 treatment. However, basal normalized P-S845 GluR1 increased 1.52 ± 0.18-fold in p35−/− mice compared to WT littermate controls, while p35 KO had no effect on total PFC GluR1 levels. These results suggest that congenital loss of p35 reduces basal PKA activity and potentiates D1DR-mediated DA efficacy in the striatum, while promoting the opposite effect in the PFC.
Discussion
Improper or delayed establishment of the mesocorticolimbic pathway may contribute to psychiatric disease, including ADHD (4, 6-8). Indeed the PFC is highly implicated in attentional and hyperactivity disorders, as therapeutic doses of MPH selectively modify neurotransmission in this area (15). Here, we have shown that p35 KO compromised layer-specificity of mPFC afferents projecting to the NAc. Additionally, an increase in PFC innervation by TH-positive fibers, increased basal PKA activity, and decreased DA degradation likely contribute to potentiated DAergic neurotransmission in the PFC of p35−/− mice. DA acts as a neuromodulator, but generally inhibits the firing of mPFC output neurons through the D2DR on pyramidal neurons and the activation of GABA interneurons (48-50). Thus, perpetually high DAergic signaling may inhibit output firing of mPFC neurons in p35−/− mice, and possibly ADHD patients.
P35−/− mice were hyperactive and exhibited hypolocomotive responses to psychostimulants. The discrepancy between the locomotor responses of WT versus p35−/− mice grew with each day of repeated dosing, as p35 KO mice were deficient in sensitization. The sensitizing effect of repeated exposure may depend upon both coordinated glutamate release in mPFC terminals of the NAc (51) and proper DAergic modulation of mPFC output neurons (52-54), which were disrupted by p35 KO. Inhibition of Cdk5 in adult rodents by pharmacological (34, 36) or transgenic means (35) has generally potentiated psychostimulant responses. In contrast, here we report that constitutive Cdk5 dysregulation via p35 KO suppressed locomotor responses, further implicating the congenital defects in motor-reward circuitry in the ADHD-like phenotype.
While MPH is presently the most widely used drug treatment for ADHD, its long-term effects on development and catecholamine metabolism are poorly understood. Chronic exposure to MPH caused significant changes in catecholamine content and degradation in p35−/− mouse PFC, while controls were unaffected. This suggests a possible role for Cdk5 in regulating catecholamine metabolism. Cdk5 may normally protect PFC neurons from the effects of typical weekday MPH dosing so that DA levels are not overly depleted. However, these data also raise the possibility that high-dose chronic MPH may deplete DA and raise 5-HT levels in the PFC if mesocorticolimbic circuitry or Cdk5 activity is significantly altered. Future studies may be warranted to evaluate these parameters in ADHD patients.
Previously we reported that Cdk5 repressed D1DR/cAMP/PKA signaling through the phosphorylation of DARPP-32 (33) and that p35 KO resulted in increased DA efficacy with regard to the phosphorylation of S845 GluR1 and T34 DARPP-32. Here we confirmed the potentiated PKA response to D1DR stimulation, but found that total levels of GluR1 were elevated and basal levels of P-T34 DARPP-32 were reduced in the striatum of p35−/− mice, raising the possibility that other factors including genetic background fluctuation may now contribute to the increased efficacy of D1DR agonists in the absence of p35. Phosphorylation of GluR1 at S845 increases the peak open probability and potentiates AMPA receptor Ca2+ conductance (55-56), while T34 phosphorylated DARPP-32 promotes phosphatase inhibition (47). MPH potentiates PKA activity in acute striatal slices while antagonizing Cdk5-dependent phosphorylation of DARPP-32 (57). Alterations in the basal phosphorylation of these sites in cortical and subcortical structures of p35−/− mice suggest that the physiological properties of PFC and striatal neurons, which are dependent on the phosphorylation state of GluR1, DARPP-32 and possibly other downstream effectors of D1DR, may contribute to hyperactivity and paradoxical responses to stimulants observed in p35−/− mice and ADHD patients. It is interesting to note that loss of another protein kinase that phosphorylates DARPP-32, casein kinase I, also results in a hyperactive phenotype (58).
