

Abstract
The intrinsic conformational preferences of a new non-proteinogenic amino acid have been explored by computational methods. This tailored molecule, named (βPro)Arg, is conceived as a replacement for arginine in bioactive peptides when the stabilization of folded turn-like conformations is required. The new residue features a proline skeleton that bears the guanidilated side chain of arginine at the Cβ position of the five-membered pyrrolidine ring, either in a cis or a trans orientation with respect to the carboxylic acid. The conformational profile of the N-acetyl-N'-methylamide derivatives of the cis and trans isomers of (βPro)Arg has been examined in the gas phase and in solution by B3LYP/6–31+G(d,p) calculations and molecular dynamics simulations. The main conformational features of both isomers represent a balance between geometric restrictions imposed by the five-membered pyrrolidine ring and the ability of the guanidilated side chain to interact with the backbone through hydrogen-bonds. Thus, both cis and trans (βPro)Arg exhibit a preference for the αL conformation as a consequence of the interactions established between the guanidinium moiety and the main-chain amide groups.
Introduction
Design of specific chemical modifications in natural amino acids is a powerful strategy to control the conformational properties of short peptides.1–2 Moreover, non-coded residues in a peptide chain may result in increased resistance to proteolytic degradation.3–7 Non-proteinogenic amino acids are useful in engineering peptide analogues with improved pharmacokinetics and medicinal applications.3–9
A non-coded amino acid can replace residues in natural peptide sequences if it does not disrupt the bioactive conformation of the preplaced segment. The resulting peptide must preserve the native shape that interacts with the receptor. When dealing with small flexible peptides, a non-coded residue can improve the targeted peptide by biasing its conformational equilibrium to a conformational set that guarantees function. Thus, the non-proteinogenic amino acid should exhibit a high preference for the conformation adopted by the natural residue that is to be replaced.
Theoretical methods can assist so that only those candidates yielding satisfactory results in silico are selected for experimental studies. This evaluation requires (i) theoretical study of the wild-type bioactive conformation (if not available experimentally); (ii) assessing which amino acid should be replaced; and (iii) design of a new non-coded amino acid adequate to replace the targeted position of the peptide. The conformational preferences of the new non-coded amino acid need to be determined before the replacement is performed. If such conformational preferences do not match those of the targeted position, the replacement will not be successful.
We are involved in a project aimed at improving the bioactivity of the pentapeptide Cys-Arg-Glu-Lys-Ala (CREKA). This peptide has biotechnological interests because it recognizes molecular markers that are present in tumor blood vessels but not in the vasculature of normal tissues,10 thus showing promising applications in cancer diagnosis and therapy. Medicinal use is however hampered by the poor stability against proteases and short half-life time typically exhibited by small and medium-size natural peptides. The conformational landscape of CREKA was explored by computational methods under different environmental conditions.11 This analysis led to a bioactive conformation exhibiting a turn motif, with the charged side chains of Arg, Glu, and Lys oriented toward the same side of the molecule.11 The peptide backbone is folded in β-turn centered at the Arg and Glu residues. Arginine occupies the first corner position of the β-turn (i+1) and adopts dihedral angles corresponding to the α-helical (αL) region of the Ramachandran map.
The first attempt to decrease the peptide sensitivity to proteases was made though by introducing unspecific chemical modifications that did not affect its overall conformational properties.12 Hence, a methyl group replaced the hydrogen atom at either N position or Cα position. This simple approach increased in vivo the half-life time of the CREKA coated nanoparticles in the tumor vessels.12 However, no effort had been made to enhance the stability of the bioactive conformation.11 Then in a second stage, we undertook the design of new non-coded amino acids that could bias the folding of CREKA towards its bioactive organization, focusing our efforts on arginine surrogates. The main goal was to incorporate the side-chain functionality of arginine, which is essential for CREKA's activity, in residue that presented clear conformational preferences for the αL region of the Ramachandran map.
A first amino acid was designed by combining a non-coded amino acid of the family 1-aminocycloalkane-1-carboxylic acids (Acnc, where n refers to the size of the cycle) and the side chain functionality of arginine (Figure 1).13 These amino acid series had been previously investigated and shown to exhibit a restricted conformational space characterized by a high propensity to adopt φ,ψ backbone angles typical of the 310-/α-helix (with some distortion in the case of Ac3c).14–18 The new amino acid (denoted as c5Arg) was built by incorporating the side chain of arginine at the β-carbon atom of Ac5c.13 The intrinsic conformational preferences of the new amino acid were studied using theoretical methods, showing that α-helical conformation was favored both in the gas phase and in solution. It was remarkable that the ability of the guanidinium moiety to form hydrogen bonds with the peptide backbone conditioned the conformational features of the parent Ac5c,17 which tends to favor the formation γ-turn based conformations before α-helix like arrangements.
Figure 1.

Structure of arginine (Arg) and some studied analog s. The first presented analog was designed upon 1-aminocyclopentane-1-carboxylic acid (Ac5c), which led to the amino acid c5Arg (ref. 7) and those based on proline structure (Pro): cis-(γPro)Arg (ref. 9), trans-(βPro)Arg and cis-(βPro)Arg, being the latter one the object of this study. The arginine surrogates are named according to the γ/β position of the proline skeleton bearing the guanidilated side chain and to the trans/cis relative orientation between this side chain and the carboxylic acid.
The latter challenge was though to achieve similar results with a backbone constitution closer to that of coded amino acids. Among proteinogenic amino acids, proline is known to impart protection against proteolytic cleavage19–22 as well as to nucleate peptide turns,23–25 with a marked propensity to occupy the i+1 position of β-turns. Accordingly, following the previous strategy we generated a new residue by attaching the arginine side chain to the proline skeleton (see the Supporting Information for details). It should be noted that the cyclic nature of proline, that includes the amine nitrogen atom in the ring constitution, facilitates a cis arrangement of the peptide bond involving the pyrrolidine nitrogen, as compared to other peptide bonds, for which the cis form is almost nonexistent.26–27 Here, however, this issue has not been addressed since the targeted arginine in wild-type CREKA presents both peptide bonds in trans5 and the new residue is therefore useful only for the latter geometry.
In a previous work,28 the guanidilated side chain was attached to the γ-carbon of the pyrrolidine ring in a cis configuration with the carboxylic acid moiety, thus giving rise to the residue denoted cis-(γPro)Arg in Figure 1. The chain length of this arginine analogue proved insufficient to reproduce the interactions observed for the guanidinium group in the bioactive conformation of CREKA.11,28 Although the addition of another methylene unit to the exocyclic guanidilated substituent was favorable, incorporation of the resulting residue into CREKA led to the disruption of the β-turn conformation of the natural peptide.28
These results led us to design a new arginine surrogate built on a proline skeleton. Here, it is the β pyrrolidine carbon that bears the guanidilated arginine side chain and the resulting residue is termed cis-(βPro)Arg, where cis refers to the position of the guanidilated substituent relative to the carboxylic acid and β denotes the carbon atom of the five-membered ring where this substituent is placed (Figure 1). Prior to testing the modified pentapeptide Cys-cis(βPro)Arg-Glu-Lys-Ala, the conformational propensities of the single amino acid have been investigated in depth by theoretical methods. As noted above, cis-(βPro)Arg presents a cis orientation between the guanidinium and carbonyl moieties –as the previously studied28 cis-(γPro)Arg– in agreement with the spatial relationship characterized for the guanilidated segment of natural arginine in CREKA.11 However, beyond the CREKA project, other peptides incorporating key arginine residues may present the guanidilated side chain oriented away form the carbonyl group in the bioactive form. This consideration prompted us to also evaluate in this work the conformational propensities of trans-(βPro)Arg (Figure 1).
