

Abstract
FOXE3 forkhead transcription factor is essential to lens development in vertebrates. The eyes of Foxe3/foxe3-deficient mice and zebrafish fail to develop normally. In humans, autosomal dominant and recessive mutations in FOXE3 have been associated with variable phenotypes including anterior segment anomalies, cataract and microphthalmia. We undertook sequencing of FOXE3 in 116 probands with a spectrum of ocular defects ranging from anterior segment dysgenesis and cataract to anophthalmia/microphthalmia. Recessive mutations in FOXE3 were found in four of 26 probands affected with bilateral microphthalmia (15% of all bilateral microphthalmia and 100% of consanguineous families with this phenotype). FOXE3-positive microphthalmia was accompanied by aphakia and/or corneal defects; no other associated systemic anomalies were observed in FOXE3-positive families. The previously reported c.720C>A (p.C240X) nonsense mutation was identified in two additional families in our sample and therefore appears to be recurrent, now reported in three independent microphthalmia families of varied ethnic backgrounds. Several missense variants were identified at varying frequencies in patient and control groups with some apparently being race-specific, which underscores the importance of utilizing race/ethnicity-matched control populations in evaluating the relevance of genetic screening results. In conclusion, FOXE3 mutations represent an important cause of nonsyndromic autosomal recessive bilateral microphthalmia.
Keywords: FOXE3, aphakia, sclerocornea, microphthalmia, isolated, nonsyndromic, recessive, consanguinity
INTRODUCTION
The Foxe3 gene was initially found to underlie the mouse dysgenetic lens (dyl) phenotype that is limited to the eye and consists of variable degrees of microphthalmia, corneal opacity, small and irregular lenses, and lenticular-corneal adhesions [Sanyal and Hawkins, 1979]. The phenotype was shown to be caused by two missense mutations in the forkhead domain of Foxe3 [Blixt et al., 2000; Brownell et al., 2000]. Ormestad et al. [2002] demonstrated that the dyl mutant Foxe3 is defective in DNA binding and thus represents a null allele. While dyl+/- heterozygotes were initially reported to be unaffected, closer examination revealed that approximately 40% of heterozygous mutant mice show an ocular phenotype resembling Peters’ anomaly, consisting of a stalk-like connection between the cornea and lens, central leukoma, cataract, or other corneal abnormalities [Ormestad et al., 2002].
The expression and function of Foxe3/foxe3/FOXE3 was found to be conserved in vertebrates and to correspond well with the reported phenotypes. During development, Foxe3 transcripts are present in the developing lens and presumptive midbrain of mouse embryos. In adult mice, expression is limited to the anterior lens epithelium [Blixt et al., 2000; Brownell et al., 2000]. Similarly, foxe3 expression was noted in the eye and brain of zebrafish embryos, with the highest levels in the lens epithelial cells, and foxe3 transcripts were also detected in the adult lens and brain [Shi et al., 2006]. Similar to Foxe3-deficient mice, zebrafish foxe3 morphants demonstrate small eyes [Shi et al., 2006]. Finally, expression studies in human embryo coronal head sections (Carnegie stage 16 and 17) found FOXE3 expressed in the developing lens only, with the strongest expression in the anterior lens epithelium [Iseri et al., 2009]; the authors note that brain expression of Foxe3 in mice is absent by the equivalent embryonic stage, thus brain expression may be present in earlier stages of human development.
Human mutations in FOXE3 (OMIM 601094) were first identified by Semina et al. [2001] in a patient with posterior embryotoxon, cataracts, and myopia (OMIM 107250). The patient and her mother, also affected with posterior embryotoxon and cataracts, both carried an autosomal dominant c.943InsG (p.L315RfsX117) mutation. In a second family, a different dominant mutation in FOXE3 was detected in a patient with Peters’ anomaly, eccentric corneal opacities, and glaucoma, but no cataract [Ormestad et al. 2002]. The c.524G>T (p.R90L) mutation resulted in an amino acid substitution in the DNA-binding domain; however, the mutant protein was able to bind DNA in vitro. Recently, Iseri and colleagues [2009] reported two additional dominant mutations in families with variable phenotypes. The first mutation, c.958T>C (p.X320RextX72), was seen in a proband with bilateral microphthalmia, right aphakia, sclerocornea, and coloboma, and left Peters’ anomaly with congenital cataract as well as in other family members affected with cataract and coloboma; the second change, c.146G>C (p.G49A, reported as p.G48A by the authors), was seen in a proband with unilateral microphthalmia/bilateral coloboma and other family members affected with cerulean or late-onset cataracts [Iseri et al., 2009].
The first case of autosomal recessive disease caused by mutations in FOXE3 was reported by Valleix et al. [2006] in a pair of siblings from a consanguineous union (coefficient of inbreeding (F) =1/8, double first cousins) affected with congenital primary aphakia (aphakia, microphthalmia, and anterior segment aplasia; OMIM 610256). Both siblings were homozygous for a c.720C>A (p.C240X) nonsense mutation. The parents were found to be heterozygous carriers of the same mutation; both had normal eye examinations [Valleix et al., 2006]. Iseri and coauthors [2009] described two additional consanguineous families affected with recessive FOXE3 mutations. The first mutation, c.244A>G (p.M82V), was found in a family with bilateral microphthalmia, aphakia, and sclerocornea in some members and anterior segment dysgenesis with glaucoma and normal eye size in others. The second homozygous mutation, c.21_24del (p.M7IfsX216), was identified in a family affected with bilateral microphthalmia, sclerocornea, and aphakia. All obligate carriers in both pedigrees were unaffected.
