

Abstract
A critical feature of the robustness of the DNA replication machinery is the ability to complete its task in the presence of interfering DNA damage. A key mechanism responsible for this task is translesion replication (also termed translesion synthesis), carried out by specialized lesion bypass DNA polymerases of the Y superfamily. Here we show that in Escherichia coli, plasmids can be replicated across a segment of foreign non-DNA material, consisting of hydrocarbon chains of 3 or 12 methylene residues. This replication is carried out by DNA polymerase V and proceeds by at least two mechanisms: (i) Editing out the foreign insert, by polymerase “hopping” across it, which can be mediated by looping out of the insert, leading to its deletion, while preserving the DNA sequence. (ii) DNA synthesis through the insert, which occurs by incorporating one or two nucleotides opposite the hydrocarbon chain, yielding a net increase in the length of the DNA sequence. The remarkable ability of DNA polymerase V to insert nucleotides opposite a hydrocarbon chain shows that DNA synthesis can occur in a region of the template strand, which lacks all fundamental features of DNA, including its purine, pyrimidine, sugar, and phosphate moieties, and its hydrophilic and ionic nature. This bypass ability reflects a striking robustness of the translesion replication apparatus and is likely to contribute to its effectiveness in maintaining genome stability.
DNA replication is characterized by very high fidelity, as required for maintaining the stability of genetic information. This high fidelity is achieved by several mechanisms, including an inherently high fidelity of replicative DNA polymerases. The high fidelity of replicative DNA polymerases is achieved via a tight active site, which accommodates the template DNA, the primer terminus, and the incoming dNTP. Both geometrical fitting and stabilizing interactions ensure the formation of correct base pairs with high precision (1–3). These fidelity factors become ineffective when encountering lesions, which distort the native structure of DNA. DNA lesions are frequently formed by intracellular agents, e.g., oxygen radicals, as well as exogenous agents, e.g., sunlight (4). Although most lesions are eliminated via DNA repair, some remain in DNA. Therefore, when replication ensues, it frequently encounters such unrepaired lesions (5). In this situation, the high accuracy of the DNA polymerases becomes an obstacle and leads to inhibition of DNA synthesis: the modified nucleotides usually strongly deviate from the canonical structure of native nucleotides, and its inability to properly fit into the active site of the polymerase may cause termination of elongation (4, 5).
To overcome replication-blocking lesions, cells use either recombinational repair (6–9) or translesion replication (TLR; also termed translesion synthesis). TLR is carried out by specialized lesion bypass DNA polymerases, which are conserved in organisms ranging from Escherichia coli to humans. The bypass DNA polymerases, which comprise the Y superfamily (10), are template-dependent DNA polymerases, with low fidelity, low processivity, and no proofreading (11–13). In this study, we report on experiments designed to probe the importance of the fundamental features of DNA, when serving as a template for TLR by E. coli DNA polymerases. Remarkably, we found that polymerase (pol) V can replicate across a chain of up to 12 methylene residues in DNA and use it as a “template” for DNA synthesis, despite the lack of any DNA features.
Materials and Methods
Materials. Oligonucleotides containing the methylene insert M3 or M12 were synthesized by Genset (Evry Cedex, France) by using hydrocarbon building blocks (phosphoramidite C3 and C12 CE phosphoramidite) obtained from Glen Research (Sterling, VA). Oligonucleotides without the hydrocarbon inserts were synthesized by Sigma. The gapped plasmids carrying the hydrocarbon inserts (GP21-M3, GP21-M12, and GP51-M12; Fig. 1) were prepared as described (14), except that the oligonucleotides with the appropriate hydrocarbon insert were used. MBP-UmuC, UmuD′, RecA, single-stranded DNA-binding protein, pol III core, and the β-subunit of pol III were purified as described (15, 16). Pol I was from Roche Molecular Biochemicals. Other proteins were generous gifts from the following researchers: pol II, Myron Goodman (University of Southern California, Los Angeles); His-tagged pol IV, Haruo Ohmori (Kyoto University, Kyoto); and the γ complex, Michael D. O'Donnell (The Rockefeller University, New York).
Fig. 1.

Structure of the hydrocarbon inserts in DNA and of the DNA sequence at the gap-lesion region. (A) The structures of the –(CH2)3-(M3) and –(CH2)12-(M12) inserts in DNA. The two flanking nucleotides are also shown. (B) The DNA sequences at the gap region in gap-lesion plasmids GP21 and GP51. X represents the hydrocarbon insert.
