

Abstract
Homocamptothecins (hCPTs) are a novel class of topoisomerase I (Top1) inhibitors with enhanced chemical stability compared with the currently used camptothecin (CPT) analogs irinotecan and topotecan. The hCPT derivative diflomotecan (BN80915) is currently in clinical trials. We established two resistant human glioblastoma cell lines, SF295/hCPT50 and SF295/BN50, by stepwise exposure of the parental SF295 line to increasing concentrations of hCPT and BN80915, respectively. The two resistant cell lines were 15- to 22-fold resistant to hCPT and BN80915 as well as 7- to 27-fold cross-resistant to other Top1 inhibitors, including CPT, topotecan, and the indenoisoquinolines MJ-III-65 (NSC 706744) and NSC 724998, but sensitive to the topoisomerase II inhibitors mitoxantrone and etoposide. Neither of the resistant cell lines displayed any detectable expression of the three major drug transporters P-glycoprotien, multidrug resistance-associated protein 1, or ATP-binding cassette, subfamily G (WHITE), member 2, as assessed by immunoblot or flow cytometry. Reduced expression of Top1 protein occurred at the transcriptional level in both of the resistant cell lines, consistent with the reduction of Top1 enzyme level as the major contribution to the resistance phenotype in SF295/hCPT50 and SF295/BN50 cells. Treatment of the resistant cell lines with the histone deacetylase inhibitor depsipeptide or the DNA methyltransferase inhibitor 5-aza-2′-deoxycytidine alone or concomitantly did not result in re-expression of Top1. Our studies suggest that selection for resistance to hCPT or BN80915 is primarily related to reduced Top1 expression at the transcriptional level, resulting in reduced enzyme levels.
Homocamptothecins (hCPTs) are a new class of camptothecin (CPT) derivatives. They are different from the conventional CPT in that they bear a seven-membered β-hydroxylactone ring instead of the naturally occurring six-membered α-hydroxylactone observed in CPT (see Fig. 1). hCPTs are therefore more stable than CPT derivatives (Lavergne et al., 2000a,b) that are readily hydrolyzed to the inactive carboxylate forms (for review, see Pommier, 2006). hCPT, like other CPT analogs, inhibits Top1 by stabilizing the covalent complex between Top1 and DNA, leading to enzyme-linked DNA strand breaks, also referred to as cleavage complexes (Pommier, 2006). Because of their increased chemical stability, hCPTs were expected to exert greater Top1 inhibition and antitumor efficacy than the current FDA-approved CPT analogs irinotecan and topotecan [for review, see Lavergne et al. (2000b); Pommier (2006)]. hCPTs showed very potent antitumor efficacy in many tumors, including brain glioblastoma mouse models. The fluorinated hCPT diflomotecan (BN80915) is one of the most active compounds in terms of antiproliferative activity in the hCPT series (Bailly et al., 1999; Lavergne et al., 2000b). BN80915 has been selected for further development as a highly active hCPT-based Top1 poison with an optimal balance between potency and stability. Oral and i.v. formulations of BN80915 are currently undergoing clinical evaluation in Europe (Gelderblom et al., 2003; Troconiz et al., 2006).
Fig. 1.
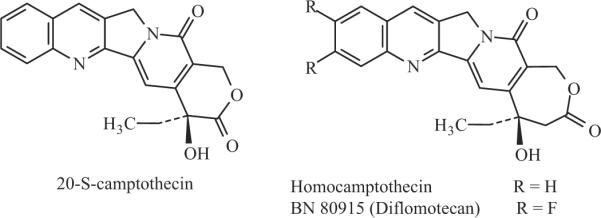
Chemical structures of camptothecin, homocamptothecin, and diflomotecan (BN80915).
Glioblastoma is the most common malignant brain tumor in adults and is among the most lethal of all cancers (Chamberlain, 2006). Chemotherapy is one of the accepted therapeutic strategies for glioblastoma multiforme (Parney and Chang, 2003; Vassal et al., 2003; Chamberlain, 2006). The Top1 inhibitor CPT-11 (irinotecan) is used as a second line drug for the treatment of glioblastoma multiforme. Response rates to this drug in glioblastoma patients have been 14 to 15% of cases, and stable disease has been achieved in 14 to 55% (Friedman et al., 1999; Chamberlain, 2002; Cloughesy et al., 2003).
To study potential mechanisms of resistance to hCPTs that may be encountered in the clinic, we established two drugresistant human glioblastoma sublines, SF295/hCPT50 and SF295/BN50 cell lines, by stepwise exposure of parental SF295 cells to increasing concentrations of hCPT and BN80915, respectively. Herein, we report that resistance to hCPTs in the SF295/hCPT50 and SF295/BN50 sublines is associated with reduced expression of Top1 protein, which occurs at the transcription level.