We have therefore performed quantum mechanics calculations on the N-acetyl-N'-methylamide derivatives of both cis-(βPro)Arg and trans-(βPro)Arg, hereafter denoted Ac-c-(βPro)Arg-NHMe and Ac-t-(βPro)Arg-NHMe, respectively. The results are compared with those reported previously28 for the analogous cis γ-substituted derivative, Ac-c-(γPro)Arg-NHMe. Additionally, parameterization of the two non-proteinogenic amino acids under study has been carried out before analyzing the conformational impact derived from their incorporation into biologically active peptides. The dynamical conformational features of the two residues have been explored in aqueous solution at room temperature using classical Molecular Dynamics (MD) simulations with explicit water molecules.
Computational Methods
Quantum mechanical calculations
Density Functional Theory (DFT) methods were applied for quantum mechanical calculations, which were performed using the Gaussian 03 computer program.29 Specifically, calculations were carried out by combining the unrestricted formalism of the B3LYP functional30,31 with the 6–31+G(d,p) basis set.32 Frequency analyses were carried out to verify the nature of the minimum state of all the stationary points obtained and to calculate the zero-point vibrational energies (ZPVE) and both thermal and entropic corrections. These statistical terms were then used to compute the conformational Gibbs free energies in the gas phase (ΔGgp) at 298K.
Figure 2 shows the backbone (ω0,φ,ψ,ω) and side chain (χi,ξi) dihedral angles that define the conformations adopted by Ac-t-(βPro)Arg-NHMe and Ac-c-(βPro)Arg-NHMe. The minimum energy structures characterized for these compounds have been denoted using a three-label code that specifies the arrangement of the peptide backbone, the puckering of the five-membered cycle and the conformation adopted by the exocyclic substituent. The first label identifies the backbone conformation type according to Perczel's nomenclature,33 which categorizes the potential energy surface E = E(φ,ψ) of α-amino acids in nine different regions: γD, δD, αD, εD, βDL, εL, αL, δL, and γL. The presence of the pyrrolidine ring in proline fixes the φ angle near −60° and, accordingly, only three of such regions can be accessed,23,24 namely, γL (γ-turn), αL (α-helix), and εL (polyproline II). Identical geometric restrictions should apply to the arginine surrogates under study since they have a proline skeleton. A trans configuration was considered for the amide bonds (ω0, ω ≈ 180°). The second label describes the down [d] or up [u] puckering of the five-membered pyrrolidine ring.23,34,35 Such conformational states are also called Cγendo and Cγexo, respectively, and correspond to those in which the Cγ atom and the carbonyl group of proline (or the proline-like residue) lie on the same and opposite sides of the plane defined by Cδ, N, and Cα. Specifically, a down puckering was assigned when χ1 and χ3 were positive while χ2 and χ4 were negative. Conversely, negative values of χ1 and χ3 and positive values of χ2 and χ4 correspond to an up-puckered pyrrolidine ring. Finally, the orientation of the polar exocyclic substituent is described by the third label, which indicates the gauche+ (g+), skew+ (s+), trans (t), skew− (s−), gauche− (g−), or cis (c) state of each ξi dihedral angle.
Figure 2.
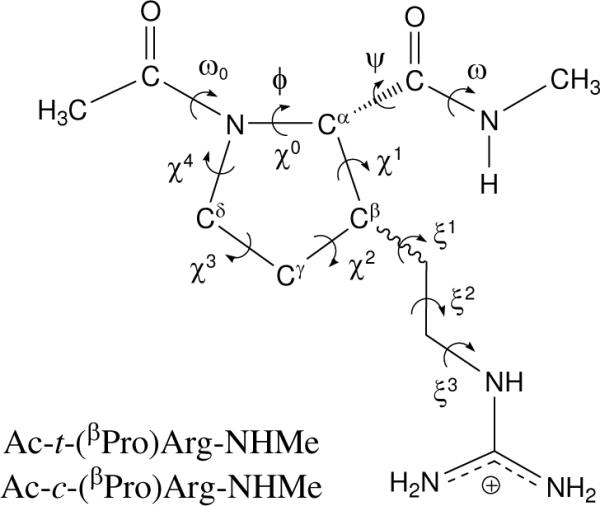
Dihedral angles used to identify the conformations of the N-acetyl-N'-methylamide derivatives of trans-(βPro)Arg and cis-(βPro)Arg studied in this work. The (φ,ψ) dihedral angles are defined by the atoms in the backbone, whereas the side-chain dihedral angles χi and ξi are given by the pyrrolidine atoms and the exocyclic side-chain atoms, respectively. In particular, φ and χ0 are defined as C(O)–N–Cα–C(O) and Cδ–N–Cα–Cβ, respectively. The dihedral angle ξ1 is given by Cα–Cβ–C(H2)–C(H2).
The conformational search was performed following the strategy used in our previous work on cis-(γPro)Arg.28 It was assumed that the two (βPro)Arg derivatives under study maintain the geometric restrictions derived from the cyclic nature of proline. Thus, the three minimum energy conformations characterized36 for Ac-Pro-NHMe with trans amide bonds, i.e. γL[d], γL[u], and αL[u], were considered as starting geometries for Ac-t-(βPro)Arg-NHMe and Ac-c-(βPro)Arg-NHMe in the present work. Regarding the substituent attached to the β-carbon of the pyrrolidine moiety, each ξi dihedral was expected to exhibit minima of the gauche+, trans and gauche− type. Accordingly, 3 (minima of Ac-Pro-NHMe16) × 3 (minima of ξ1) × 3 (minima of ξ2) × 3 (minima of ξ3) = 81 minima were anticipated for the potential energy hypersurface E = E(φ,ψ,χi,ξi) of each (βPro)Arg derivative. All these structures were used as starting points for subsequent full geometry optimizations.
The influence of the solvent on the conformational preferences of the compounds under study was quantified by performing Self-Consistent Reaction Field (SCRF) calculations on the optimized geometries. Under this formalism, the solute is treated at the quantum mechanical level, while the solvent is represented as a dielectric continuum. In particular, we used the Polarizable Continuum Model (PCM) developed by Tomasi and co-workers to describe the bulk solvent.37–40 PCM calculations were performed following the standard protocol and considering the dielectric constants of carbon tetrachloride (ε = 2.228), chloroform (ε = 4.9), and water (ε = 78.4). The conformational free energies in solution (ΔGsol, where sol refers to the solvent) were computed using the classical thermodynamics scheme, i.e. for each minimum, the free energy of solvation provided by the PCM model was added to the ΔGgp value.