The bacterial strains used in this study were: E. coli ZTR10, same as AB1157 (argE3, hisG4, leuB6, proA2, thr1, ara14, galK2, lacY1, mtl1, xyl5, thi1, tsx33, rpsL31, and supE44), except tna300::Tn10; E. coli WBY100, same as AB1157, but also ΔumuDC; E. coli RW118, same as AB1157, except that it is leuB+, araD139, sulA211; E. coli AR30, same as RW118, but also ΔdinB61::Ble strain (a generous gift from Roger Woodgate, National Institutes of Health, Bethesda); E. coli JM109, e14– (McrA–), recA1, endA1, gyrA96, thi1, hsdR17, ( ,
, 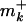 ), supE44, relA1, Δ(lacproAB), (F′ traD36, proAB, laclq, and ZΔM15).
), supE44, relA1, Δ(lacproAB), (F′ traD36, proAB, laclq, and ZΔM15).
In Vivo Replication of Gapped Plasmids with Hydrocarbon Inserts in the Single-Stranded DNA Region. The gapped plasmids carrying the oligomethylene groups (GP21-M3, GP21-M12, and GP51-M12) and the respective control plasmids without the hydrocarbons (GP20 or GP50) were used to transform UV-irradiated E. coli cells, as described (9, 15). The cells were UV-irradiated at 20 Jm–2, followed by a recovery period of 30 min at 37°, after which they were transformed with the gapped plasmid (Fig. 2A). Survival was calculated by dividing the number of transformants obtained with the gap-lesion plasmid by the number of transformants obtained with the gapped plasmid without the lesion. Cultures were grown from individual colonies and their plasmid contents extracted and subjected to automated DNA sequence analysis, performed by the Biological Services Department of the Weizmann Institute of Science.
Fig. 2.

Outline of in vivo and in vitro gap-filling assays. (A) The in vivo gap-filling assay involves transformation of UV-irradiated E. coli cells with the gap-lesion plasmid and selection on plates containing kanamycin. (B) In vitro gap-filling DNA synthesis analysis was performed by using a radiolabeled gap-lesion plasmid and a purified DNA polymerase. The reaction products were restricted with MspA1I and Asp-700, fractionated by 15% PAGE–urea, followed by phosphorimaging analysis, as described in Materials and Methods. The full rectangle represents the hydrocarbon insert. The asterisk represents a radiolabeled phosphate. See text for details.
In Vitro Gap-Filling Replication Assay. The in vitro gap-filling replication reaction was performed as described (15, 16), with minor changes (Fig. 2B). The standard reaction mixture (25 μl) contained 20 mM Tris·HCl, pH 7.5; 8 μg/ml BSA; 5 mM DTT; 0.1 mM EDTA; 4% glycerol; 1 mM ATP; 10 mM MgCl2; 0.1 mM each of dATP, dGTP, dTTP, and dCTP; and 0.1 μg (2 nM) of gapped plasmid. Reactions were performed with one of the following DNA polymerases: pol I (90 nM), pol II (90 nM), pol III core (108 nM), pol IV (as His-DinB, 100 nM), or pol V (as MBP-UmuC, 100 nM). Reactions with pol V were carried out with 50 nM single-stranded DNA-binding protein, 2 μM RecA, and 400 nM UmuD′, and either with or without the processivity proteins, namely the β-subunit of pol III (40 nM as dimer) and the γ complex clamp loader (20 nM). Reactions were performed at 37°C for 5–15 min. The DNA samples were fractionated by 15% PAGE–urea, followed by phosphorimaging analysis (Fuji BAS 2500). TLR was calculated by dividing the amount of bypass products (primers extended past the lesion; 30-mer) by the total amount of DNA in the reaction.
DNA Sequence Analysis of in Vitro Gap-Filling Replication Products. To analyze the gap-filling reaction products, a gap-filling TLR reaction was performed as described, with either pol V or IV at 100 nM, in the presence of the β-subunit and γ complex. Reaction products were used to transform a recA strain, which is unable to induce the SOS response and cannot perform replication across replication-blocking lesions (15, 17). A gapped plasmid that was subjected to the same treatment, but without a DNA polymerase, was used as a control. Cultures were grown from individual transformant colonies and their plasmid contents extracted and subjected to automated DNA sequence analysis, performed by the Biological Services Department of the Weizmann Institute of Science.