In order to further define the spectrum of phenotypes associated with mutations in FOXE3, we undertook screening of its coding region in patients affected with various ocular disorders.
METHODS
Human subjects
The study was approved by the Institutional Review Boards of the Children’s Hospital of Wisconsin and Albert Einstein Healthcare Network with informed consent obtained from every subject. The screening included 116 probands with the following phenotypes: 26 patients with bilateral microphthalmia (13 isolated and 13 with non-ocular anomalies), 14 with unilateral microphthalmia (nine isolated and five with non-ocular anomalies), 19 with anophthalmia in at least one eye (four bilateral anophthalmia/microphthalmia (A/M) isolated, 14 bilateral A/M with non-ocular anomalies, and one unilateral anophthalmia with non-ocular anomalies), 29 with anterior segment dysgenesis without microphthalmia including 15 with Peters’ anomaly (12 isolated and 17 with extra-ocular anomalies), four with glaucoma with normal anterior segment and no extra-ocular anomalies (one congenital and three adult-onset), five with coloboma with no extra-ocular anomlies, eight with isolated cataract with no extra-ocular anomalies (nine congenital/juvenile and one adult-onset), and 11 with other ocular defects. Extraocular defects were highly variable and included developmental delay and craniofacial anomalies (abnormal ear shape or position, microcephaly, or dysmorphic facial features) in half of such cases. Other frequently associated features included genitourinary defects, short stature, structural brain abnormalities, and heart defects.
Eight patients with previously identified SOX2 mutations were excluded from this screening (one patient with bilateral microphthalmia, two with unilateral microphthalmia, and five with anophthalmia). Of the 116 probands, 78 were Caucasian, 15 were Hispanic, five were African American, 13 were Asian, two were Caucasian/Asian, and three were unreported.
FOXE3 mutation screen
The FOXE3 coding region was examined by direct DNA sequencing of PCR products, as previously described [Reis et al., 2008] and using Mutation Surveyor (SoftGenetics, State College, PA) to analyze sequences; the following primers were used to generate PCR products: set 1 forward, 5’-TGGGAGAGGAAATTAGAGGG-3’, and reverse, 5’-ACCTTGACGAAGCAGTCGTT-3’, and set 2 forward, 5’-CGCAAGTGGCAGAACAGCAT-3’, and reverse, 5’-TAGCAGGAGTTTGAGTCCAG-3’. The Reference sequence NM_012186.2 was used.
To obtain control data for normal variation in FOXE3 in these ethnic populations, we determined exact gene sequence from 127/161 Caucasian, 80/84 Hispanic, 79/83 African American, and 55/46 Asian control individuals for FOXE3 sets 1/2, correspondingly, using the above approach. The Caucasian control DNA samples were received from the European Collection of Cell Cultures (Salisbury, UK) while the African American, Hispanic, and Asian control panels were received from the Coriell Institute for Medical Research (Camden, NJ). All control individuals were reported as unaffected at the time of sample collection; however, it should be noted that since detailed phenotypic information for control persons is not available and age at sample collection is variable, it is possible that some individuals with mild or late-onset ocular conditions are included in these control populations.
RESULTS
A number of variants were identified in patients and controls (Table I). Homozygous or compound heterozygote variants which were not seen in control populations were identified in four families. All heterozygous variants identified in patients were also seen in control populations, thus no dominant disease-causing mutations in FOXE3 were identified in probands.
Table I.
Summary of FOXE3 sequencing results in patient and control samples (this study).
| Nucleotide substitutions | Protein effect | Caucasian patients | Caucasian controls | Hispanic patients | Hispanic controls | African American patients | African American controls | Asian patients | Asian controls | Total patients** | Total controls |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Disease-causing mutations | |||||||||||