Reports of changes in ADHD patient glucose metabolism conflict (59-61). As a noninvasive metabolic imaging technique with clinical acceptance, FDG-PET demonstrated clear perturbations in glucose metabolism, prompting our further study of the effects of p35 KO on mesocorticolimbic circuitry and function. The hypermetabolic brain activity of p35−/− mice may result from reduced Cdk5/p35-dependent inhibition of neuronal excitability (62-63). We also revealed that p35 KO caused increased mesolimbic connectivity and PFC neurotransmitter content possibly necessitating an increase in supportive energy expenditure. Increased glucose metabolism could also serve as an essential compensatory response, given the extent of anatomical defects resulting from p35 KO. Although the effects of anesthesia are controlled for in WT animals, these results must be interpreted carefully as isoflurane nonspecifically lowers brain metabolism (64) and the glucose uptake of p35−/− mice could be differently affected by this drug.
The pathoetiology of ADHD remains poorly understood and a number of independent genetic, epigenetic, and environmental factors may cumulatively contribute to the composite human disorder. Here we present evidence implicating Cdk5 dysregulation in ADHD. Interestingly, exposure to neurotoxins such as organophosphates, which can dysregulate Cdk5 (65), has also been linked to a higher ADHD incidence in humans (66). Combining the results of this study with the known role of Cdk5 in corticogenesis and regulation of the DA signal, we conclude that its dysfunction during development may contribute to ADHD etiology.
Supplementary Material
Acknowledgements
We thank Donald Cooper for helpful advice and discussion, Li-Huei Tsai for the p35−/− mice, Shari Birnbaum for assistance with behavioral studies, and Laurent Couque, Nathan DeCarolis, Mehreen Kouser, and Guiyang Hao for technical assistance. J.M.D. was the recipient of a Green Fellowship from UT Dallas and a Summer Undergraduate Research Fellowship from UT Southwestern. Support was also provided by a NARSAD Young Investigator Award (K.H.), and U.S. National Institutes of Health Grants to M.L. (K08 MH084058-1A1, UL1RR024923, RL1 DK081182, and RL1 DK081185) and J.A.B. (MH79710 and DA016672).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Financial Disclosures: All authors report no biomedical financial interests or potential conflicts of interest.
REFERENCES
- 1.American Psychiatric Association . Diagnostic and Statistical Manual of Mental Disorders. 4th ed. American Psychiatric Press; Washington, DC: 2000. [Google Scholar]
- 2.Zametkin AJ, Nordahl TE, Gross M, King AC, Semple WE, Rumsey J, et al. Cerebral glucose metabolism in adults with hyperactivity of childhood onset. N Engl J Med. 1990;323(20):1361–6. doi: 10.1056/NEJM199011153232001. [DOI] [PubMed] [Google Scholar]
- 3.Giedd JN, Castellanos FX, Casey BJ, Kozuch P, King AC, Hamburger SD, et al. Quantitative morphology of the corpus callosum in attention deficit hyperactivity disorder. Am J Psychiatry. 1994;(151):665–9. doi: 10.1176/ajp.151.5.665. [DOI] [PubMed] [Google Scholar]
- 4.Casey BJ, et al. Implication of right fronto-striatal circuitry in response inhibition and attention-deficit/hyperactivity disorder. J Am Acad Child Adolesc Psych. 1997;36:374–383. doi: 10.1097/00004583-199703000-00016. [DOI] [PubMed] [Google Scholar]