Results
In Vivo Translesion Replication Across Hydrocarbon Inserts in Gapped Plasmids. The experimental design in this study included the construction of gapped plasmids, which carried an intact kanamycin-resistance gene and a hydrocarbon chain, inserted within the single-stranded DNA region opposite the gap (the gap-lesion plasmid; Fig. 1). A kanamycin-sensitive E. coli strain was transformed with such a gap-lesion plasmid and seeded on selective plates containing kanamycin (Fig. 2 A). Kanamycin-resistant colonies were expected to form only if the cells had managed to replicate the gap-lesion plasmid and maintained it in intact form. In the absence of a complementary strand, the foreign insert cannot be removed by excision repair, and in the absence of a homologous DNA, it cannot be tolerated by recombinational repair. Thus, before the plasmid can properly replicate, the hydrocarbon chain must be bypassed via TLR. The cells were UV-irradiated before transformation to activate their SOS response, which causes the induction of several cellular processes, which collectively help the cell to overcome genotoxic lesions that cause replication blocks (4).
Table 1 shows the survival of SOS-induced E. coli ZTR10 “wild-type” cells, which were transformed to kanamycin resistable with gapped plasmid GP21-M3 (Fig. 1B), carrying the –(CH2)3-insert. Survival, normalized to the gapped plasmid without the insert, was 3.8%, indicating considerable toxicity of the insert. A slightly lower survival (2.1%; Table 1) was obtained with plasmid GP51-M3, in which the M3 lesion is inserted within a different DNA sequence context (Fig. 1). We assayed also two other gapped plasmids, GP21-M12 and GP51-M12. The two are similar to substrates GP21-M3 and GP51-M3, except that they contained a longer hydrocarbon insert of 12 melthylene residues (Fig. 1 A). Transformation with GP21-M12 yielded a survival of 4.5% (Table 1), whereas the survival with GP51-M12 was lower (0.9%). These results suggest that, whereas the hydrocarbon chain impairs the ability of the plasmid to replicate, its effect is not totally blocking, and a small percentage of the plasmid molecules manage to survive and replicate in their host cells, enabling further proliferation of the bacteria. Overall, the cytotoxicity of the hydrocarbon lesions is similar to that of an abasic site (15, 18, 19), one of the most common lesions in DNA (20).
Table 1. Survival and DNA sequence analysis of plasmids carrying hydrocarbon chain inserts.
| GP21-M3 -(CH2)3- | GP51-M3 -(CH2)3- | GP21-M12 -(CH2)12-insert | GP51-M12 -(CH2)12- | |
|---|---|---|---|---|
| Survival of cells harboring plasmid, % | ||||
| E. coli strain | ||||
| Wild type | 3.8 (±2) | 2.1 (±0.9) | 4.5 (±0.4) | 0.9 |
| ΔumuDC | 0.05 | 0.06 | 0.05 | 0.05 |
| ΔdinB | 2.7 | ND | ND | 0.6 |
| Number of occurrences | ||||
| DNA sequence | ||||
| Base additions | ||||
| AA | - | - | 4 | 1 |
| AT | - | - | - | 1 |
| A | 29 | 20 | 10 | 4 |
| G | 5 | - | - | - |
| C | - | - | - | 1 |
| Deletions | ||||
| M | - | 1 | 1 | 49 |
| M + 1 | - | - | 21 | 1 |
| M+3 to M+7 | - | - | - | 1 |
| Complex deletions | - | - | 4 | 1 |
| Total colonies analyzed | 34 | 21 | 40 | 59 |
The in vivo analysis was performed by introducing the gapped plasmid carrying the hydrocarbon chain [(CH2)3 or (CH2)12] into UV-irradiated E. coli ZTR10 (wild type), E. coli WBY100 (ΔumuDC), or E. coli AR30 (ΔdinB) cells. Survival was calculated by dividing the number of transformants by that obtained with gapped plasmid without the lesion. The DNA sequence opposite the lesion obtained for individual E. coli ZTR10 clones is shown, transformed with the indicated gap-lesion plasmid. M, hydrocarbon insert, ND, not determined.
Overcoming replication obstacles frequently requires the local intervention of a lesion bypass DNA polymerase, primarily pol V (11, 12, 16, 21). When the transformation experiments were repeated with an E. coli WBY100ΔumuDC mutant lacking pol V, survival was drastically reduced by 18- to 90-fold, down to a background level of 0.05% (Table 1). In contrast, survival was marginally affected in E. coli AR30ΔdinB cells, lacking pol IV, a mutagenic DNA polymerase (22) that, like pol V, is a member of the Y superfamily of DNA polymerases (10) (Table 1). This implies that the replication and survival of the plasmid depended on the assistance of the pol V-based TLR.
Sequence Specificity of Replication Across Hydrocarbon Chains in Vivo. To further characterize the events occurring during in vivo replication across the hydrocarbon insert, we performed DNA sequence analysis of descendants of the gap-lesion plasmids, obtained from kanamycin-resistant colonies. Analysis of descendants of plasmid GP21-M3 carrying the -(CH2)3-insert revealed that all isolates contained a purine nucleotide, mostly an A (29/34; 85%), opposite the site corresponding to the M3 lesion. A similar DNA sequence specificity was found among descendants of GP51-M3, where 20/21 isolates contained an A (Table 1).