| c.244A>G* | p.M82V | 0.6% (1/156) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0.4% (1/232) | 0 |
| c.557delT | p.F186SfsX38 | 1% (2/156) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0.9% (2/232) | 0 |
| c.705delC | p.E236SfsX71 | 0.6% (1/156)) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0.4% (1/232) | 0 |
| c.720C>A* | p.C240X | 1% (2/156) | 0 | 0 | 0 | 0 | 0 | 8% (2/26) | 0 | 1.7% (4/232) | 0 |
| Variants | |||||||||||
| c.16G>A | p.D6N | 0 | 0 | 0 | 0 | 0 | 3% (5/158) | 0 | 0 | 0 | 0.7% (5/682) |
| c.135G>A | p.A45A | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1% (1/110) | 0 | 0.1% (1/682) |
| c.146G>C*† | p.G49A | 0 | 0 | 0 | 0 | 10% (1/10) | 2.5% (4/158) | 0 | 0 | 0.4% (1/232) | 0.6% (4/682) |
| c.158C>T | p.P53P | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2.7% (3/110) | 0 | 0.4% (3/682) |
| c.276C>T | p.L92L | 0 | 0 | 0 | 0 | 0 | 0.6% (1/158) | 0 | 0 | 0 | 0.1% (1/682) |
| c.423G>A | p.K141K | 0 | 0 | 0 | 0 | 0 | 3.6% (6/166) | 0 | 0 | 0 | 0.8% (6/748) |
| c.445C>T | p.P149S | 0 | 0 | 0 | 0.6% (1/168) | 0 | 0 | 0 | 0 | 0 | 0.1% (1/748) |
| c.510C>T* (rs34082359) | p.A170A | 38% (60/156) | 42% (135/322) | 37% (11/30) | 18% (30/168) | 40% (4/10) | 22% (37/166) | 31% (8/26) | 34% (31/92) | 37% (86/232) | 31% (233/748) |
| c.527C>T | p.A176V | 0 | 0.3% (1/322) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0.1% (1/748) |
| c.601G>A | p.V201M | 0 | 0 | 10% (3/30) | 3% (5/168) | 0 | 0 | 4% (1/26) | 0 | 1.7% (4/232) | 0.7% (5/748) |
| c.618C>G* | p.A206A | 2% (3/156) | 0.6% (2/322) | 0 | 1% (2/168) | 0 | 0.6% (1/166) | 8% (2/26) | 3% (3/92) | 2.6% (6/232) | 1.6% (12/748) |
| c.783C>G | p.L261L | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1% (1/92) | 0 | 0.1% (1/748) |
| c.831C>T | p.G277G | 0 | 0 | 0 | 0 | 0 | 0.6% (1/166) | 0 | 0 | 0 | 0.1% (1/748) |
| c.898A>G* | p.S300G | 2% (3/156) | 0.3% (1/322) | 0 | 0 | 0 | 0 | 0 | 0 | 1.3% (3/232) | 0.1% (1/748) |
| c.929G>A | p.G310D | 0 | 0 | 0 | 0.6% (1/168) | 0 | 0.6% (1/166) | 0 | 0 | 0 | 0.3% (2/748) |
Number of chromosomes was used in all calculations.
Reference SNP (rs) numbers are shown for all variants included in the dbSNP database (http://www.ncbi.nlm.nih.gov/projects/SNP/)
= Previously reported substitutions (Semina et al., 2000; Valliex et al., 2006; Iseri et al., 2009)
= Previously reported as disease-causing mutation (Iseri et al., 2009)
= Includes three patients with unreported race/ethnicity and two with mixed racial background
Patient 1 is a 2½-year-old Caucasian female with severe bilateral microphthalmia and sclerocornea (Fig 1, Table II). The iris, lens, and optic disc could not be visualized. MRI of the head showed small optic nerves and globes, but normal brain structures. Renal ultrasound at 2 months showed mild right pelvic dilation but voiding cystourethrogram was normal. Development was normal. This patient was the only child born to these parents, who were reported to be unaffected but were not available for detailed ophthalmological examination. There was no history of consanguinity. The patient was found to be a compound heterozygote with a c.244A>G (p.M82V) missense mutation, previously reported [Iseri et al., 2009], and a c.705delC (p.E236SfsX71) frameshift mutation (Supplemental Fig 1). The c.244A>G mutation results in a predicted substitution of a highly conserved residue located in the DNA-binding forkhead domain (Figure 2), while the c.705delC (p.E236SfsX71) is predicted to cause C-terminal truncation of normal protein sequence and frameshift with 70 additional amino acids in the new reading frame. Examination of parental samples identified the presence of the c.244A>G (p.M82V) mutation in the reportedly unaffected mother and the c.705delC (p.E236SfsX71) mutation in the reportedly unaffected father.
Figure 1. Patient photographs.
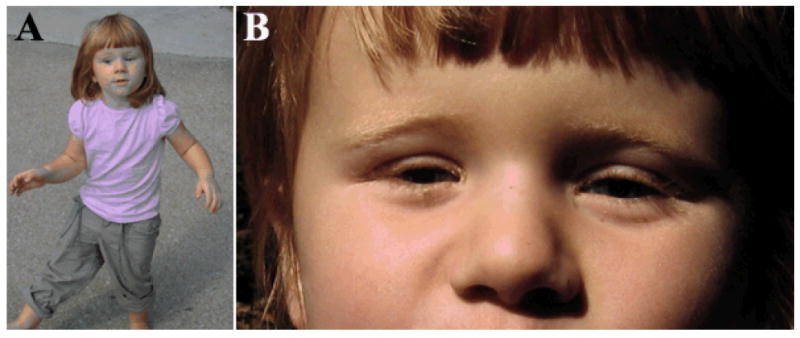
A) Patient 1 at 2-½-years-old with ocular prosthetics. Note absence of facial dysmorphology and other defects. B) Patient 1 with prosthetics removed demonstrating bilateral severe microphthalmia.
Table II.
Summary of FOXE3 mutations reported to date and associated phenotypes.