Materials and Methods
Chemicals
Homocamptothecin and BN80915 were obtained from Dr. Paola Principe (Beaufour-Ipsen, Paris, France). NSC724998 and MJ-III-65 were from the National Cancer Institute Anticancer Drug Screen (Bethesda, MD). Topotecan was from LKT Laboratories (St. Paul, MN). Mitoxantrone, camptothecin, etoposide, and bromodeoxyuridine were purchased from Sigma (St. Louis, MO). Calcein-AM was from Invitrogen Corporation (Carlsbad, CA). Pheophorbide A was purchased from Frontier Scientific (Logan, UT). Fumitremorgin C was isolated by Thomas McCloud, Developmental Therapeutics Program, Natural Products Extraction Laboratory, National Institutes of Health (Bethesda, MD).
Cell Lines
The parental SF295 human glioblastoma cell line was obtained from the National Institutes of Health Anticancer Drug Screen and was maintained in RPMI 1640 medium supplemented with 10% fetal calf serum with penicillin/streptomycin. The drug-selected SF295/BN50 and SF295/hCPT50 sublines were developed by stepwise selection and are maintained in 50 nM BN80915 or hCPT, respectively. Human breast carcinoma MCF-7 cell lines were maintained in Richter's medium with 10% fetal calf serum and penicillin/streptomycin. ABCG2-overexpresing MCF-7/FLV1000 cells (Robey et al., 2001b) and multidrug resistance-associated protein 1 (MRP1)-over-expressing cells (Schneider et al., 1994) were additionally maintained in 1.0 μM flavopiridol or 4.0 μM etoposide, respectively. P-glycoprotein (Pgp)-overexpressing MCF-7/Tx200 cells are maintained in 200 ng/ml paclitaxel (Robey et al., 2004). Human embryonic kidney (HEK) 293 cells transfected with MDR1 (ABCB1), MRP1 (ABCC1), or ABCG2 were maintained in Eagle's minimum essential medium with 10% fetal calf serum and penicillin/streptomycin (Robey et al., 2006). Expression of transporter proteins was enforced by addition of 2 mg/ml G418.
Cytotoxicity Assays
Four-day cytotoxicity assays were performed using the sulforhodamine B assay (Robey et al., 2006). In brief, cells were plated in flat-bottomed, 96-well plates at a density of 2500 cells per well and allowed to attach for 24 h at 37°C in 5% CO2. Chemotherapeutic agents at various concentrations were added to the cells, and the plates were allowed to incubate for 96 h at 37°C in 5% CO2. Cells were subsequently fixed in 50% trichloroacetic acid at 4°C for 1 h, after which the plates were washed in water and allowed to dry. The plates were subsequently stained with sulforhodamine B solution [0.4% sulforhodamine B (w/v) in 1% acetic acid] and washed in 1% acetic acid in water. Sulforhodamine B was then solubilized and optical densities were read on a plate reader (Bio-Rad Laboratories, Hercules, CA) at an absorbance of 540 nm. Each concentration was tested in quadruplicate and controls were performed in replicates of eight.
DNA-Protein Cross-Links Analysis
DPCs induced by CPT were measured by alkaline elution assays (Kohn, 1996) in SF295 and SF295/BN cells. Proliferating cells in log phase were labeled with [3H]thymidine (0.02 μCi/ml; PerkinElmer Life and Analytical Sciences, Waltham, MA) for 48 h, chased for 4 h in isotope-free medium, and exposed to CPT for 1 h at indicated concentrations. Equal number of cells were loaded onto polyvinyl chloride/acrylic copolymer membrane filters (2-μm pore size, 25-mm diameter; Nucleopore Corporation, Livermore, CA), lysed, and subjected to alkaline elution. Cells were lysed with 5 ml of SDS-lysis solution (0.1 M glycine, 2% SDS, and 0.025 M Na2EDTA, pH 10.0). After washing with 0.02 M EDTA, pH 10.0, cell lysates were eluted by 40 ml of an eluting solution containing 0.02 M H4EDTA, 2% tetrapropyl ammonium hydroxide (Sigma), pH 12.1. Five fractions were collected with approximately 5 ml in each. After collection, filters were placed in scintillation vials to which 0.4 ml of 1 N HCl was added in each vial. The vials were sealed and heated at 60°C for 1 h to depurinate the DNA. After removing the vials from the oven, 2.5 ml of 0.4 N NaOH was added for 1 h at room temperature to release the labeled DNA from the filters. Radioactivity in the elution fractions was counted by adding 10 ml of Aquassure (PerkinElmer Life and Analytical Sciences) containing 0.7% glacial acetic acid into each vial. Filter retention rate was calculated and plotted versus elution time. The formula is: DPC = [(1 - r)-/-1 - (1 - r0)-/-1 ] × 3000, where r0 is the retention for drug-treated cells and r0 is the retention for cells that have been irradiated with 3000 rads (control cells).