Analysis of descendants of plasmids GP21-M12 and GP51-M12, each containing the M12 insert, revealed two significant classes of products. The major class involved elimination of the M12 insert, either with (GP21-M12) or without (GP51-M12) a neighbor nucleotide (Table 1). Thus, the TLR machinery has the ability to edit out the longer hydrocarbon chain from DNA. Most remarkably, the second, minor class of descendants contained one or two extra nucleotides at the position opposite to the M12 segment in the parent plasmid (Table 1); the main addition was of a single A (10/14 addition events in GP21-M12 and 4/7 addition events in GP51-M12). However, additions of two tandem nucleotides were also obtained: AA in GP21-M12 (4/14 base additions) and AA and AT in GP51-M12 (2/7 base additions).
In Vitro Ability of Purified E. coli DNA Polymerases to Replicate Across Hydrocarbon Chains in DNA. The in vivo results described above suggest that a key step that enables replication and propagation of gapped plasmids carrying hydrocarbon inserts is TLR by pol V. This possibility was tested by using an in vitro assay system (Fig. 2B), which is based on the same gapped plasmid used in the in vivo experiments, except that it contained an internal radioactive phosphate in the primer strand (15, 16, 23). On addition of a purified DNA polymerase, the primer terminus is extended up to or beyond the hydrocarbon insert. To facilitate their analysis, reaction products were restricted with MspA1I and Asp-700, leading to the generation of short oligonucleotides, whose length is determined by the type of replicative event (arrest at the lesion, or bypass; Fig. 2B). The experiments were done with each of the five known DNA polymerases of E. coli, in both the presence and absence of the processivity proteins, the β-subunit sliding DNA clamp, and the γ complex clamp loader.
As can be seen in Fig. 3A, the M3 insert completely blocked pol I–III, as indicated by the lack of replication products longer than 30 nt. The addition of the processivity proteins did not affect the bypass activity of pol I or II (Fig. 3A, lanes 3 and 5); however, it did endow pol III with a limited ability (4%) to bypass the M3 lesion (Fig. 3A, lane 7). The situation was totally different with DNA pol IV and V. Even in the absence of the processivity proteins, these polymerases were able to bypass the M3 insert (Fig. 3A, lanes 8 and 10). In the presence of the processivity proteins, both polymerases exhibited a significant extent of TLR, amounting to 48% and 14% for pol IV and V, respectively (Fig. 3A, lanes 9 and 11). Interestingly, pol IV's bypass products were shorter than pol V's bypass products (Fig. 3A, compare lanes 9 and 11), suggesting a different mode of bypass (see below).
Fig. 3.

In vitro DNA synthesis across hydrocarbon chain inserts in gapped plasmids. (A) Replication across (CH2)3 in GP21-M3. (B) Replication across (CH2)12 in GP21-M12. (C) Replication across (CH2)12 in GP51-M12. The reactions were carried out with purified DNA polymerases and accessory processivity proteins, as indicated. All reactions with pol V included also UmuD′, RecA, and SSB. The reaction times were 8 min (A), 5 min (B), and 8 and 15 min (C). The in vitro reaction products were fractionated by 15% PAGE–urea followed by phosphorimager analysis. The 19-mer is the unextended primer; a 30-mer represents replication arrest at the hydrocarbon insert; products 31–47 nt long represent replication across the hydrocarbon insert (bypass products). TLR was calculated by dividing the amount of bypass products by the total amount of DNA in the reaction. nd, not detectable.
Experiments performed with the dodecamethylene M12 insert showed that it completely blocked the classical DNA polymerases (pol I–III), both in the presence or absence of processivity proteins (Fig. 3 B, lanes 2–7, and C, lanes 2–7). In contrast, both pol IV and V were able to bypass the M12 insert and did it more effectively in the presence of the processivity proteins. Interestingly, despite the increased length of the hydrocarbon chain, in one sequence context (GP21-M12), bypass across the M12 insert was more effective than across the M3 insert (notice that the TLR extents for GP21-M3 are for 8 min, whereas the TLR extents for GP21-M12 are for 5 min). In another sequence context (GP51-M12), the extents of TLR were lower, and pol V was more effective than pol IV (Fig. 3C, lanes 9, 11, 12, and 13). Thus, in vitro, both pol IV and V replicate through the M3 and M12 segments, whereas in vivo the task is carried out by pol V (Table 1). This is similar to the case of a furanyl abasic site, which is bypassed in vitro both by pol V and IV, whereas in vivo it is bypass by pol V (24).