- 5.Amen DG, Carmichael BD. High-resolution brain SPECT imaging in ADHD. Ann Clin Psychiatry. 1997;9(2):81–6. doi: 10.1023/a:1026201218296. [DOI] [PubMed] [Google Scholar]
- 6.Castellanos FX, Lee PP, Sharp W, Jeffries NO, Greenstein DK, Clasen LS, et al. Developmental trajectories of brain volume abnormalites in children and adolescents with attention-deficit/hyperactivity disorder. JAMA. 2002;288(14):1740–1748. doi: 10.1001/jama.288.14.1740. [DOI] [PubMed] [Google Scholar]
- 7.Qiu A, Crocetti D, Adler M, Mahone EM, Denckla MB, Miller MI, et al. Basal ganglia volume and shape in children with attention deficit hyperactivity disorder. Am J Psychiatry. 2009;166(1):74–82. doi: 10.1176/appi.ajp.2008.08030426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shaw P, Eckstrand K, Sharp W, Blumenthal J, Lerch JP, Greenstein D, et al. Attention-deficit/hyperactivity disorder is characterized by a delay in cortical maturation. Proc Nat Acad Sci. 2007;104(49):19649–54. doi: 10.1073/pnas.0707741104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tripp G, Wickens J. Neurobiology of ADHD. Neuropharmacology. 2009;57(7-8):579–89. doi: 10.1016/j.neuropharm.2009.07.026. [DOI] [PubMed] [Google Scholar]
- 10.Pan D, Gatley SJ, Dewey SL, Chen R, Alexoff DA, Ding YS, et al. Binding of bromine-substituted analogs of methylphenidate to monoamine transporters. Eur J Pharmacol. 1994;264(2):177–82. doi: 10.1016/0014-2999(94)00460-9. [DOI] [PubMed] [Google Scholar]
- 11.Ding YS, Fowler JS, Volkow ND, Logan J, Gatley SJ, Sugano Y. Carbon-11-d-threo-methylphenidate binding to dopamine transporter in baboon brain. J Nuclear Medicine. 1995;36(12):2298–305. [PubMed] [Google Scholar]
- 12.Vijayraghavan S, Wang M, Birnbaum SG, Williams GV, Arnsten AF. Inverted-U dopamine D1 receptor actions on prefrontal neurons engaged in working memory. Nat Neuroscience. 2007;10(3):376–84. doi: 10.1038/nn1846. [DOI] [PubMed] [Google Scholar]
- 13.Granon S, Passetti F, Thomas KL, Dalley JW, Everitt BJ, Robbins TW. Enhanced and impaired attentional performance after infusion of D1 dopaminergic receptor agents into rat prefrontal cortex. J Neurosci. 2000;20(3):1208–15. doi: 10.1523/JNEUROSCI.20-03-01208.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chudasama Y, Robbins TW. Dopaminergic modulation of visual attention and working memory in the rodent prefrontal cortex. Neuropsychopharmacology. 2004;29(9):1628–36. doi: 10.1038/sj.npp.1300490. [DOI] [PubMed] [Google Scholar]
- 15.Berridge CW, Devilbiss DM, Andrzejewski ME, Arnsten AFT, Kelley AE, Schmeichel B, et al. Methylphenidate preferentially increases catecholamine neurotransmission within the prefrontal cortex at low doses that enhance cognitive function. Biol Psych. 2006;60(10):1111–20. doi: 10.1016/j.biopsych.2006.04.022. [DOI] [PubMed] [Google Scholar]
- 16.Zahn TP, Rapoport JL, Thompson CL. Autonomic and behavioral effects of dextroamphetamine and placebo in normal and hyperactive prepubertal boys. J Abnorm Child Psychology. 1980;8(2):145–60. doi: 10.1007/BF00919060. [DOI] [PubMed] [Google Scholar]