| Reference | Mutation | Protein Change | Ocular Features Bilateral unless indicated | Other Features | Family History | Consanguinity |
|---|---|---|---|---|---|---|
| HOMOZYGOUS/ COMPOUND HETEROZYGOUS MUTATIONS | ||||||
| Patient 1, this study | heterozygous c.244A>G; c.705delC | p.M82V; p.E236SfsX71 | Severe microphthalmia, sclerocornea (anterior segment not visualized) | Mild pelvic dilation with normal VCUG | Unaffected parents both heterozygotes | No |
| Patient 2, this study | homozygous c.557delT | p.F186SfsX38 | Microphthalmia, aphakia, sclerocornea, | None | Parents both heterozygotes, mother with unilateral corneal ‘cloudiness’ | Yes |
| Patient 3, this study | homozygous c.720C>A | p.C240X | Microphthalmia, aphakia, corneal opacity, glaucoma | Autism, developmental delay | Parents unaffected, DNA not available | Yes |
| Patient 4, this study | homozygous c.720C>A | p.C240X | Microphthalmia, coloboma, glaucoma, corneal opacity (anterior segment not visualized | Chair 1 malformation, umbilical hernia | Affected brother (below), Unaffected parents both heterozygotes | Yes |
| Patient 5, this study | homozygous c.720C>A | p.C240X | Microphthalmia, glaucoma, corneal opacity (anterior segment not visualized) | Hypertrichosis | Sibling of above | Yes |
| Case 1, Iseri et al., 2009 | Homozygous c.244A>G | p.M82V | Microphthalmia, aphakia, sclerocornea | None | Large consanguineous pedigree, multiple affected | Yes |
| Case 2, Iseri et al., 2009 | Homozygous c.244A>G | p.M82V | Anterior segment dysgenesis and congenital glaucoma | None | Member of large pedigree (above) | Yes |
| Case 3, Iseri et al., 2009 | Homozygous c.244A>G | p.M82V | Microphthalmia, sclerocornea | None | Member of large pedigree (above) | Yes |
| Case 4, Iseri et al., 2009 | Homozygous c.244A>G | p.M82V | Anterior segment dysgenesis, congenital glaucoma | None | Member of large pedigree (above) | Yes |
| Case 5, Iseri et al., 2009 | Homozygous c.244A>G | p.M82V | Microphthalmia, sclerocornea | None | Member of large pedigree (above) | Yes |
| Case 6, Iseri et al., 2009 | Homozygous c.244A>G | p.M82V | Aphakia, sclerocornea, absent anterior chamber, central nodular corneal degeneration | None | Member of large pedigree (above) | Yes |
| Case 7, Iseri et al., 2009 | Homozygous c.21_24del | p.M7IfsX216 | Microphthalmia, aphakia, sclerocornea, aniridia | None | Two affected cousins | Yes |
| Case 8, Iseri et al., 2009 | Homozygous c.21_24del | p.M7IfsX216 | Aphakia, sclerocornea | None | Cousin of above | Yes |
| Case 9, Iseri et al., 2009 | Homozygous c.21_24del | p.M7IfsX216 | Congenital blindness | Developmental delay, pregnancy complications | Cousin of above | Yes |
| Valleix et al., 2006 | homozygous c.720C>A | p.C240X | Aphakia, asymmetric microphthalmia, right megalocornea, elevated IOP | None | Two affected siblings; unaffected parents both carry mutation | Yes |
| Valleix et al., 2006 | homozygous c.720C>A | p.C240X | Microphthalmia, aphakia, sclerocornea | None | Sibling of above | Yes |
| HETEROZYGOUS MUTATIONS | ||||||
| Semina et al., 2001 | heterozygous c.943InsG | p.L315RfsX117 | Posterior embryotoxon, cataract, myopia | None | Affected mother also carries mutation | No |
| Semina et al., 2001 | heterozygous c.943InsG | p.L315RfsX117 | Posterior embryotoxon, cataract | None | Mother of above | No |
| Ormestad et al., 2002 | heterozygous c.524G>T | p.R90L | Peters’ anomaly, glaucoma | None | Large autosomal dominant pedigree; no other family members tested | No |
| Case 10, Iseri et al., 2009 | Heterozygous c.958T>C | p.X320RextX72 | R microphthalmia, aphakia, sclerocornea, coloboma; L Peters’ anomaly, microphthalmia, congenital cataract | Prominent incisors | Family members with coloboma, cataract | No |
| Iseri et al., 2009 | Heterozygous c.958T>C | p.X320RextX72 | Nuclear sclerosis, cataracts | None | Mother of Case 10 | No |
| Iseri et al., 2009 | Heterozygous c.958T>C | p.X320RextX72 | Partial iris coloboma, cataracts | None | Maternal uncle of Case 10 | No |
| Iseri et al., 2009 | Heterozygous c.958T>C | p.X320RextX72 | Early adult onset cataracts (20s) | None | Maternal aunt of Case 10 | No |
| Iseri et al., 2009 | Heterozygous c.958T>C | p.X320RextX72 | Cataract extraction age 40 | None | Maternal grandmother of Case 10 | No |
| Case 11, Iseri et al., 2009 | Heterozygous c.146G>C* | p.G49A* | R microphthalmia, microcornea, coloboma; L coloboma | Mild behavioral difficulties | Family members with cataract | No |
| Iseri et al., 2009 | Heterozygous c.146G>C* | p.G49A* | Cerulean cataract | None | Mother of Case 11 | No |
| Iseri et al., 2009 | Heterozygous c.146G>C* | p.G49A* | Cerulean cataract | None | Brother of Case 11 | No |
| Iseri et al., 2009 | Heterozygous c.146G>C* | p.G49A* | Cataract extraction age 70 | None | Maternal grandmother of Case 11 | No |
Highlighted rows indicate probands/unrelated affected individuals
indicates probable race-specific variant (please see Table I)
Figure 2. FOXE3 mutations.