Specificity of Nucleotide Insertion During TLR in Vitro. We further analyzed the in vitro bypass by pol IV and V by analyzing the DNA sequence at the site corresponding to the M3 and M12 segments. Bypass of the M3 insert by pol V led almost exclusively to the insertion of a purine base opposite the M3 segment (26/27), with a preference for A over G (Table 2). In contrast, bypass by pol IV led to the deletion of the M3 segment, along with an accompanying nucleotide (19/19; Table 2). These results are in agreement with the product length analysis, which showed that the main pol IV bypass product is slightly shorter than the main pol V bypass product (Fig. 3A, lanes 9 and 11). The pol V specificity of TLR across the hydrocarbon chains is similar to the specificity of in vivo TLR, strengthening the notion that pol V (and not pol IV) is responsible for bypass in vivo.
Table 2. DNA sequence analysis of products synthesized during in vitro replication across hydrocarbon chains by IV and D pol V holoenzyme.
| GP21-M3 -(CH2)3-
|
GP21-M12 -(CH2)12-
|
GP51-M12 -(CH2)12-
|
||||
|---|---|---|---|---|---|---|
| Pol IV | Pol V | Pol IV | Pol V | Pol IV | Pol V | |
| Number of occurrences | ||||||
| DNA sequence | ||||||
| Base additions | ||||||
| AA | - | - | - | - | - | 1 |
| GA | - | - | - | 1 | - | - |
| AT | - | - | - | - | - | - |
| A | - | 16 | 3 | - | 1 | 8 |
| G | - | 10 | - | - | - | - |
| T | - | - | - | - | - | 1 |
| C | - | - | - | - | - | - |
| Deletions | ||||||
| M | - | 1 | - | 2 | - | 22 |
| M+1 | 19 | - | 3 | 11 | 13 | 2 |
| M+2 | - | - | - | 1 | 2 | - |
| M+3 to M+7 | - | - | 12 | 1 | 4 | 2 |
| Complex deletions | - | - | 15 | 6 | 30 | 20 |
| Others | - | - | - | 1 | - | - |
| Total colonies analyzed | 19 | 27 | 33 | 23 | 50 | 56 |
DNA sequence analysis of products synthesized in vitro by pol IV and pol V was conducted by transforming an E. coli JM109 recA strain with the DNA synthesis reaction products. The DNA sequence opposite the lesion obtained for individual clones is shown. Complex deletions refer to deletions accompanied by at least one additional mutation.
A major class of products formed during in vitro bypass across the M12 segment consisted of deletion of the M12 insert, either without or with a neighboring nucleotide (and sometimes more than one nucleotide). For pol V, experiments with GP51–12 yielded primarily the precise elimination of the M12 insert (Table 2; 22/56 isolates), whereas experiments with substrate GP21-M12 yielded primarily the elimination of the M12 insert along with an adjacent nucleotide (Table 2; 11/23 isolates). The one-nucleotide difference in the length of the products stems from a DNA sequence context effect. As will be discussed below, GP51-M12 represents a general case, whereas in GP21-M12, a special template sequence seems to enable misalignment of the primer terminus during DNA synthesis, leading to the elimination of the additional nucleotide. Thus, the more general case involves precise elimination of the hydrocarbon insert. Most remarkably, pol V was also able to insert one or two nucleotides opposite the M12 insert, albeit at a low frequency (Table 2). With template GP51-M12, we obtained eight occurrences of A, one T, and one AA (Table 2). An additional occurrence of GA was observed with substrate GP21-M12, suggesting that pol V accepted the M12 insert as a template, despite its totally different chemistry and structure.
Discussion
Taken together, our results show a remarkable ability of pol V to cope with segments of completely foreign material in DNA. Most remarkable is its ability to overcome a chain of 12 methylene residues, with efficiency comparable to overcoming an abasic site, one of the most common lesions in DNA. The main bypass event involves an editing reaction, which leads to the precise elimination of the M12 insert. This is clearly seen with substrate GP51-M12, where the M12 insert was accurately eliminated in 83% (49/59) of the colonies (Table 1). A possible mechanism for this reaction involves looping out of the hydrocarbon chain, followed by polymerase hopping across the insert, such that the next nucleotide added to the primer is complementary to template A, 5′ to the insert (Fig. 4A, the “Looping-out” pathway). This polymerase hopping mechanism is similar to the previously reported ability of DNA polymerases to jump across hairpins (25) or between templates (26) during PCR and the ability of RNA polymerase to bypass gaps (27).
Fig. 4.