Western Blot for Top1, Top2α, Top2β, and Drug Transporter Expression
Cells were lysed at 4°C in buffer containing 1% SDS, 1 mM sodium vanadate, and 10 mM Tris-HCl, pH 7.4, supplemented with protease inhibitors (Complete; Roche Diagnostics, Indianapolis, IN) and phosphatase inhibitors (Sigma). Viscosity of the samples was reduced by brief sonication, and 30 μg of protein was incubated in loading buffer (125 mM Tris-HCl, pH 6.8, 10% β-mercaptoethanol, 4.6% SDS, 20% glycerol, and 0.003% bromphenol blue), separated by SDS-polyacrylamide gel electrophoresis (SDS-PAGE), and transferred to polyvinylidene difluoride membrane (Immobilon-P, Millipore, MA). After blocking nonspecific binding sites for 1 h with 5% milk in PBS-T (phosphate-buffered saline, 0.5% Tween 20), the membrane was incubated for 1 h with primary antibody under the following conditions: mouse monoclonal anti-Top1 (C21 antibody from Dr. Yung-Chi Cheng, Yale University, New Haven, CT) at 1:1000 dilution, rabbit anti-Top2α (Abcam, Cambridge, MA) at 1/10,000 dilution, mouse anti-Top2β (BD Biosciences, San Jose, CA) at 1/1000 dilution, or mouse anti-β-actin (Abcam) at 1:5000 dilution. After three washes in PBS-T, the membrane was incubated with horseradish peroxidase-conjugated goat anti-rabbit (1:5000 dilution) or anti-mouse (1:5000 dilution) antibody (GE Healthcare, Chalfont St. Giles, Buckinghamshire, UK) for 1 h and then washed three times in PBS-T. Immunoblot was performed using an enhanced chemiluminescence detection kit (Pierce, Rockford, IL) by autoradiography.
For determination of drug transporter expression by immunoblot, microsomal membrane protein (30 μg) was obtained by nitrogen cavitation, separated by 7.5% (w/v) SDS-PAGE, and transferred to a polyvinylidene difluoride membrane. The membrane was then probed with the anti-Pgp antibody C219 (Signet Laboratories, Deadham, MA) and subjected to enhanced chemiluminescence detection. After stripping with 0.2 M NaOH, the blot was subsequently probed with the anti-MRP1 antibody MRPm6 (Kamiya Biomedical, Seattle, WA) and the anti-ABCG2 antibody BXP-21 (Kamiya Biomedical) in the same manner.
Flow Cytometry Determination of Drug Transporter Expression
Expression of drug transporters was determined by flow cytometry as described previously (Robey et al., 2004). In brief, cells were incubated for 30 min in complete medium (phenol red-free Richter's medium with 10% fetal calf serum) containing 0.5 μM rhodamine with or without 3 μg/ml valspodar, 200 nM calcein-AM with or without 50 μM MK-571; or 10 μM pheophorbide A with or without 10 μM fumitremorgin C (FTC) to determine P-glycoprotein, MRP1, or ABCG2 expression, respectively. Subsequently, cells were washed and incubated for 1 h in substrate-free medium continuing with or without inhibitor. Intracellular fluorescence of rhodamine, calcein, or pheophorbide A was measured with a FACSort flow cytometer (BD Biosciences, San Jose, CA) equipped with an argon laser at 488 nm and a red diode laser at 635 nm. Fluorescence histograms were generated with CellQuest Software (BD Biosciences). At least two independent experiments were performed.