- 17.Spencer TJ, Biederman J, Madras BK, Dougherty DD, Bonab AA, Livni E, et al. Further evidence of dopamine transporter dysregulation in ADHD: A controlled PET imaging study using altropane. Biol Psych. 2007;62(9):1059–1061. doi: 10.1016/j.biopsych.2006.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jucaite A, Fernell E, Halldin C, Forssberg H, Farde L. Reduced midbrain dopamine transporter binding in male adolescents with attention-deficit/hyperactivity disorder: association between striatal dopamine markers and motor hyperactivity. Biol Psych. 2005;57(3):229–38. doi: 10.1016/j.biopsych.2004.11.009. [DOI] [PubMed] [Google Scholar]
- 19.Seaman MI, Fisher JB, Chang F, Kidd KK. Tandem duplication polymorphism upstream of the dopamine D4 receptor gene (DRD4) Am J Med Genetics. 1999;88(6):705–9. doi: 10.1002/(sici)1096-8628(19991215)88:6<705::aid-ajmg22>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 20.McCracken JT, Smalley SL, McGough JJ, Crawford L, Del’Homme M, Cantor RM, et al. Evidence for linkage of a tandem duplication polymorphism upstream of the dopamine D4 receptor gene (DRD4) with attention deficit hyperactivity disorder (ADHD) Molecular Psychiatry. 2000;5(5):531–6. doi: 10.1038/sj.mp.4000770. [DOI] [PubMed] [Google Scholar]
- 21.Kustanovitch V, Ishii J, Crawford L, Yang M, McGough JJ, McCracken JT, et al. Transmission disequilibrium testing of dopamine-related candidate gene polymorphisms in ADHD: confirmation of association with DRD4 and DRD5. Molecular Psychiatry. 2004;9(7):711–7. doi: 10.1038/sj.mp.4001466. [DOI] [PubMed] [Google Scholar]
- 22.Kieling C, Roman T, Doyle AE, Hutz MH, Rohde LA. Association between DRD4 gene and performance of children with ADHD in a test of sustained attention. Biol Psych. 2006;60(10):1163–5. doi: 10.1016/j.biopsych.2006.04.027. [DOI] [PubMed] [Google Scholar]
- 23.Kim B, Koo MS, Jun JY, Park IH, Oh DY, Cheon KA. Association between Dopamine D4 Receptor Gene Polymorphism and scores on a continuous performance test in Korean children with attention deficit hyperactivity disorder. Psychiatry Investigations. 2009;6(3):216–21. doi: 10.4306/pi.2009.6.3.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tsai LH, Delalle I, Caviness VS, Chae T, Harlow E. p35 is a neural-specific regulatory subunit of cyclin-dependent kinase 5. Nature. 1994;371:419–423. doi: 10.1038/371419a0. [DOI] [PubMed] [Google Scholar]
- 25.Lew J, Huang QQ, Qi Z, Winkfein RV, Aebersold R, Hunt T, et al. A brain-specific activator of cyclin-dependent kinase 5. Nature. 1994;371:423–6. doi: 10.1038/371423a0. [DOI] [PubMed] [Google Scholar]
- 26.Cai XH, Tomizawa K, Tang D, Lu YF, Moriwaki A, Tokuda M, et al. Changes in the expression of novel Cdk5 activator messenger RNA (p39nck5ai mRNA) during rat brain development. Neurosci Res. 1997;28:355–60. doi: 10.1016/s0168-0102(97)00063-1. [DOI] [PubMed] [Google Scholar]
- 27.Gilmore EC, Ohshima T, Goffinet AM, Kulkarni AB, Herrup K. Cyclin-dependent kinase 5-deficient mice demonstrate novel developmental arrest in cerebral cortex. J Neurosci. 1998;18(16):6370–7. doi: 10.1523/JNEUROSCI.18-16-06370.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ohshima T, Ward JM, Huh CG, Longenecker G, Veeranna, Pant HC, et al. Targeted disruption of the cyclin-dependent kinase 5 gene results in abnormal corticogenesis, neuronal pathology and perinatal death. Proc Nat Acad Sci. 1996;93:11173–8. doi: 10.1073/pnas.93.20.11173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chae T, Kwon YT, Bronson R, Dikkes P, Li E, Tsai LH. Mice lacking p35, a neuronal specific activator of Cdk5, display cortical lamination defects, siezures, and adult lethality. Neuron. 1997;18(1):29–42. doi: 10.1016/s0896-6273(01)80044-1. [DOI] [PubMed] [Google Scholar]