Suggested mechanisms for translesion replication across the dodecamethylene insert in DNA. See text for details. The curved line within the DNA sequence represents the -(CH2)12-insert.
In GP21-M12, the main event is the elimination of the M12 insert along with a single adjacent nucleotide. We suggest that the extra nucleotide deleted in the case of GP21-M12 is due to the specific DNA sequence context in this plasmid, 5′-CTGXG-3′ (X = insert; Fig. 1B). After a C is inserted opposite the template G preceding the M12 insert (Fig. 4B, box 1), it misaligns and pairs with the template G past the M12 insert. If extended, this will lead to the deletion of the M12 insert along with the adjacent G (Fig. 4B, box 2). We suggest this is the main event that occurs during gap-filling in plasmid GP21-M12. Thus, whereas in GP51-12 the foreign insert is precisely edited out, in GP21-12, the misalignment causes the net deletion of a nucleotide from the original sequence. Notice that in vivo, the survival of GP21-12 was 5-fold higher than GP51-12 (Table 1). This increased survival is caused most likely by more effective bypass by pol V in this DNA sequence context (Fig. 3). Thus, although misalignment is potentially mutagenic, it might assist the DNA polymerase in overcoming replication blocks by stabilization of bypass intermediates. The importance of DNA misalignment mechanisms in lesion bypass was previously reported for a number of DNA polymerases (28–32).
The most striking finding in this study is what appears to be an ability of pol V to incorporate one or even two nucleotides opposite a hydrocarbon chain, which has none of the typical features of DNA. Is pol V really able to treat the hydrocarbon chain as a “template” and misinsert opposite it? In GP21-M12, a misalignment event can lead to the same product as misinsertion opposite the hydrocarbon chain, without the need to incorporate a nucleotide opposite the hydrocarbon segment. Such a mechanism is shown in Fig. 4B. After a C is inserted opposite the template G preceding the M12 insert (Fig. 4B, box 1), it misaligns and pairs with the template G past the M12 insert. Next the primer is extended by a single A, which pairs with the next template T (Fig. 4B, “Addition”). If now, instead of further extension, the primer terminus realigns, the A will be placed opposite the hydrocarbon chain (Fig. 4B, “Re-alignment”). Subsequently, partial looping out of the hydrocarbon chain followed by extension will lead to a replication product with an extra A opposite the hydrocarbon chain (Fig. 4B, box 3). An identical product would have been formed by direct incorporation of an A opposite the hydrocarbon chain.
In contrast to GP21-12, the appearance of an extra A, or two As in tandem, in GP51-12 cannot be explained by misalignment, due to the different DNA sequence context. In this case, it seems that pol V indeed treats the hydrocarbon chain as a template. Thus, an extra A in some GP51-12 descendants can be explained by insertion of an A opposite the M12 insert, used as a template, followed by partial looping out of the insert and further extension (Fig. 4A, box 3). Moreover, the presence of plasmid isolates in which two nucleotides were inserted across the M12 insert (in both substrates) strongly indicates that the polymerase accepted the M12 segment as a template and polymerized nucleotides opposite it (Fig. 4A, box 4).
The smaller M3 insert is treated as a single nucleotide by the replication machinery. The critical parameter here seems to be the length of the insert, which is similar to that of a single nucleotide in DNA (Fig. 1 A). The nucleotides inserted opposite the M3 insert are exclusively purines, with a preference for the insertion of an A. This may be dictated primarily by spatial considerations (3). The M3 insert occupies a space much smaller than any nucleotide (Fig. 1 A), and therefore the polymerase inserts opposite it a nucleotide with a larger size, namely a purine. This behavior resembles the A rule, suggested based on the preferential insertion of A opposite abasic sites (31, 33, 34).
DNA synthesis by pol V, like other template-dependent DNA polymerases, is expected to require specific interactions in its active site with the DNA template, the primer terminus, and the incoming dNTP. Based on the structures of other Y family DNA polymerases (13, 35, 36), both pol IV and V are expected to have a spacious and flexible active site, with enough room to accommodate two nucleotides, which correspond roughly to the length of the (CH2)12 segment (Fig. 1 A). Although at this point it is not clear how the active site responds to the presence of the (CH2)12 segment, one possibility is that the polymerase switches to a mode of a local template-independent deoxynucleotidyl transferase activity and polymerizes one or two dNTPs, while ignoring the hydrocarbon segment of the template. This mode of activity resembles terminal deoxynucleotidyl transferase (TdT), which is completely template-independent (37). However, unlike TdT, pol V is a template-dependent DNA polymerase (16, 21), and it is unable to extend the termini of synthetic single-stranded oligos or linear double-stranded DNA with blunt ends or with 3′ overhangs (N. B. Reuven and Z.L., unpublished results). Therefore, we suggest that pol V is not a TdT, but rather a template-dependent DNA polymerase, which becomes locally and temporarily TdT-like when facing the non-DNA insert. Furthermore, we suggest that the polymerase must maintain contact with the DNA flanking the insert, and that misinsertion will be more effective when this binding is tighter (e.g., when a longer portion of the flanking DNA is in contact with the enzyme). Consistent with this model is the finding that misinsertion opposite the M3 insert in GP51-M3 is 2.1%, 20-fold higher than opposite M12 in the same DNA sequence context (GP51-M12; only 0.1%; 7/59 occurrences multiplied by 0.9%; Table 1). Because the M3 insert is shorter than the M12 insert, it is expected that the flanking DNA in contact with the polymerase will be longer than with the M12 insert. The larger interaction surface is expected to stabilize the DNA–enzyme complex, which can explain the higher misinsertion efficiency opposite the M3 insert.