Real-Time Quantitative PCR and Semiquantitative RT-PCR
Cells were lysed and total RNA was extracted using RNAqueous-4PCR (Ambion, Austin, TX). Total RNA was reverse-transcribed using RETROscript kit (Ambion). Real-time quantitative PCR was done using ABsolute QPCR Mixes (Abgene, Rochester, NY) on an ABI 7900 real-time PCR instrument (AME Bioscience, Chicago, IL). Thermal cycling conditions were 50°C for 2 min, 95°C for 15 min, then 40 cycles of 95°C for 15 s and 60°C for 1 min. Primers and probe sequences used for Top1 were TGACAGCCCCGGATGAGA (sense), TGCAACAGCTCGATTGGC (antisense), 5-carboxyfluorescein- CATCCCAGCGAAGATCCTTTCTTATAACCG-5-carboxytetramethylrhodamine (probe), and for 18S RNA were GATTAAGTCCCTGCCCTTTGTACA (sense), GATCCGAGGGCCTCACTAAAC (antisense), 5-carboxyfluorescein-CGCCCGTCGCTACTACCGATTGG-carboxytetramethylrhodamine (probe). Gene expression was analyzed using Sequence Detection Systems software, version 1.7 (Prism; Applied Biosystems, Foster City, CA). mRNA levels of Top1 were normalized to the 18S RNA internal standard. For semiquantitative RT-PCR, amplification of cDNA was done using primers specific for Top1 and GAPDH. Primers specific for Top1 cDNA amplification were 5′-AGCCCAGACGGAAGC-3′ (forward) and 5′-TCCAGGAAACCAGCCA-3′ (reverse). The primer pair specific for GAPDH amplification was 5′-ACCACAGTCCATGCCATCAC (forward) and 5′-TCCACCACCCTGTTGCTGTA (reverse). Amplification of GAPDH cDNA served as an internal control. PCR amplification for the Top1 and GAPDH mRNA was performed at an annealing temperature of 55°C for 25 cycles to yield 320- and 440-base pair products, respectively. The PCR products were resolved on 2% agarose gel, stained with ethidium bromide, and quantitated.
BrdU Incorporation and Cell Cycle Analysis
After a 30-min pulse with 50 μM BrdU, the culture medium was removed, and the cells were washed with PBS and incubated with fresh medium for the indicated times. Cells were then harvested by trypsinization and washed twice with PBS. The pellet containing 2 to 4 × 106 cells was suspended in 200 μl of PBS and fixed with 5 ml of ice-cold 70% ethanol. After overnight storage at -20°C, cells were centrifuged and the pellet was suspended in 3 ml of 2 N HCl. After a 30-min incubation at room temperature, the medium was neutralized by the addition of 6 ml of 0.1 M sodium borate, pH 8.5. Cells were centrifuged and the pellet was washed twice in PBS containing 0.5% Tween 20 and 0.5% bovine serum albumin. Cells were incubated with 15 μl of anti BrdU-fluorescein isothiocyanate (BD Biosciences) for 1 h in the dark at room temperature. To determine DNA content, 500 μl of staining solution containing 20 μg/ml propidium iodide and 50 units of RNase A in PBS was added to the pellet. Cells were analyzed with a FACScan flow cytometer (BD Biosciences) using the CellQuest software (BD Biosciences).
Results
Establishment and Characterization of hCPT- and BN80915-Resistant Cells
hCPT- and BN80915-resistant cells were generated from SF295 glioblastoma cells by a stepwise increase in exposure to hCPT and BN80915, respectively. These cell lines were maintained in medium containing 50 nM hCPT or BN80915. The resistant cell lines were maintained in constant culture with selecting drug for at least 4 months before characterization by cytotoxicity assay; thus, we considered them to be a homogeneous population of cells.
To determine whether the drug-selected cell lines were cross-resistant to other Top1 inhibitors, 4-day cytotoxicity assays were performed with other Top1 inhibitors as well as Top2 inhibitors. As seen in Table 1, the parental SF295 cells were 35-fold more sensitive to hCPT and 8-fold more sensitive to BN80915 compared with topotecan, although they were comparably resistant to CPT. SF295/hCPT50 cells were comparably resistant to hCPT and BN80915 (21- and 22-fold, respectively); similarly, SF295/BN50 cells were comparably resistant to both hCPT and BN80915 (15- and 21-fold, respectively). Both resistant sublines also demonstrated 9- to 20-fold cross-resistance to the indenoisoquinoline non-CPT Top1 inhibitors NSC 724998 and MJ-III-65 (NSC 706744) (Pommier, 2006) (Table 1). On the other hand, both SF295/hCPT50 and SF295/BN50 cells showed 3-fold higher sensitivity to the Top2 inhibitor mitoxantrone. SF295/BN50 cells only showed slightly increased sensitivity to etopside (VP-16), whereas SF295/hCPT50 was slightly resistant to etoposide (VP-16) (Table 1).
TABLE 1.