Force-field parameterization
The stretching, bending, torsion and van der Waals interactions of trans-(βPro)Arg and cis-(βPro)Arg were described classically by extrapolating the force-field parameters contained in the AMBER libraries41 for proline and arginine. For selected minimum energy conformations, electrostatic atomic centered charges were calculated by fitting the UHF/6–31G(d) quantum mechanical and the Coulombic Molecular Electrostatic Potentials (MEPs) to a large set of points placed outside the nuclear region. The electrostatic parameters derived at this level of theory are fully compatible with the current parameters of the AMBER force-field.41 Electrostatic force-field parameters for the two (βPro)Arg isomers were obtained by applying to such atomic charges a strategy based on a Boltzmann distribution of multiple conformations, which was originally proposed by different authors42–44 and has been shown to be specially suitable for non-proteinogenic residues.13,16,17,43,45 Moreover, this strategy provides conformationally independent electrostatic parameters.
Force-field calculations
MD simulations in water solution were performed using the NAMD program.46 The Ac-t-(βPro)Arg-NHMe or Ac-c-(βPro)Arg-NHMe molecules were placed in the center of a cubic simulation box (a = 30.6 Å) filled with 955 explicit water molecules, which were represented using the TIP3 model.47 Negatively charged chloride atoms were added to reach electron neutrality. Before the production runs, the simulation box was equilibrated for each compound. Thus, 0.5 ns of NVT-MD at 500K were used to homogeneously distribute the solvent and ions in the box. Next, 0.5 ns of NVT-MD at 298K (thermal equilibration) and 0.5 ns of NPT-MD at 298K (density relaxation) were carried out. The last snapshot of the NPT-MD was used as the starting point for production NVT-MD runs at standard conditions.
The energy was calculated using the AMBER potential.41 Atom pair distance cutoffs were applied at 12.0 Å to compute the van der Waals and electrostatic interactions. In order to avoid discontinuities in the potential energy function, non-bonding energy terms were slowly converged to 0 by applying a smoothing factor from a distance of 10.0 Å. Both temperature and pressure were controlled using the weak coupling method48 applying a time constant for heat bath coupling and a pressure relaxation time of 1 ps. Bond lengths were constrained using the SHAKE algorithm49 with a numerical integration step of 2 fs.
Results and Discussion
A total of 21 minimum energy conformations were found and characterized for Ac-t-(βPro)Arg-NHMe in the gas phase. The conformational parameters of those with relative energies (ΔEgp) below 5.0 kcal/mol are listed in Table 1 (the complete list is provided as Supporting Information). In the global minimum (γL[u]s−g+t, Figure 3a), the terminal acetyl CO and methylamide NH groups are linked by a hydrogen bond [dH…O= 1.793 Å, <N−H…O= 151.3°] closing a seven-membered cycle (γ-turn or C7 conformation), and the pyrrolidine ring adopts an up puckering. The orientation of the exocyclic guanidilated side chain, which is defined by the skew−, gauche+ and trans arrangement of ξ1, ξ2 and ξ3, respectively, enables the formation of a strong hydrogen bond between the carbonyl oxygen of the trans-(βPro)Arg residue and the guanidinium NH site [dH…O= 1.628 Å, <N−H…O= 176.2°]. The second (γL[u]g−g−s+, Figure 3b) and third (γL[u]g−g−s−, Figure 3c) minima also exhibit the seven-membered hydrogen-bonded ring typical of a C7 conformation and an up-puckered pyrrolidine moiety, while they differ from the global minimum in the orientation of the guanidilated side chain. In the (γL[u]g−g−s+ conformer, the side chain…backbone interaction involves an NH2 group in the guanidinium substituent instead of the NH site. The less favorable arrangement of the exocyclic side chain in these two conformers produces a destabilization of 1.1–1.5 kcal/mol with respect to the global minimum. A similar but more pronounced effect is observed for the last minimum listed in Table 1 (γL[u]s−tg−, Figure 3g). This conformer presents identical shapes for both the peptide backbone and the pyrrolidine moiety to those described above for the first, second and third minima, but a much higher energy (ΔEgp = 3.7 kcal/mol). This destabilization should be attributed to the unfavorable steric interactions produced within the methylene groups in the side chain to allow the formation of a hydrogen bond between the trans-(βPro)Arg CO and the guanidinium NH2.
Table 1.
Dihedral angles (see Figure 2; in degrees), pseudorotational parameters of the pyrrolidine ring (A, P; in degrees), and relative energy (ΔEgp; in kcal/mol) of the minimum energy conformations with ΔEgp < 5.0 kcal/mol characterized for Ac-t-(βPro)Arg-NHMe at the B3LYP/6–31+G(d,p) level.
| Conformer | ω 0 | φ | ψ | ω | A, Pa | ξ 1 | ξ 2 | ξ 3 | Δ Egp |
|---|---|---|---|---|---|---|---|---|---|
| γL[u]s−g+t | −169.6 | −77.8 | 54.8 | 178.2 | 40.1, 92.5b | −127.0 | 61.1 | 162.8 | 0.0c |
| γL[u]g−g−s+ | −170.3 | −79.9 | 59.5 | 179.1 | 39.0, 98.3d | −84.2 | −77.5 | 117.9 | 1.1 |
| γL[u]−g−s− | −170.3 | −80.5 | 56.1 | 179.3 | 38.0, 103.3e | −59.8 | −49.6 | −115.6 | 1.5 |
| αL[u]s−g+t | −169.7 | −89.7 | −3.5 | 176.5 | 36.0, 112.0f | −92.4 | 61.2 | 156.5 | 2.1 |
| αL[u]g+g−t | −170.2 | −73.1 | −20.2 | 179.0 | 40.5, 76.2g | 39.5 | −77.7 | −173.7 | 3.0 |
| αL[u]g−tg− | −169.9 | −76.5 | −16.0 | 177.2 | 38.0, 85.5h | −88.4 | 155.2 | −86.1 | 3.5 |
| γL[u]s−tg− | −169.4 | −73.5 | 44.0 | 177.3 | 40.8, 85.5i | −126.2 | 157.4 | −87.1 | 3.7 |
See ref. 16 for definition.
χ0 = −1.7°, χ1 = −22.3°, χ2 = 37.5°, χ3 = −38.2°, χ4 = 25.2°.
E = −856.5567162 a.u.
χ0 = −5.7°, χ1 = −18.4°, χ2 = 34.9°, χ3 = −38.0°, χ4 = 27.5°.
χ0 = −8.7°, χ1 = −15.1°, χ2 = 32.5°, χ3 = −37.3°, χ4 = 29.1°.
χ0 = −13.5°, χ1 = −9.2°, χ2 = 27.4°, χ3 = −35.4°, χ4 = 30.7°.
χ0 = 9.7°, χ1 = −30.7°, χ2 = 40.2°, χ3 = −34.5°, χ4 = 15.5°.
χ0 = 3.0°, χ1 = −24.6°, χ2 = 36.8°, χ3 = −35.1°, χ4 = 20.0°.