- 30.Kwon YT, Tsai LH. A novel disruption of cortical development in p35 (−/−) mice distinct from reeler. J Comp Neurol. 1998;395:510–522. doi: 10.1002/(sici)1096-9861(19980615)395:4<510::aid-cne7>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 31.Nikolic M, Dudek H, Kwon YT, Ramos YF, Tsai LH. The cdk5/p35 kinase is essential for neurite outgrowth during neuronal differentiation. Genes Dev. 1996;18:816–825. doi: 10.1101/gad.10.7.816. [DOI] [PubMed] [Google Scholar]
- 32.Wenzel HJ, Tamse CT, Schwartzkroin PA. Dentate development in organotypic hippocampal slice cultures from p35 knockout mice. Developmental Neuroscience. 2007;(29):99–112. doi: 10.1159/000096215. [DOI] [PubMed] [Google Scholar]
- 33.Bibb JA, Snyder G, Nishi A, Yan Z, Meijer L, Fienberg AA, et al. Phosphorylation of DARPP-32 by Cdk5 modulates dopamine signalling in neurons. Nature. 1999;402:669–671. doi: 10.1038/45251. [DOI] [PubMed] [Google Scholar]
- 34.Bibb JA, Chen J, Taylor JR, Svenningsson P, Nishi A, Snyder GL, et al. Effects of chronic exposure to cocaine are regulated by the neuronal protein cdk5. Nature. 2001;410:376–380. doi: 10.1038/35066591. [DOI] [PubMed] [Google Scholar]
- 35.Benavides DR, Quinn JJ, Zhong P, Hawasli AH, DiLeone RJ, Kansy JW, et al. Cdk5 modulates cocaine reward, motivation, and striatal neuron excitability. Journal of Neuroscience. 2007;27(47):12967–76. doi: 10.1523/JNEUROSCI.4061-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Taylor JR, Lynch WJ, Sanchez H, Olausson P, Nestler EJ, Bibb JA. Inhibition of Cdk5 in the nucleus accumbens enhances the locomotor-activating and incentive-motivational effects of cocaine. Proc Nat Acad Sci. 2007;104:4147–4152. doi: 10.1073/pnas.0610288104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lindskog M, Svenningsson P, Pozzi L, Kim Y, Feinberg AA, Bibb JA, et al. Involvement of DARPP-32 phosphorylation in the stimulant action of caffeine. Nature. 2002;418:774–778. doi: 10.1038/nature00817. [DOI] [PubMed] [Google Scholar]
- 38.Mani SK, Feinberg A, O’Callaghan JP, Snyder GL, Allen PB, Dash PK, et al. Requirement for DARPP-32 in progesterone-facilitated sexual receptivity in female rats and mice. Science. 2000;287(5455):1053–6. doi: 10.1126/science.287.5455.1053. [DOI] [PubMed] [Google Scholar]
- 39.Chergui K, Svenningsson P, Greengard P. Cyclin-dependent kinase 5 regulates dopaminergic and glutamatergic transmission in the striatum. Proc Nat Acad Sci. 2004;101:2191–2196. doi: 10.1073/pnas.0308652100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moy LY, Tsai LH. Cyclin-dependent kinase 5 phosphorylates serine-31 of tyrosine hydroxylase and regulates its stability. J Biol Chem. 2004;279:54487–54493. doi: 10.1074/jbc.M406636200. [DOI] [PubMed] [Google Scholar]