In summary, pol V is permissive in an unprecedented manner with respect to the chemical and conformational features of the template that occupies the active site. Essentially none of the fundamental features of DNA is required and can be substituted, at least partially, by a hydrocarbon chain. The use of hydrocarbon chains was aimed for mechanistic probing of the activity of DNA polymerases, and we are unaware of any data indicating that in nature, foreign molecules may be inserted into the DNA backbone. However, it is possible that, rarely, non-DNA segments may be accidentally inserted into the DNA backbone during transaction that involve cutting and joining of DNA segments, such as recombination and the evolution of genes and genomes (38, 39), V(D)J recombination (40), and unscrambling of ciliate genes (41). The ability to replicate across or edit out such non-DNA segments by specialized DNA polymerases might rescue DNA replication and help maintain genome stability.
Acknowledgments
We thank Haruo Ohmori for his generous gift of pol IV, Michael O'Donnell for his generous gift of the γ complex, Myron Goodman for his generous gift of pol II, and Roger Woodgate for his generous gift of strains E. coli RW118 and AR30. Z.L. is an Incumbent of the Maxwell Ellis Professorial Chair in Biomedical Research. This research was supported by a grant from the United States–Israel Binational Science Foundation (no. 96-00448).
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: TLR, translesion replication; pol n, polymerase η.
References
- 1.Echols, H. & Goodman, M. F. (1991) Annu. Rev. Biochem. 60, 477–511. [DOI] [PubMed] [Google Scholar]
- 2.Kunkel, T. A. & Bebenek, K. (2000) Annu. Rev. Biochem. 69, 497–526. [DOI] [PubMed] [Google Scholar]
- 3.Kool, E. T. (2002) Annu. Rev. Biochem. 71, 191–219. [DOI] [PubMed] [Google Scholar]
- 4.Friedberg, E. C., Walker, G. C. & Siede, W. (1995) DNA Repair and Mutagenesis (Am. Soc. Microbiol., Washington, DC).
- 5.Livneh, Z., Cohen-Fix, O., Skaliter, R. & Elizur, T. (1993) CRC Crit. Rev. Biochem. Mol. Biol. 28, 465–513. [DOI] [PubMed] [Google Scholar]
- 6.Rupp, W. D., Wilde III, C. E., Reno, D. L. & Howard-Flanders, P. (1971) J. Mol. Biol. 61, 25–44. [DOI] [PubMed] [Google Scholar]
- 7.Eggleston, A. K. & West, S. C. (1996) Trends Genet. 12, 20–26. [DOI] [PubMed] [Google Scholar]
- 8.Cox, M. M. (1998) Genes Cells 3, 65–78. [DOI] [PubMed] [Google Scholar]
- 9.Berdichevsky, A., Izhar, L. & Livneh, Z. (2002) Mol. Cell 10, 917–924. [DOI] [PubMed] [Google Scholar]
- 10.Ohmori, H., Friedberg, E. C., Fuchs, R. P. P., Goodman, M. F., Hanaoka, F., Hinkle, D., Kunkel, T. A., Lawrence, C. W., Livneh, Z., Nohmi, T., et al. (2001) Mol. Cell 8, 7–8. [DOI] [PubMed] [Google Scholar]
- 11.Goodman, M. F. (2000) Trends Biochem. Sci. 25, 189–195. [DOI] [PubMed] [Google Scholar]
- 12.Livneh, Z. (2001) J. Biol. Chem. 276, 25639–25642. [DOI] [PubMed] [Google Scholar]
- 13.Prakash, S. & Prakash, L. (2002) Genes Dev. 16, 1872–1883. [DOI] [PubMed] [Google Scholar]
- 14.Tomer, G. & Livneh, Z. (1999) Biochemistry 38, 5948–5958. [DOI] [PubMed] [Google Scholar]