χ0 = 3.2°, χ1 = −26.6°, χ2 = 39.6°, χ3 = −37.4°, χ4=21.5°.
Figure 3.

Selected minimum energy conformations of Ac-t-(βPro)Arg-NHMe obtained from B3LYP/6–31+G(d,p) calculations (ΔEgp < 5.0 kcal/mol, see Table 1): (a) γL[u]s−g+t; (b) γL[u]g−g−s+; (c) γL[u]g−g−s−; (d) αL[u]s−g+t; (e) αL[u]g+g−t; (f) αL[u]g−tg−; and (g) γL[u]s−tg−. Intramolecular hydrogen bonds are indicated by dashed lines (H…O distances and N–H…O angles are given).
The most stable structure in Table 1 exhibiting a backbone conformation other than a γ-turn is αL[u]s−g+t, which is unfavored by 2.1 kcal/mol with respect to the global minimum. This is noteworthy, since the most stable αL conformation with trans amide bonds characterized for Ac-Pro-NHMe exhibits a ΔEgp value of 4.9 kcal/mol.36 The αL[u]s−g+ t minimum of Ac-t-(βPro)Arg-NHMe (Figure 3d) presents no hydrogen-bonding interaction within the backbone amide groups, but is stabilized by a strong backbone…side chain hydrogen bond. The two additional αL conformers in Table 1, αL[u]g+g−t (Figure 3e) and αL[u]g−tg− (Figure 3f), exhibit similar arrangements for the peptide backbone and the pyrrolidine ring, while differing in the orientation of the guanidilated substituent and the topology of the associated backbone…side chain interaction. Local repulsions within the aliphatic segment in this exocyclic side chain produce a destabilization of 0.9 and 1.4 kcal/mol, respectively, relative to the most stable αL conformer.
Comparison with the results previously reported28 for Ac-c-(γPro)Arg-NHMe provides evidence for the higher flexibility of the β-substituted derivative Ac-t-(βPro)Arg-NHMe studied in the present work. This is due to the presence of an additional exocyclic methylene unit in the latter case (Figure 1), which broadens the conformational space that may be explored by the guanidilated side chain. Yet, the number of energetically accessible conformers is small in both cases, as expected from the restrictions imposed by the proline skeleton. The two compounds share the main structural features of the global minimum, which belongs to the γL[u] category and exhibits identical patterns for both the backbone···backbone and side chain···backbone hydrogen-bonding interactions. However, a highly stable γL[d] conformer was located28 for Ac-c-(γPro)Arg-NHMe at only 0.4 kcal/mol, whereas no γL[d] structure appears in Table 1 for Ac-t-(βPro)Arg-NHMe. Indeed, the only γL[d] minimum located for the latter compound presents ΔEgp = 19.0 kcal/mol (see Supporting Information). Another important difference is the presence of αL conformers in Table 1, whereas no Ac-c-(γPro)Arg-NHMe minima28 presented this backbone conformation.
Table 2 gives the conformational parameters of the five minima characterized for Ac-c-(βPro)Arg-NHMe with ΔEgp values below 5.0 kcal/mol (see the Supporting Information for a complete list of minima). Interestingly, the backbone shape preferred by this compound corresponds to the α-helix, whereas the γ-turn is unfavored by at least 2.0 kcal/mol. This is in sharp contrast with that described above for the trans-(βPro)Arg derivative, as well as with the behavior observed before for the analogous γ-substituted compound28 [Ac-c-(γPro)Arg-NHMe] and for proline itself36 (Ac-Pro-NHMe). Indeed, the most stable α-helical minimum characterized for Ac-Pro-NHMe with trans amide bonds lies 4.9 kcal/mol above the preferred γ-turn conformer,36 and no minimum energy structure was characterized in the α-helix region for Ac-c-(γPro)Arg-NHMe.28 This comparative analysis provides evidence for the enormous impact that the incorporation of a functionalized side chain able to establish hydrogen-bonding interactions with the main-chain amide groups may have on the conformational preferences of the peptide backbone. It also illustrates the fact that the conformational profile of arginine analogues bearing a proline skeleton depends dramatically on the specific position of the guanidilated side chain, that is, the pyrrolidine carbon bearing it and its relative orientation with respect to the carboxyl terminus.
Table 2.
Dihedral angles (see Figure 2; in degrees), pseudorotational parameters of the pyrrolidine ring (A, P; in degrees), and relative energy (ΔEgp; in kcal/mol) of the minimum energy conformations with ΔEgp < 5.0 kcal/mol characterized for Ac-c-(βPro)Arg-NHMe at the B3LYP/6–31+G(d,p) level.
| Conformer | ω 0 | φ | ψ | ω | A,Pa | ξ 1 | ξ 2 | ξ 3 | Δ Egp |
|---|---|---|---|---|---|---|---|---|---|
| αL[d]g+g−t | −169.7 | −86.3 | −11.6 | 175.3 | 39.6, −108.3b | 75.3 | −71.4 | 178.9 | 0.0c |
| αL[d]g+ts+ | −169.2 | −90.1 | −4.3 | 174.5 | 40.1, −111.7d | 69.2 | −172.0 | 95.2 | 1.3 |
| γL[d]s−g+t | −171.1 | −84.1 | 75.5 | −176.7 | 39.8, −117.0e | −116.3 | 70.4 | 164.2 | 2.0 |
| γL[d]s−ts− | −170.3 | −83.1 | 66.4 | −178.5 | 40.0, −113.6f | −104.8 | 168.1 | −93.4 | 3.1 |
| γL[u]s+g−t | −168.7 | −66.8 | 31.8 | 174.5 | 43.7, 78.2g | 127.7 | −65.4 | 178.3 | 3.9 |
See ref. 16 for definition.
χ0 = −12.4°, χ1 = 31.7°, χ2 = −39.6°, χ3 = 32.0°, χ4 = −12.2°.
E = −856,554209 a.u.
χ0 = −14.8°, χ1 = 33.5°, χ2 = −40.1°, χ3 = 31.0°, χ4 = −10.0°.
χ0 = −18.1°, χ1 = 35.0°, χ2 = −39.5°, χ3 = 28.3°, χ4 = −6.3°.
χ0 = −16.0°, χ1 = 34.1°, χ2 = −40.0°, χ3 = 30.0°, χ4 = −8.8°.
χ0= 8.9°, χ1 = −32.1°, χ2 = 43.4°, χ3 = −38.2°, χ4= 18.1°.
As expected for an α-helical conformer, the lowest energy minimum of Ac-c-(βPro)Arg-NHMe (αL[d]g+g−t, Figure 4a) exhibits no hydrogen-bonding interaction involving the backbone amide groups. However, a very strong hydrogen bond is established between the cis-(βPro)Arg CO and the guanidinium NH sites [dH···O= 1.595 Å, <N−H···O= 173.4°]. The αL backbone conformation and the down pyrrolidine puckering are also present in the second minimum (αL[d]g+ts+, Figure 4b). However, the less favorable backbone···side chain interaction in this case, which involves a guanidinium NH2 moiety, produces a destabilization of 1.3 kcal/mol.