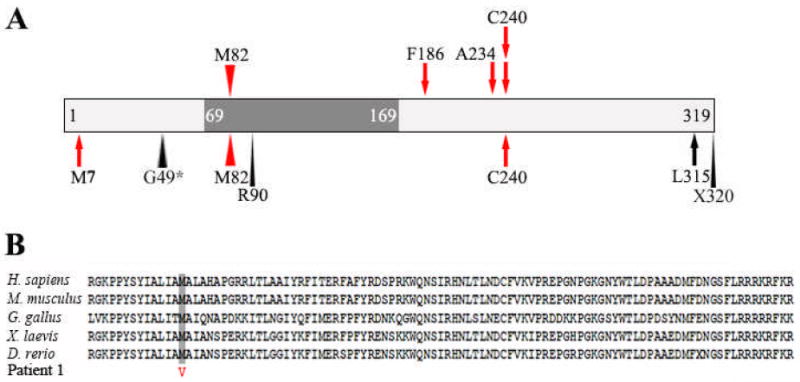
A) Schematic drawing demonstrating the relative positions of FOXE3 mutations identified in our study (top) and previously reported (bottom). The FOXE3 forkhead domain is shown as a dark grey box. Missense mutations are denoted by arrowheads while stop codon/frameshift mutations are indicated with arrows; recessive mutations are shown in red. B) Alignment of FOXE3 forkhead domain sequences showing conservation of the M82 amino acid in vertebrates. Sequences were obtained from NCBI Protein Database: Homo sapiens (NP_036318.1), Mus musculus (NP_056573.1), Gallus gallus (NP_990337.1), Xenopus laevis (NP_001079202.1), and Danio rerio (NP_001073150.1).
Patient 2 is an 11-month-old Caucasian male (United Arab Emirates) with bilateral microphthalmia, aphakia, and abnormal anterior segment with sclerocornea and dysplastic irides (Table II). He also had elevated intraocular pressure with probable optic nerve head cupping. Brain imaging studies were not available. He had no extraocular anomalies and his development was appropriate for age. The patient had two brothers with no ocular defects. The parents were first cousins (F=1/16) and the father was reported to be unaffected. The mother was reported to have decreased visual acuity and unilateral corneal ‘cloudiness’ with no other details available. Neither parent was available for detailed ophthalmological examination. This patient was found to have a homozygous c.557delT (p.F186SfsX38) mutation predicted to result in frameshift with 37 erroneous amino acids after the frameshift (Supplemental Figure 1). Parental testing showed that both parents carry a single copy of the same mutation seen in their child. No other mutations in FOXE3 were seen in either parent.
Patient 3 is a seven-year-old Asian male (Bangladesh) with bilateral asymmetric microphthalmia (the left eye is more severely affected than the right), aphakia, corneal opacity, and glaucoma. Cranial CT also demonstrated possible optic disc drusen and coloboma of the right optic nerve along with normal brain structures. In addition, the patient had autism and global developmental delay (Table II). There was termination of a sibling pregnancy after sonographic detection of holoprosencephaly. A chromosomal anomaly, 46,XX,del(7)(q34), was found in the aborted fetus. There was an additional younger sibling who was unaffected. The parents were first cousins (F=1/16) and were reported to be unaffected but were not available for detailed ophthalmological examination. The patient was found to have an apparently homozygous c.720C>A (p.C240X) mutation (Supplemental Figure 1), previously reported [Valleix et al., 2006]. Parental samples were not available for testing. A deletion of the second allele could not be ruled out, but is unlikely given the history of consanguinity.
Patient 4 is a Caucasian female (Kuwait) with bilateral microphthalmia with glaucoma, coloboma, and opaque corneas with no view of the anterior segment (Table II). Severe central ectasia was noted with central scarring and complete corneal neovascularization of the right and almost no central perforation and staphylomatous malformation of the left cornea. MRI of the head showed Chiari 1 malformation as well as attenuation of the optic nerves, chiasm, and optic tract. She also had nystagmus and an umbilical hernia. Development was normal. There was an affected brother, Patient 5, with bilateral microphthalmia, glaucoma, and corneal ectasia with central scarring and complete corneal neovascularization in both eyes (Table II). The anterior segment was not visible. He also had nystagmus and hypertrichosis on his back. Development was normal; brain imaging studies were not available. There were two unaffected siblings. The parents were first cousins (F=1/16) and were also reported to be unaffected but were not available for detailed ophthalmological examination. The patient and her affected brother were found to have a homozygous c.720C>A (p.C240X) mutation (Supplemental Fig 1), as seen in Patient 3 and previously reported [Valleix et al., 2006]. Testing of parental samples showed both parents carry a single copy of this mutation.
Screening of different control groups did not identify the above described variants in either homozygous or heterozygous state in normal populations (with a total of 341 and 374 control individual sequences obtained for FOXE3 set 1 and set 2, respectively).
In addition to these apparently disease-causing mutations, we identified several variants in our patient and control populations at varying frequencies (Table I). Even though some variants seem to show enrichment in patient versus control populations (c.146G>C (p.G49A), c.601G>A (p.V201M) and c.898A>G(p.S300G)), the differences are likely to be attributed to size/local population variations between the groups rather than disease associations. Of special interest is the c.146G>C (p.G49A) change that was previously reported as a disease-causing mutation [Iseri et al. 2009]. We observed this variant in one African American patient and four control individuals of African American descent. Our patient carrying the c.146G>C variant has been previously described [Patient 8; Reis et al., 2008]. Briefly, his phenotype shows overlap with Peters-plus syndrome, consisting of unilateral Peters’ anomaly and microphthalmia, bilateral brachydactyly of the hands, bilateral cleft lip and palate, bilateral ear tags, hydrocephaly, and dysmorphic facial features but normal growth. The patient’s brother, who is unaffected, was also found to carry this variant; his unaffected mother does not carry the variant, and the father was not available for testing. The c.146G>C variant was not seen in cases or controls of Caucasian, Hispanic, or Asian descent, suggesting it is a variant specific to the African American population.