- 41.Kansy JW, Daubner SC, Nishi A, Sotogaku N, Lloyd MD, Nguyen C, et al. Identification of tyrosine hydroxylase as a physiological substrate for Cdk5. J Neurochemistry. 2004;91(2):374–84. doi: 10.1111/j.1471-4159.2004.02723.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Frank-Cannon TC, Tran T, Ruhn KA, Martinez TN, Hong J, Marvin M, et al. Parkin deficiency increases vulnerability to inflammation-related nigral degeneration. J Neurosci. 2008;28(43):10825–34. doi: 10.1523/JNEUROSCI.3001-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sahin B, Hawasli A, Greene RW, Molkentin JD, Bibb JA. Negative regulation of cyclin-dependent kinase 5 targets by protein kinase C. Eur J Pharmacol. 2008;581(3):270–5. doi: 10.1016/j.ejphar.2007.11.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schmued LC, Fallon JH. Fluoro-Gold: a fluorescent retrograde axonal tracer with numerous unique properties. Brain Research. 1986;(377):147–154. doi: 10.1016/0006-8993(86)91199-6. [DOI] [PubMed] [Google Scholar]
- 45.Crawford CA, McDougall SA, Meier TL, Collins RL, Watson JB. Repeated methylphenidate treatment induces behavioral sensitization and decreases protein kinase A and dopamine-stimulated adenylyl cyclase activity in the dorsal striatum. Psychopharmacology. 1998;136:34–43. doi: 10.1007/s002130050536. [DOI] [PubMed] [Google Scholar]
- 46.Roche KW, O’Brien R, Mammen AL, Bernhardt J, Huganir RL. Characterization of multiple phosphorylation sites on the AMPA receptor GluR1 subunit. Neuron. 1996;16(6):1179–88. doi: 10.1016/s0896-6273(00)80144-0. [DOI] [PubMed] [Google Scholar]
- 47.Hemmings HC, Greengard P, Tung HYL, Cohen P. DARPP-32, a dopamine-regulated neuronal phosphoprotein, is a potent inhibitor of protein phosphatase-1. Nature. 1984;(310):503–5. doi: 10.1038/310503a0. [DOI] [PubMed] [Google Scholar]
- 48.Sesack SR, Bunney BS. Pharmacological characterization of the receptor mediating electrophysiological responses to dopamine in the rat medial prefrontal cortex: A microiontophoretic study. Journal of Pharmacology and Experimental Therapeutics. 1989;248(3):1323–33. [PubMed] [Google Scholar]
- 49.Law-Tho D, Hirsch JC, Crepel F. Dopamine modulation of synaptic transmission in rat prefrontal cortex: An in vitro electrophysiological study. Neurosci Res. 1994;21(2):151–60. doi: 10.1016/0168-0102(94)90157-0. [DOI] [PubMed] [Google Scholar]
- 50.Grobin AC, Deutch AY. Dopaminergic regulation of extracellular y-aminobutyric acid levels in the prefrontal cortex of the rat. Journal of Pharmacology and Experimental Therapeutics. 1998;285(1):350–7. [PubMed] [Google Scholar]
- 51.Kalivas PW, Stewart J. Dopamine transmission in the initiation and expression of drug- and stress-induced sensitization motor activity. Brain Research Reviews. 1991;16(3):223–44. doi: 10.1016/0165-0173(91)90007-u. [DOI] [PubMed] [Google Scholar]
- 52.Sorg BA, Li N, Wu W. Dopamine D1 receptor activation in the medial prefrontal cortex prevents the expression of cocaine sensitization. Journal of Pharmacology and Experimental Therapeutics. 2001;297(2):501–8. [PubMed] [Google Scholar]
- 53.Banks KE, Gratton A. Possible involvement of medial prefrontal cortex in amphetamine-induced sensitization of mesolimbic dopamine function. Eur J Pharmacol. 1995;282(1-3):157–67. doi: 10.1016/0014-2999(95)00306-6. [DOI] [PubMed] [Google Scholar]
- 54.Beyer CE, Steketee J. Cocaine sensitization: modulation by dopamine D2 receptors. Cereb Cortex. 2002;12(5):526–35. doi: 10.1093/cercor/12.5.526. [DOI] [PubMed] [Google Scholar]