Figure 4.

Selected minimum energy conformations of Ac-c-(βPro)Arg-NHMe obtained from B3LYP/6–31+G(d,p) calculations (ΔEgp < 5.0 kcal/mol, see Table 2): (a) αL[d]g+g−t; (b) αL[d]g+ts+; (c) γL[d]s−g+t; (d) γL[d]s−ts−; and (e) γL[u]s+g−t. Intramolecular hydrogen bonds are indicated by dashed lines (H…O distances and N–H…O angles are given).
The remaining Ac-c-(βPro)Arg-NHMe minima in Table 2 exhibit a γ-turn conformation stabilized by the corresponding hydrogen bond connecting the acetyl CO and methylamide NH groups. The two most stable conformers of this type, γL[d]s−g+t (Figure 4c) and γL[d]s−ts− (Figure 4d), retain the down pyrrolidine puckering observed in the helical minima and differ from each other in the guanidinium site (NH/NH2) that is hydrogen-bonded to the carbonyl group of cis-(βPro)Arg. Comparison of their relative energies (2.0 and 3.1 kcal/mol, respectively) suggests that the guanidinium NH provides a better geometry for hydrogen bonding to the main chain and therefore produces a higher stabilizing effect, as observed before for the helical conformers (Figures 4a and 4b). It should be noted that the most stable αL and γL conformers characterized for the trans-(βPro)Arg derivative (Table 1) also present backbone···side chain interactions involving the NH guanidinium site. Therefore, this seems to be a general trend of (βPro)Arg, independently of the cis/trans relative orientation of the guanidilated side chain.
The trans-(βPro)Arg and cis-(βPro)Arg derivatives investigated in this work not only differ in their respective preferences to accommodate γ-turn or α-helical backbone arrangements. These compounds also exhibit different conformational propensities in their five-membered ring. Thus, all minima in Table 1 exhibit an up-puckered pyrrolidine unit, whereas the most stable Ac-c-(βPro)Arg-NHMe minima (Table 2) present a down puckering. Moreover, the preference for a particular arrangement of the five-membered ring is much more pronounced in the former case as evidenced by the fact that the first up-puckered minimum of cis-(βPro)Arg lies 3.9 kcal/mol above the global minimum (Table 2) whereas no minima exhibiting a conformation other than up was identified below 7.0 kcal/mol for the trans isomer (see Supporting Information). Indeed, arrangements of the pyrrolidine ring rarely observed in proline and proline-like residues were found to be preferred over the down conformation for this compound (see Supporting Information). The contrast between the puckering tendencies of the five-membered ring in trans-(βPro)Arg and cis-(βPro)Arg becomes most evident when minima in the γL region are compared. Thus, for the cis isomer (Table 2), the most stable γL[d] and γL[u] minima are separated by an energy gap of 1.9 kcal/mol only, whereas the corresponding energy difference for the trans derivative amounts to 19.0 kcal/mol (Table 1 and Supporting Information). This distinct behavior should be ascribed to the different spatial relative disposition between the (βPro)Arg carbonyl and guanidinium groups in the compounds under study. In both cases, the pyrrolidine puckering providing optimal geometry for the establishment of hydrogen-bonding interactions between the two groups mentioned is preferred. For cis-(βPro)Arg, both substituents lie on the same face of the five-membered cycle and are therefore close enough to interact for any puckering state. In contrast, the two interacting groups exhibit a trans relative orientation in trans-(βPro)Arg, and it is the up (but not the down) arrangement of the pyrrolidine ring that brings them in close proximity, thus allowing for strong hydrogen bonding. For the trans isomer, the up puckering is particularly favorable in the case of γL conformers, because positive ψ values make the carbonyl oxygen of trans-(βPro)Arg point in the opposite direction to where the guanidilated side chain is located. It should be noted that no hydrogen bond between the guanidinium group and the backbone exists in the only γL[d] minimum characterized for this compound.
Table 3 displays the free energies in the gas phase (ΔGgp) at 298K for the Ac-t-(βPro)Arg-NHMe and Ac-c-(βPro)Arg-NHMe minima described above. Consideration of the ZPVE, thermal, and entropic corrections to transform ΔEgp into ΔGgp affects substantially the relative energy order of the minimum energy conformations characterized for the trans-(βPro)Arg derivative. Specifically, αL[u]s−g+t becomes the most stable conformer, with γL[u]s−g+t being destabilized by 0.9 kcal/mol. Regarding the cis isomer, the relative stability of the αL[d]g+g−t minimum is enhanced upon addition of these statistical contributions. The ΔGgp values in Table 3 therefore indicate that αL is the preferred backbone arrangement for both compounds in the gas phase. Assuming a Boltzmann distribution, the population of αL conformers at room temperature is about 80% and 100% for the trans and cis compound, respectively. In contrast, minima of the γL type showed the lowest ΔGgp value for both Ac-c-(γPro)Arg-NHMe,28 and Ac-Pro-NHMe,36 thus indicating a substantially higher tendency to adopt conformations in the αL region for the arginine surrogates investigated in the present work, especially, the cis isomer.
Table 3.
Relative free energya in the gas phase (ΔGgp) and in carbon tetrachloride, chloroform and aqueous solutions (ΔGCCl4, ΔGCHCl3, and ΔGH2O, respectively) at 298K for selectedb minimum energy conformations of Ac-t-(βPro)Arg-NHMe and Ac-c-(βPro)Arg-NHMe at the B3LYP/6–31+G(d,p) level.
| Conformer | Δ Ggp | Δ GCCl4 | Δ GCHCl3 | Δ GH2O |
|---|---|---|---|---|
| Ac-t-(βPro)Arg-NHMe | ||||
| γL[u]s−g+t | 0.9 | 1.2 | 2.5 | 6.5 |
| γL[u]g−g−s+ | 2.6 | 0.6 | 0.3 | 2.6 |
| γL[u]g−g−s− | 2.0 | 1.5 | 1.9 | 4.9 |
| αL[u]s−g+t | 0.0c | 0.0 | 0.5 | 2.7 |
| αL[u]g+g−t | 3.1 | 3.2 | 3.6 | 4.9 |
| αL[u]g−tg− | 4.1 | 1.2 | 0.0 | 0.0 |
| γL[u]s−tg− | 5.1 | 2.4 | 1.9 | 4.2 |
|
| ||||
| Ac-c-(βPro)Arg-NHMe | ||||
| αL[d]g+g−t | 0.0d | 0.0 | 1.1 | 2.1 |
| αL[d]g+ts+ | 3.0 | 0.2 | 0.0 | 0.0 |
| γL[d]s−g+t | 3.9 | 4.4 | 6.5 | 9.4 |
| γL[d]s−ts− | 5.9 | 3.6 | 4.1 | 5.7 |
| γL[u]s+g−t | 5.8 | 6.0 | 7.9 | 10.2 |
The effect of solvation was next evaluated by performing single point calculations on the optimized structures through the PCM method. The presence of chlorinated solvents results in the stabilization of several Ac-t-(βPro)Arg-NHMe minima (Table 3), in particular, γL[u]g−g−s+ and αL[u]g−tg−. The relative stability of the latter notably increases with the polarity of the solvent. Indeed, it becomes the preferred structure in chloroform, even if two other minima exhibit relative free energies within a 0.5 kcal/mol interval, and is the only accessible conformation at room temperature in aqueous solution. Regarding Ac-c-(βPro)Arg-NHMe, only αL structures are predicted to be populated either in the gas phase or in the different solvents considered (Table 3). For this compound, solvation seems to affect mainly the arrangement of the exocyclic substituent, with the g+ts+ disposition being favored with increasing polarity. Accordingly, αL[d]g+ts+ becomes the preferred conformation in chloroform and, to a larger extent, in aqueous solution.