DISCUSSION
Based on our results, FOXE3 coding region mutations appear to be a significant cause of bilateral microphthalmia (15%: 4/26 in the examined SOX2-negative population or 4/27 of all collected bilateral microphthalmia cases). FOXE3-associated microphthalmia demonstrates an autosomal recessive mode of inheritance and, therefore, the mutations were primarily observed in consanguineous unions, with one compound heterozygote also identified. The previously reported c.720C>A (p.C240X) nonsense mutation [Valleix et al. 2006] was identified in two additional unrelated families of Asian and Caucasian descent in our sample and therefore seems to be a recurrent mutation (Figure 2). Also, the c.244A>G (p.M82V) mutation that was previously reported in one consanguineous pedigree [Iseri et al. 2009] was identified in one proband in our study (Patient 1; Table II) together with a second FOXE3 mutation.
Mutations in FOXE3 appear to result in nonsyndromic ocular disease since the majority of affected individuals demonstrate no extraocular defects (Table II). Of the 16 cases with recessive mutations and 12 cases with dominant mutations, three were reported with developmental delay\behavioral difficulties (one possibly related to pregnancy complications) and four with other mild extra-ocular anomalies; the observed systemic anomalies reveal no consistent patterns and, for consanguineous families, may be related to the increased likelihood of homozygozity at other loci. In terms of ocular features, the majority of patients carrying two recessive mutations in FOXE3 are affected with microphthalmia and significant corneal defects (Table II); eight patients were diagnosed with aphakia, but the anterior segment could not be visualized in five of the remaining probands due to the corneal opacity or lack of availability for examination; interestingly, affected relatives in two families were reported with aphakia and normal eye size along with homozygous FOXE3 mutations [Iseri et al., 2009]. FOXE3 does not appear to contribute to microphthalmia with normal anterior segment structures nor to anophthalmia.
Since FOXE3 mutations are associated with the recessive form of microphthalmia, we analyzed our population for pedigrees suggestive of recessive inheritance and identified eight families. Four families demonstrated consanguinity, each with a coefficient of inbreeding (F) of 1/16 (= first cousins) or greater; three of these families were found to have FOXE3 mutations (Families 2-4) and the only consanguineous family in which mutations in FOXE3 were not identified was a family affected with clinical anophthalmia, rather than microphthalmia. Therefore FOXE3 mutations were found in 100% (3/3) of consanguineous pedigrees with bilateral microphthalmia. In addition to the consanguineous families, there were four non-consanguineous pedigrees with healthy parents and affected sibling pairs who did not carry mutations in FOXE3; one sibling pair was affected with microphthalmia, aphakia, and anterior segment dysgenesis, two pairs with microphthalmia and cataract, and the final pair with microphthalmia (and glaucoma in one sibling). Since these are small pedigrees, it is impossible to definitively establish the mode of inheritance without identification of causative mutations. The occurrence of multiple affected siblings can be indicative of recessive inheritance or may be due to gonosomal mosaicism of a dominant mutation in one of the parents, as was shown for other anophthalmia/microphthalmia genes [Faivre et al., 2006; Ragge et al., 2005; Schneider et al., 2008]. Mutations in other regions of FOXE3 (regulatory regions that have yet to be identified) or other genetic factors ought to be responsible for the phenotype in the remaining cases.
While multiple genes have been linked to syndromic forms of autosomal recessive microphthalmia, only three other loci have been reported for isolated autosomal recessive microphthalmia (OMIM: 251600, 610093, 611038), each accounting for a small proportion of patients with anophthalmia/microphthalmia. Homozygous mutations in VSX2 (formerly CHX10, OMIM: 142993) have been identified in several consanguineous families of Middle-Eastern origin affected with nonsyndromic anophthalmia/microphthalmia, sometimes associated with cataract, anterior segment anomalies, or coloboma (2% of initial cohort of patients with anophthalmia/microphthalmia) [Ferda Percin et al., 2000; Bar-Yosef et al., 2004; Faiyaz-Ul-Haque et al., 2007], but no mutations were identified in a cohort of 198 children with anophthalmia/ microphthalmia or coloboma from Scotland [Morrison et al., 2002]. Two probands with nonsyndromic clinical anophthalmia (one unilateral, one bilateral) were reported with compound heterozygous mutations in RAX (OMIM: 601881) (1% of initial cohort of patients with anophthalmia/ microphthalmia) [Voronina et al., 2004; Lequeux et al., 2008]. In addition to these known genetic factors, the 14q32 region (OMIM: 251600) has also been identified as a locus for recessive microphthalmia in a large consanguineous family from Pakistan [Bessant et al., 1999]; the phenotype, consisting of nonsyndromic bilateral microphthalmia with severe corneal defects but no evidence of lenticular anomalies, overlaps that seen in patients with FOXE3 mutations.