- 15.Reuven, N. B., Tomer, G. & Livneh, Z. (1998) Mol. Cell 2, 191–199. [DOI] [PubMed] [Google Scholar]
- 16.Reuven, N. B., Arad, G., Maor-Shoshani, A. & Livneh, Z. (1999) J. Biol. Chem. 274, 31763–31766. [DOI] [PubMed] [Google Scholar]
- 17.Avkin, S., Adar, S., Blander, G. & Livneh, Z. (2002) Proc. Natl. Acad. Sci. USA 99, 3764–3769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lawrence, C. W., Borden, A., Banerjee, S. K. & LeClerc, J. E. (1990) Nucleic Acids Res. 18, 2153–2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Henderson, P. T., Delaney, J. C., Gu, F., Tannenbaum, S. R. & Essigmann, J. M. (2002) Biochemistry 41, 914–921. [DOI] [PubMed] [Google Scholar]
- 20.Lindahl, T. (1993) Nature 362, 709–715. [DOI] [PubMed] [Google Scholar]
- 21.Tang, M., Shen, X., Frank, E. G., O'Donnell, M., Woodgate, R. & Goodman, M. F. (1999) Proc. Natl. Acad. Sci. USA 96, 8919–8924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wagner, J., Gruz, P., Kim, S. R., Yamada, M., Matsui, K., Fuchs, R. P. P. & Nohmi, T. (1999) Mol. Cell 4, 281–286. [DOI] [PubMed] [Google Scholar]
- 23.Tomer, G., Reuven, N. B. & Livneh, Z. (1998) Proc. Natl. Acad. Sci. USA 95, 14106–14111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maor-Shoshani, A., Hayashi, K., Ohmori, H. & Livneh, Z. (2003) DNA Rep. 2, 1227–1238. [DOI] [PubMed] [Google Scholar]
- 25.Viswanathan, V. K., Krcmarik, K. & Cianciotto, N. P. (1999) BioTechniques 27, 508–511. [DOI] [PubMed] [Google Scholar]
- 26.Paabo, S., Irwin, D. M. & Wilson, A. C. (1990) J. Biol. Chem. 265, 4718–4721. [PubMed] [Google Scholar]
- 27.Liu, J. & Doetsch, P. W. (1996) Biochemistry 35, 14999–15008. [DOI] [PubMed] [Google Scholar]
- 28.Kunkel, T. A. (1990) Biochemistry 29, 8003–8011. [DOI] [PubMed] [Google Scholar]
- 29.Shibutani, S. & Grollman, A. P. (1993) J. Biol. Chem. 268, 11703–11710. [PubMed] [Google Scholar]
- 30.Koffel-Schwartz, N. & Fuchs, R. P. (1995) J. Mol. Biol. 252, 507–513. [DOI] [PubMed] [Google Scholar]
- 31.Shibutani, S., Takeshita, M. & Grollman, A. P. (1997) J. Biol. Chem. 272, 13916–13922. [DOI] [PubMed] [Google Scholar]
- 32.Efrati, E., Tocco, G., Eritja, R., Wilson, S. H. & Goodman, M. F. (1997) J. Biol. Chem. 272, 2559–2569. [DOI] [PubMed] [Google Scholar]
- 33.Strauss, B. S. (1991) BioEssays 13, 79–84. [DOI] [PubMed] [Google Scholar]
- 34.Paz-Elizur, T., Takeshita, M. & Livneh, Z. (1997) Biochemistry 36, 1766–1773. [DOI] [PubMed] [Google Scholar]
- 35.Beard, W. A. & Wilson, S. H. (2001) Structure (Cambridge, U.K.) 9, 759–764. [DOI] [PubMed] [Google Scholar]
- 36.Friedberg, E. C., Fischhaber, P. L. & Kisker, C. (2001) Cell 107, 9–12. [DOI] [PubMed] [Google Scholar]
- 37.Kornberg, A. & Baker, T. (1991) DNA Replication (Freeman, New York).
- 38.Arber, W. (2000) FEMS Microbiol. Rev. 24, 1–7. [DOI] [PubMed] [Google Scholar]
- 39.Copley, R. R., Letunic, I. & Bork, P. (2002) Curr. Opin. Chem. Biol. 6, 39–45. [DOI] [PubMed] [Google Scholar]
- 40.Gellert, M. (2002) Annu. Rev. Biochem. 71, 101–132. [DOI] [PubMed] [Google Scholar]
- 41.Prescott, D. M. (1999) Nucleic Acids Res. 27, 1243–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]