Comparison of the solvation effects described above with those observed before12 for the γ-substituted compound Ac-c-(γPro)Arg-NHMe provides further evidence for the higher stability of the αL conformation in the arginine surrogates investigated in the present work. Indeed, minima of the γL type showed the lowest ΔG value for the cis-(γPro)Arg derivative not only in the gas phase but also in carbon tetrachloride and chloroform solutions.28 In water, conformations devoid of intramolecular hydrogen bonds between the backbone amide groups are usually favored for small peptides like the ones considered and, accordingly, an εL conformer became the most populated structure for Ac-c-(γPro)Arg-NHMe in this solvent.28 Interestingly, Ac-Pro-NHMe was predicted36 to prefer the α-helical structure in water. The latter point shows that the conformational preferences of proline in aqueous solution are retained to a larger extent when the arginine side-chain is attached to the β position of the pyrrolidine moiety.
As stated in the Introduction, trans-(βPro)Arg and cis-(βPro)Arg are conceived as arginine substitutes in biologically active peptides. The conformational consequences arising from the incorporation of these arginine surrogates into such peptides may be performed by methods like molecular dynamics (MD) simulations. For this purpose, previous parameterization of the non-proteinogenic residues is necessary. A specific set of force-field parameters was developed for trans-(βPro)Arg and cis-(βPro)Arg to describe the inter- and intramolecular interactions within the classical formalism. Our previous work showed that there is no special electronic effect that might condition the conformational preferences of proline upon addition of the arginine side chain28 and, therefore, the stretching, bending, torsional, and van der Waals parameters for trans-(βPro)Arg and cis-(βPro)Arg were transferred directly from the AMBER force-field.41 Accordingly, electrostatic charges were the only force-field parameters specifically developed for these non-proteinogenic residues.
Atomic charges were calculated by fitting the HF/6–31G(d) quantum mechanical and the Coulombic MEPs to a large set of points placed outside the nuclear region. The electrostatic parameters were obtained by weighting the charges calculated for the low-energy conformers of each compound according to a Boltzmann distribution.42–45 The latter was estimated with the ΔGgp values listed in Table 3. The αL[u]s−g+t, γL[u]s−g+t and γL[u]g−g−s− structures were considered for the trans isomer, whereas αL[d]g+g−t was the only conformer used for the cis derivative since all the local minima are destabilized by at least 3.0 kcal/mol. The electrostatic parameters obtained for trans-(βPro)Arg and cis-(βPro)Arg are given in Figure 5.
Figure 5.
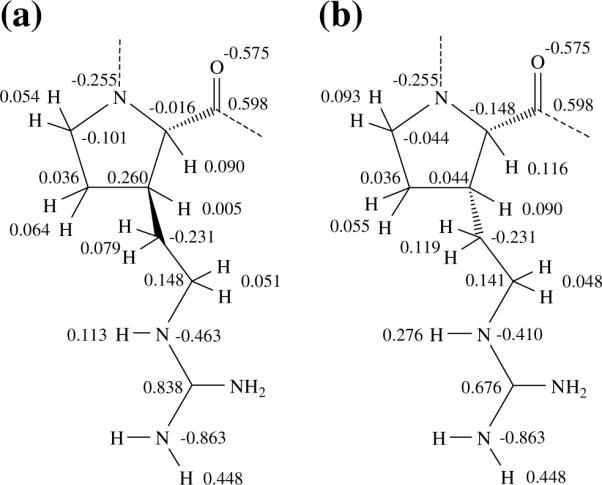
Electrostatic parameters determined for the (a) trans-(βPro)Arg and (b) cis-(βPro)Arg residues.
To check the validity of classical MD simulations in describing the conformational properties of the arginine analogues under study, MD with explicit solvent molecules were performed on Ac-t-(βPro)Arg-NHMe and Ac-c-(βPro)Arg-NHMe in aqueous solution at 298K. For each compound, the lowest-energy conformation was used as the starting point of a 10 ns trajectory. Figure 6 represents the accumulated Ramachandran plot obtained for each derivative. In both cases, the most populated backbone structure corresponds to αL, which is visited much more frequently than the γL region during the trajectory. This finding is in excellent agreement with the results displayed in Table 3, which indicate that αL is the most favored conformation in aqueous solution for both compounds.
Figure 6.

Accumulated Ramachandran plots for (a) Ac-t-(βPro)Arg-NHMe and (b) Ac-c-(βPro)Arg-NHMe derived from MD simulations in aqueous solution.
Conclusions
Two isomers of an arginine surrogate have been built by attaching the arginine side chain to the proline β-carbon in either a trans or a cis disposition relative to the carboxylic acid. The resulting amino acids, respectively denoted trans-(βPro)Arg and cis-(βPro)Arg, combine the conformational restrictions associated with the cyclic nature of proline with the side-chain functionality of arginine. Quantum mechanics calculations on the N-acetyl-N'-methylamide derivatives of these arginine surrogates show that the conformational space available is highly restricted, as expected from their proline-like character. Their conformational preferences are essentially determined by their cyclic structure and the capacity of the guanidilated side chain to establish hydrogen-bonding interactions with the peptide backbone. The latter factor is especially significant for the αL conformation, which is stabilized with respect to natural proline36 and is predicted to be the most populated structure for both (βPro)Arg isomers not only in the gas phase but also in aqueous solution. MD simulations show that the restricted flexibility and the preference for α-helical conformations are kept when thermal agitation is included.
The two non-coded amino acids studied in the present work are suitable candidates to replace arginine in bioactive peptides when the natural residue is found in the αL region, and more specifically, occupying the i+1 position of a β-turn of type I. In particular, cis-(βPro)Arg may be an excellent replacement for arginine in CREKA, and more appropriate than the arginine surrogate considered in a previous work.28 Thus, cis-(βPro)Arg is expected not only to increase resistance to proteases but also to greatly stabilize the type I β-turn found for CREKA. For other biologically relevant peptides, either the cis or the trans isomer of (βPro)Arg may be adequate to replace arginine depending on the orientation attained by the guanidilated side chain in the bioactive conformation.