No autosomal dominant mutations in FOXE3 were observed within our proband population that included 26 patients with Peters’ anomaly and/or cataract, consistent with the previously reported dominant FOXE3 phenotypes. It is interesting that both heterozygous and homozygous mutations in FOXE3 can result in ocular disease, yet heterozygous carriers of the homozygous mutations described in this paper, by Valleix et al. [2006], and by Iseri et al. [2009] are unaffected, with the exception of the mother of Patient 2, who carries a single copy of the c.557delT mutation and is reported to have unilateral corneal ‘cloudiness’ and decreased visual acuity. The heterozygous FOXE3 disease-causing mutations may operate through a dominant-negative mechanism; the previously reported mutations which cause dominant disease are missense or late truncating mutations, while the recessive FOXE3 mutations are mainly nonsense/frameshift mutations earlier in the coding region (Figure 2). However, the mother of Patient 1 carries a missense mutation (M82V) that changes a highly conserved amino acid in the FOXE3 DNA-binding forkhead domain, similar to the previously reported dominantly inherited Peters’ anomaly variant (R90L) (Figure 2) and is unaffected. Another possible explanation for the phenotypic variability of FOXE3 mutations is the presence of modifying mutations at other genetic loci that can vary between families/individuals. Further research is needed to determine why some heterozygous mutations result in disease while others only manifest symptoms when present in homozygous (or compound heterozygote) form.
Interestingly, several FOXE3 variants appear to be race-specific, underscoring the importance of utilizing race/ethnicity-matched control populations in evaluating the relevance of genetic screening results. The c.601G>A (p.V201M) variant was observed in patients and controls of Hispanic background and one patient of Asian background, but not in other groups; the c.898A>G (p.S300G) substitution was seen in patients and controls of Caucasian origin only. Finally, the c.146G>C (p.G49A) variant was only found in African American patients and controls (Table I). The c.146G>C change has been previously described by Iseri and colleagues [2009] in a family with a highly variable phenotype ranging from unilateral microphthalmia and coloboma in the proband to late-onset cataract in the maternal grandmother. The race/ethnicity of this family and control samples included in the study was not reported. In our study, the c.146G>C change was detected in an African-American patient affected with a Peters plus-like condition (involving multiple extraocular features), his unaffected brother, and four control individuals of African American descent. Although it is possible that this variant results in a highly variable phenotype which includes some very mild and/or late-onset conditions, therefore explaining the presence of this variant in an unaffected brother and race-matched control individuals, overall it appears to be more plausible that this change represents a normal race-specific variation in FOXE3 gene. Further functional studies and mutation analysis of affected and unaffected individuals of varied racial/ethnic backgrounds is needed to define what role, if any, this and other FOXE3 variants play in ocular disease.
In conclusion, our results demonstrate an important role for FOXE3 in nonsyndromic bilateral microphthalmia. FOXE3-associated microphthalmia is accompanied by aphakia and/or corneal defects and demonstrates an autosomal recessive mode of inheritance. Mutations in FOXE3 do not appear to be involved in anophthalmia, microphthalmia with normal anterior segment structures, or syndromic eye anomalies.
Supplementary Material
Pedigree structure and DNA sequencing chromatograms are shown for patients 1 (A), 2 (B), 3 (C) and 4-5 (D). Mutation positions are indicated with arrows; genotypes identified for different family members are shown.
Acknowledgments
We are grateful to the families for their participation in this study and to our funding sources. This project was supported by awards EY013606 and EY015518 from the National Eye Institute and a grant from the Children’s Research Institute Foundation at Children’s Hospital of Wisconsin to EVS, GCRC Grant M01-RR00058 from NIH, as well as two grants from Mellon Mid-Atlantic Charitable Trusts, the Albert B. Millett Memorial Fund and the Rae S. Uber Trust, and a grant from the Gustavus and Louis Pfeiffer Research Foundation.
Footnotes
Web Resources
The URLs for data presented herein are as follows:
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/omim
NCBI Protein Database, http://www.ncbi.nlm.nih.gov/protein/
References
- Bar-Yosef U, Abuelaish I, Harel T, Hendler N, Ofir R, Birk OS. CHX10 mutations cause nonsyndromic microphthalmia/anophthalmia in Arab and Jewish kindreds. Hum Genet. 2004;115:302–309. doi: 10.1007/s00439-004-1154-2. [DOI] [PubMed] [Google Scholar]