- 55.Keller BU, Hollman M, Heinemann S, Konnerth A. Calcium influx through subunits GluR1/GluR3 of kainate/AMPA receptor channels is regulated by cAMP dependent protein kinase. EMBO J. 1992;11:891–6. doi: 10.1002/j.1460-2075.1992.tb05127.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Banke TG, Bowie D, Lee H, Huganir RL, Schousboe A, Traynelis SF. Control of GluR1 AMPA receptor function by cAMP-dependent protein kinase. J Neurosci. 2000;20(1):89–102. doi: 10.1523/JNEUROSCI.20-01-00089.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fukui R, S. P, Matsuishi T, Higashi H, Nairn AC, Greengard P, Nishi A. Effect of methylphenidate on dopamine/DARPP signalling in adult, but not young, mice. Journal of Neurochemistry. 2003;87(6):1391–1401. doi: 10.1046/j.1471-4159.2003.02101.x. [DOI] [PubMed] [Google Scholar]
- 58.Zhou M, Rebholz H, Brocia C, Warner-Schmidt JL, Fienberg AA, Nairn AC, et al. Forebrain overexpression of CK1delta leads to down-regulation of dopamine receptors and altered locomotor activity reminiscent of ADHD. Proc Natl Acad Sci. 2010;107(9):4401–6. doi: 10.1073/pnas.0915173107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zametkin AJ, Nordahl TE, Gross M, King AC, Semple WE, Rumsey J, et al. Cerebral glucose metabolism in adults with hyperactivity of childhood onset. N Engl J Med. 1990;323(20):1361–6. doi: 10.1056/NEJM199011153232001. [DOI] [PubMed] [Google Scholar]
- 60.Zametkin AJ, Liebenauer LL, Fitzgerald GA, King AC, Minkunas DV, Herscovitch P, et al. Brain metabolism in teenagers with attention-deficit hyperactivity disorder. Arch Gen Psychiatry. 1993;50(5):333–40. doi: 10.1001/archpsyc.1993.01820170011002. [DOI] [PubMed] [Google Scholar]
- 61.Ernst M, Liebenauer LL, King AC, Fitzgerald GA, Cohen RM, Zametkin AJ. Reduced brain metabolism in hyperactive girls. Journal of Am Acad of Child and Adol Psy. 1994;33(6):858–68. doi: 10.1097/00004583-199407000-00012. [DOI] [PubMed] [Google Scholar]
- 62.Hawasli AH, Benavides D, Nguyen C, Kansy J, Hayashi K, Chambon P, et al. Cyclin-dependent kinase 5 governs learning and synaptic plasticity via control of NMDAR degredation. Nat Neuroscience. 2007;10:880–886. doi: 10.1038/nn1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hawasli AH, Koovakkattu D, Hayashi K, Anderson AE, Powell CM, Sinton CM, et al. Regulation of hippocampal and behavioral excitability by cyclin-dependent kinase 5. PLoS One. 2009;4(6) doi: 10.1371/journal.pone.0005808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Alkire MT, Haier RJ, Shah NK, Anderson CT. Positron emission tomography study of regional cerebral metabolism in humans during isoflurane anesthesia. Anesthesiology. 1997;86(3):549–557. doi: 10.1097/00000542-199703000-00006. [DOI] [PubMed] [Google Scholar]
- 65.Wang YP, Mou DL, Song JF, Rao ZR, Li D, Ju G. Aberrant activation of CDK5 is involved in the pathogenesis of OPIDN. Journal of Neurochemistry. 2006;99(1):186–97. doi: 10.1111/j.1471-4159.2006.04027.x. [DOI] [PubMed] [Google Scholar]
- 66.Bouchard MF, Bellinger DC, Wright RO, Weisskopf MG. Attention-Deficit/Hyperactivity Disorder and Urinary Metabolites of Organophosphate Pesticides. Pediatrics. 2010;125(6):1270–7. doi: 10.1542/peds.2009-3058. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.