Supplementary Material
Acknowledgements
Gratitude is expressed to the Centre de Supercomputació de Catalunya (CESCA). We also acknowledge the National Cancer Institute for partial allocation of computing time and staff support at the Advanced Biomedical Computing Center of the Frederick Cancer Research and Development Center. Classical calculations were partially carried out using the high-performance computational capabilities of the Biowulf PC/Linux cluster at the National Institutes of Health, Bethesda, MD (http://biowulf.nih.gov). Financial support from the Ministerio de Educación y Ciencia (Ramon y Cajal contract for D.Z.; project CTQ2007-62245), Gobierno de Aragón (research group E40) and Generalitat de Catalunya (research group 2009 SGR 925; XRQTC; ICREA Academia prize for excellence in research to C.A.) is gratefully acknowledged. This project has been funded in whole or in part with Federal funds from the National Cancer Institute, National Institutes of Health, under contract number HHSN261200800001E. The content of this publication does not necessarily reflect the view of the policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organization imply endorsement by the U.S. Government. This research was supported [in part] by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
Footnotes
Supporting Information All minimum energy conformations characterized for Ac-t-(βPro)Arg-NHMe and Ac-c-(βPro)Arg-NHMe at the B3LYP/6–31+G(d,p) level are provided. Some methodological details about the design of the non-coded amino acids studied are given. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Venkatraman J, Shankaramma SC, Balaram P. Design of Folded Peptides. Chem.Rev. 2001;101:3131–3152. doi: 10.1021/cr000053z. [DOI] [PubMed] [Google Scholar]
- 2.Toniolo C, Crisma M, Formaggio F, Peggion C. Control of Peptide Conformation by the Thorpe-Ingold Effect (Cα-Tetrasubstitution) Biopolymers (Pept. Sci.) 2001;60:396–419. doi: 10.1002/1097-0282(2001)60:6<396::AID-BIP10184>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 3.Nestor JJ. The Medicinal Chemistry of Peptides. Curr. Med. Chem. 2009;16:4399–4418. doi: 10.2174/092986709789712907. [DOI] [PubMed] [Google Scholar]
- 4.Horne WS, Gellman SH. Foldamers with Heterogeneous Backbones. Acc. Chem. Res. 2008;41:1399–1408. doi: 10.1021/ar800009n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chatterjee J, Gilon C, Hoffman A, Kessler H. N-Methylation of Peptides: A New Perspective in Medicinal Chemistry. Acc. Chem. Res. 2008;41:1331–1342. doi: 10.1021/ar8000603. [DOI] [PubMed] [Google Scholar]
- 6.Lelais G, Seebach D. β2-Amino Acids – Syntheses, Occurrence in Natural Products, and Components of β-Peptides. Biopolymers(Pept. Sci.) 2004;76:206–243. doi: 10.1002/bip.20088. [DOI] [PubMed] [Google Scholar]
- 7.Adessi C, Soto C. Converting a Peptide into a Drug: Strategies to Improve Stability and Bioavailability. Curr. Med. Chem. 2002;9:963–978. doi: 10.2174/0929867024606731. [DOI] [PubMed] [Google Scholar]
- 8.Jensen KJ, editor. Peptide and Protein Design for Biopharmaceutical Applications. John Wiley & Sons; Chichester: 2009. [Google Scholar]
- 9.Nielsen PE, editor. Pseudo-Peptides in Drug Discovery. Wiley-VCH; Weinheim: 2004. [Google Scholar]
- 10.Simberg D, Duza T, Park JH, Essler M, Pilch J, Zhang L, Derfus AM, Yang M, Hoffman RM, Bathia S, Sailor MJ, Ruoslahti E. Biomimetic Amplification of Nanoparticle Homing to Tumors. Proc. Natl. Acad. Sci. USA. 2007;104:932–936. doi: 10.1073/pnas.0610298104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zanuy D, Flores-Ortega A, Casanovas J, Curcó D, Nussinov R, Alemán C. The Energy Landscape of a Selective Tumor-Homing Pentapeptide. J. Phys. Chem. B. 2008;112:8692–8700. doi: 10.1021/jp711477k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Agemy L, Sugahara KN, Kotamraju VR, Gujraty K, Girard OM, Kono Y, Mattrey RF, Park JH, Sailor MJ, Jimenez AI, Cativiela C, Zanuy D, Sayago FJ, Aleman C, Nussinov R, Ruoslahti E. Nanoparticle-induced vascular blockade in human prostate cancer. Blood. 2010 doi: 10.1182/blood-2010-03-274258. DOI 10.1182/blood-2010-03-274258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Revilla-Lopez G, Torras J, Jimenez AI, Cativiela C, Nussinov R, Aleman C. Side-Chain to Backbone Interactions Dictate the Conformational Preferences of a Cyclopentane Arginine Analogue. J. Org. Chem. 2009;74(6):2403–2412. doi: 10.1021/jo802704h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alemán C. Conformational properties of alpha-amino acids disubstituted at the alpha-carbon. J. Phys. Chem. B. 1997;101:5046–5050. [Google Scholar]
- 15.Alemán C, Jiménez AI, Cativiela C, Perez JJ, Casanovas J. Influence of the phenyl side chain on the conformation of cyclopropane analogues of phenylalanine. J. Phys. Chem. B. 2002;106:11849–11858. [Google Scholar]
- 16.Casanovas J, Zanuy D, Nussinov R, Alemán C. Identification of the intrinsic conformational properties of 1-aminocyclobutane-1-carboxylic acid. Chem. Phys. Lett. 2006;429:558–562. [Google Scholar]
- 17.Alemán C, Zanuy D, Casanovas J, Cativiela C, Nussinov R. Backbone Conformational Preferences and Pseudorotational Ring Puckering of 1-Aminocyclopentane-1-carboxylic Acid. J. Phys. Chem. B. 2006;110:21264–21271. doi: 10.1021/jp062804s. [DOI] [PubMed] [Google Scholar]
- 18.Rodriguez-Ropero F, Zanuy D, Casanovas J, Nussinov R, Alemán C. Application of 1-Aminocyclohexane Carboxylic Acid to Protein Nanostructure Computer Design. J. Chem. Inf. Model. 2008;48:333–343. doi: 10.1021/ci700291x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Markert Y, Köditz J, Ulbrich-Hofmann R, Arnold U. Proline versus Charge Concept for Protein Stabilization against Proteolytic Attack. Protein Eng. 2003;16:1041–1046. doi: 10.1093/protein/gzg136. [DOI] [PubMed] [Google Scholar]
- 20.Walker JR, Altman RK, Warren JW, Altman E. Using Protein-Based Motifs to Stabilize Peptides. J. Pept. Res. 2003;62:214–226. doi: 10.1034/j.1399-3011.2003.00085.x. [DOI] [PubMed] [Google Scholar]