- Bessant DA, Anwar K, Khaliq S, Hameed A, Ismail M, Payne AM, Mehdi SQ, Bhattacharya SS. Phenotype of autosomal recessive congenital microphthalmia mapping to chromosome 14q32. Br J Ophthalmol. 1999;83:919–922. doi: 10.1136/bjo.83.8.919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blixt A, Mahlapuu M, Aitola M, Pelto-Huikko M, Enerback S, Carlsson P. A forkhead gene, FoxE3, is essential for lens epithelial proliferation and closure of the lens vesicle. Genes Dev. 2000;14:245–254. [PMC free article] [PubMed] [Google Scholar]
- Brownell I, Dirksen M, Jamrich M. Forkhead Foxe3 maps to the dysgenetic lens locus and is critical in lens development and differentiation. Genesis. 2000;27:81–93. doi: 10.1002/1526-968x(200006)27:2<81::aid-gene50>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- Faivre L, Williamson KA, Faber V, Laurent N, Grimaldi M, Thauvin-Robinet C, Durand C, Mugneret F, Gouyon JB, Bron A, Huet F, Hayward C, Heyningen V, Fitzpatrick DR. Recurrence of SOX2 anophthalmia syndrome with gonosomal mosaicism in a phenotypically normal mother. Am J Med Genet A. 2006;140:636–639. doi: 10.1002/ajmg.a.31114. [DOI] [PubMed] [Google Scholar]
- Faiyaz-Ul-Haque M, Zaidi SH, Al-Mureikhi MS, Peltekova I, Tsui LC, Teebi AS. Mutations in the CHX10 gene in nonsyndromic microphthalmia/anophthalmia patients from qatar. Clin Genet. 2007;72:164–166. doi: 10.1111/j.1399-0004.2007.00846.x. [DOI] [PubMed] [Google Scholar]
- Ferda Percin E, Ploder LA, Yu JJ, Arici K, Horsford DJ, Rutherford A, Bapat B, Cox DW, Duncan AM, Kalnins VI, Kocak-Altintas A, Sowden JC, Traboulsi E, Sarfarazi M, McInnes RR. Human microphthalmia associated with mutations in the retinal homeobox gene CHX10. Nat Genet. 2000;25:397–401. doi: 10.1038/78071. [DOI] [PubMed] [Google Scholar]
- Iseri SU, Osborne RJ, Farrall M, Wyatt AW, Mirza G, Nurnberg G, Kluck C, Herbert H, Martin A, Hussain MS, Collin JR, Lathrop M, Nurnberg P, Ragoussis J, Ragge NK. Seeing clearly: The dominant and recessive nature of FOXE3 in eye developmental anomalies. Hum Mutat. 2009;30:1378–1386. doi: 10.1002/humu.21079. [DOI] [PubMed] [Google Scholar]
- Lequeux L, Rio M, Vigouroux A, Titeux M, Etchevers H, Malecaze F, Chassaing N, Calvas P. Confirmation of RAX gene involvement in human anophthalmia. Clin Genet. 2008;74:392–395. doi: 10.1111/j.1399-0004.2008.01078.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison D, FitzPatrick D, Hanson I, Williamson K, van Heyningen V, Fleck B, Jones I, Chalmers J, Campbell H. National study of microphthalmia, anophthalmia, and coloboma (MAC) in scotland: Investigation of genetic aetiology. J Med Genet. 2002;39:16–22. doi: 10.1136/jmg.39.1.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ormestad M, Blixt A, Churchill A, Martinsson T, Enerback S, Carlsson P. Foxe3 haploinsufficiency in mice: A model for peters’ anomaly. Invest Ophthalmol Vis Sci. 2002;43:1350–1357. [PubMed] [Google Scholar]
- Ragge NK, Brown AG, Poloschek CM, Lorenz B, Henderson RA, Clarke MP, Russell-Eggitt I, Fielder A, Gerrelli D, Martinez-Barbera JP, Ruddle P, Hurst J, Collin JR, Salt A, Cooper ST, Thompson PJ, Sisodiya SM, Williamson KA, Fitzpatrick DR, van Heyningen V, Hanson IM. Heterozygous mutations of OTX2 cause severe ocular malformations. Am J Hum Genet. 2005;76:1008–1022. doi: 10.1086/430721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reis LM, Tyler RC, Abdul-Rahman O, Trapane P, Wallerstein R, Broome D, Hoffman J, Khan A, Paradiso C, Ron N, Bergner A, Semina EV. Mutation analysis of B3GALTL in Peters Plus Syndrome. Am J Med Genet A. 2008;146A:2603–2610. doi: 10.1002/ajmg.a.32498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanyal S, Hawkins RK. Dysgenetic lens (dyl)--a new gene in the mouse. Invest Ophthalmol Vis Sci. 1979;18:642–645. [PubMed] [Google Scholar]
- Semina EV, Brownell I, Mintz-Hittner HA, Murray JC, Jamrich M. Mutations in the human forkhead transcription factor FOXE3 associated with anterior segment ocular dysgenesis and cataracts. Hum Mol Genet. 2001;10:231–236. doi: 10.1093/hmg/10.3.231. [DOI] [PubMed] [Google Scholar]
- Schneider A, Bardakjian TM, Zhou J, Hughes N, Keep R, Dorsainville D, Kherani F, Katowitz J, Schimmenti LA, Hummel M, Fitzpatrick DR, Young TL. Familial recurrence of SOX2 anophthalmia syndrome: Phenotypically normal mother with two affected daughters. Am J Med Genet A. 2008;146A:2794–2798. doi: 10.1002/ajmg.a.32384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi X, Luo Y, Howley S, Dzialo A, Foley S, Hyde DR, Vihtelic TS. Zebrafish foxe3: Roles in ocular lens morphogenesis through interaction with pitx3. Mech Dev. 2006;123:761–782. doi: 10.1016/j.mod.2006.07.004. [DOI] [PubMed] [Google Scholar]
- Valleix S, Niel F, Nedelec B, Algros MP, Schwartz C, Delbosc B, Delpech M, Kantelip B. Homozygous nonsense mutation in the FOXE3 gene as a cause of congenital primary aphakia in humans. Am J Hum Genet. 2006;79:358–364. doi: 10.1086/505654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voronina VA, Kozhemyakina EA, O’Kernick CM, Kahn ND, Wenger SL, Linberg JV, Schneider AS, Mathers PH. Mutations in the human RAX homeobox gene in a patient with anophthalmia and sclerocornea. Hum Mol Genet. 2004;13:315–322. doi: 10.1093/hmg/ddh025. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Pedigree structure and DNA sequencing chromatograms are shown for patients 1 (A), 2 (B), 3 (C) and 4-5 (D). Mutation positions are indicated with arrows; genotypes identified for different family members are shown.