- 21.Vanhoof G, Goossens F, De Meester I, Hendriks D, Scharpé S. Proline Motifs in Peptides and Their Biological Processing. Faseb J. 1995;9:736–744. [PubMed] [Google Scholar]
- 22.Frenken LGJ, Egmond MR, Batenburg AM, Verrips CT. Pseudomonas-Glumae Lipase: Increased Proteolytic Stability by Protein Engineering. Protein Eng. 1993;6:637–642. doi: 10.1093/protein/6.6.637. [DOI] [PubMed] [Google Scholar]
- 23.Chakrabarti P, Pal D. The Interrelationships of Side-Chain and Main-Chain Conformations in Proteins. Prog. Biophys. Mol. Biol. 2001;76:1–102. doi: 10.1016/s0079-6107(01)00005-0. [DOI] [PubMed] [Google Scholar]
- 24.MacArthur MW, Thornton JM. Influence of Proline Residues on Protein Conformation. J. Mol. Biol. 1991;218:397–412. doi: 10.1016/0022-2836(91)90721-h. [DOI] [PubMed] [Google Scholar]
- 25.Rose GD, Gierasch LM, Smith JA. Turns in Peptides and Proteins. Adv. Protein Chem. 1985;37:1–109. doi: 10.1016/s0065-3233(08)60063-7. [DOI] [PubMed] [Google Scholar]
- 26.Che Y, Marshall GR. Impact of Cis-Proline Analogs on Peptide Conformation. Biopolymers. 2006;81:392–406. doi: 10.1002/bip.20431. [DOI] [PubMed] [Google Scholar]
- 27.Sahai MA, Fejer SN, Viskolcz B, Pai EF, Csizmadia IG. First-Principle Computational Study on the Full Conformational Space of L-Threonine Diamide, the Energetic Stability of Cis and Trans Isomers. J. Phys. Chem. A. 2006;110:11527–11536. doi: 10.1021/jp0680488. [DOI] [PubMed] [Google Scholar]
- 28.Zanuy D, Flores-Ortega A, Jiménez AI, Calaza MI, Cativiela C, Nussinov R, Ruoslahti E, Alemán C. In Silico Molecular Engineering for a Targeted Replacement in a Tumor-Homing Peptide. J. Phys. Chem. B. 2009;113:7879–7889. doi: 10.1021/jp9006119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JA, Vreven T, Jr., Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, C. Strain M, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA. Gaussian 03. Revision B.02 Gaussian, Inc.; Pittsburgh PA: 2003. [Google Scholar]
- 30.Becke AD. A New Mixing of Hartree-Fock and Local Density-Functional Theories. J. Chem. Phys. 1993;98:1372–1377. [Google Scholar]
- 31.Lee C, Yang W, Parr RG. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron-Density. Phys. Rev. 1988;37:785–789. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- 32.McLean AD, Chandler GS. Contracted Gaussian Basis Sets for Molecular Calculations. I. Second Row Atoms, Z=11–18. J. Chem. Phys. 1980;72:5639–5648. [Google Scholar]
- 33.Perczel A, Angyán JG, Kajtar M, Viviani W, Rivail J-L, Marcoccia J-F, Csizmadia IG. Peptide Models. 1. Topology of Selected Peptide Conformational Potential Energy Surfaces (Glycine and Alanine Derivatives) J. Am. Chem. Soc. 1991;113:6256–6265. [Google Scholar]
- 34.Milner-White EJ, Bell LH, Maccallum PH. Pyrrolidine Ring Puckering in cis and trans-Proline Residues in Proteins and Polypeptides – Different Puckers are Favoured in Certain Situations. J. Mol. Biol. 1992;228:725–734. doi: 10.1016/0022-2836(92)90859-i. [DOI] [PubMed] [Google Scholar]
- 35.Némethy G, Gibson KD, Palmer KA, Yoon CN, Paterlini G, Zagari A, Rumsey S, Scheraga HA. Energy Parameters in Polypeptides. 10. Improved Geometrical Parameters and Nonbonded Interactions for Use in the ECEPP/3 Algorithm, with Application to Proline-Containing Peptides. J. Phys. Chem. 1992;96:6472–6484. [Google Scholar]
- 36.Flores-Ortega A, Jiménez AI, Cativiela C, Nussinov R, Alemán C, Casanovas J. Conformational Preferences of α-Substituted Proline Analogues. J. Org. Chem. 2008;73:3418–3427. doi: 10.1021/jo702710x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tomasi J, Mennucci B, Cammi R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005;105:2999–3093. doi: 10.1021/cr9904009. [DOI] [PubMed] [Google Scholar]
- 38.Tomasi J, Persico M. Molecular Interactions in Solution: An Overview of Methods Based on Continuous Distributions of the Solvent. Chem. Rev. 1994;94:2027–2094. [Google Scholar]
- 39.Miertus S, Tomasi J. Approximate Evaluations of the Electrostatic Free Energy and Internal Energy Changes in Solution Processes. Chem. Phys. 1982;65:239–245. [Google Scholar]
- 40.Miertus M, Scrocco E, Tomasi J. Electrostatic Interaction of a Solute with a Continuum. A Direct Utilization of Ab Initio Molecular Potentials for the Prevision of Solvent Effects. Chem. Phys. 1981;55:117–129. [Google Scholar]
- 41.Cornell WD, Cieplak P, Bayly CI, Gould IR, Merz KM, Ferguson DM, Spellmeyer DC, Fox T, Caldwell JW, Kollman PA. A Second Generation Force Field for the Simulation of Proteins, Nucleic Acids, and Organic Molecules. J. Am. Chem. Soc. 1995;117:5179–5197. [Google Scholar]
- 42.Cieplak P, Cornell WD, Bayly C, Kollman PA. Application of the Multimolecule and Multiconformational RESP Methodology to Biopolymers: Charge Derivation for DNA, RNA, and Proteins. J. Comput. Chem. 1995;16:1357–1377. [Google Scholar]
- 43.Alemán C, Casanovas J. Ab Initio SCF and Force-Field Calculations on Low-Energy Conformers of 2-Acetylamino-2, N-Dimetylpropanamide. J. Chem. Soc. Perkin Trans. 1994;2:563–568. [Google Scholar]
- 44.Reynolds CA, Essex JW, Richards WG. Atomic Charges for Variable Molecular Conformations. J. Am. Chem. Soc. 1992;114:9075–9079. [Google Scholar]
- 45.Casanovas J, Zanuy D, Nussinov R, Alemán C. Intrinsic Conformational Characteristics of α,α-Diphenylglycine. J. Org. Chem. 2007;72:2174–2181. doi: 10.1021/jo0624905. [DOI] [PubMed] [Google Scholar]
- 46.Kalé L, Skeel R, Bhandarkar M, Brunner R, Gursoy A, Krawetz N, Phillips J, Shinozaki A, Varadarajan K, Schulten K. NAMD2: Greater Scalability for Parallel Molecular Dynamics. J. Comput. Phys. 1999;151:283–312. [Google Scholar]
- 47.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983;79:926–935. [Google Scholar]
- 48.Berendsen HJC, Postma JPM, van Gunsteren WF, DiNola A, Haak JR. Molecular Dynamics with Coupling to an External Bath. J. Chem. Phys. 1984;81:3684–3690. [Google Scholar]
- 49.Ryckaert JP, Ciccotti G, Berendsen HJC. Numerical Integration of the Cartesian Equations of Motion of a System with Constraints: Molecular Dynamics of n-Alkanes. J. Comput. Phys. 1977;23:327–341. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.