Drug sensitivity of parent (SF295) and BN80915 or hCPT-resistant (SF295/BN50 or SF295/hCPT50) human glioblastoma cancer cell lines to various anticancer drugs Drug sensitivity was determined by the SRB assay as outlined under Materials and Methods. Degree of resistance (DR) was calculated by dividing the IC50 value of the resistant lines by that of the corresponding parental lines.
| Drug | n | IC50 |
DR | IC50 SF295/hCPT50 | DR | |
|---|---|---|---|---|---|---|
| SF295 | SF295/BN50 | |||||
| BN80915 | 3 | 0.3 ± 0.06 | 6.3 ± 4.0 | 101 | 6.5 ± 4.3 | 22 |
| hCPT | 3 | 1.3 ± 0.6 | 20 ± 10 | 15 | 27 ± 15 | 21 |
| TPT | 3 | 10.7 ± 3.8 | 70 ± 10 | 7 | 83.3 ± 15.3 | 8 |
| CPT | 4 | 1.1 ± 0.3 | 26.3 ± 7.5 | 24 | 30 ± 8.1 | 27 |
| NSC 724998 | 3 | 0.08 ± 0.03 | 1.2 ± 0.7 | 15 | 0.7 ± 0.3 | 9 |
| MJ-III-65 | 2 | 0.006 ± 0.005 | 0.13 ± 0.11 | 22 | 0.08 ± 0.03 | 13 |
| MX | 3 | 4.7 ± 1.5 | 1.6 ± 0.8 | 0.3 | 1.7 ± 1.0 | 0.3 |
| Etoposide | 3 | 0.7 ± 0.3 | 0.5 ± 0.2 | 0.7 | 1.1 ± 0.1 | 1.4 |
TPT, topotecan; MX, mitoxantrone.
Lack of Expression of Drug Transporters in the Resistant Cells
Several groups have shown that the ATP-binding cassette (ABC) proteins P-glycoprotein (Pgp), multidrug resistance-associated protein 1 (MRP1), and ABCG2 can mediate multidrug resistance to cytotoxic drugs such as CPTs (Chen et al., 1991, 1999; Hoki et al., 1997; Chen et al., 1999) and to a lesser extent to hCPTs (Chauvier et al., 2002; Bates et al., 2004). Using MCF-7/FLV1000 (expressing ABCG2), MCF-7/VP (expressing MRP1), and MCF-7/TX200 (expressing Pgp) cells as positive controls, we observed no detectable expression of MRP1, ABCG2, or Pgp in either parental SF295 line or either of the drug-selected lines (Fig. 2A). Protein loading is shown by Ponceau staining in Fig. 2B.
Fig. 2.

Analysis of drug transporter protein expression in human glioblastoma SF295 and drug-resistant SF295/BN50 and SF295/hCPT50 cells. A, for qualitative analysis of the three drug transporter proteins expression in these cell lines, 30 μg of microsomal proteins were separated by SDS-PAGE and probed with anti-Pgp, -MRP1, and -ABCG2 antibodies as described under Materials and Methods. B, Ponceau staining of membrane shown in A. C, SF295 parental and resistant sublines from A were incubated with 0.5 μg/ml rhodamine 123 in the presence (dashed line) or absence (solid line) of 3 μg/ml valspodar (column 1); 200 nM calcein AM in the presence (dashed line) or absence (solid line) of 50 μM MK571 (second column); or 10 μM pheophorbide A in the presence (dashed line) or absence (solid line) of 10 μM FTC (last column) to detect Pgp, MRP1, or ABCG2, respectively. ABCB1-, ABCC1-, or ABCG2-transfected HEK293 cells served as positive controls for Pgp, MRP1, and ABCG2, respectively (+ Control, top row of histograms). Representative results from one of two independent experiments are shown.
We next measured Pgp, MRP1, and ABCG2 expression by a flow cytometry-based functional assay, which, we have concluded, can detect small changes in transporter expression that may not be detected by immunoblot analysis. As shown in column 1 of Fig. 2C, Pgp-overexpressing HEK293 cells readily transport rhodamine 123, as shown by the difference between the histograms depicting cells incubated without (solid line) or with (dashed line) 3 μg/ml valspodar, a Pgp inhibitor. Pgp function was not detected in the SF295 parental cells, given that the solid and dashed histograms overlap. Pgp expression in the drug-selected BN50 and hCPT50 sublines was similar to that of the parental cells, suggesting that Pgp expression was not increased in the sublines. Likewise, in the second column of Fig. 2C, MRP1-overexpressing HEK293 cells readily transport the fluorescent compound calcein AM when incubated without (solid line) or with 25 μM MK571, a MRP1 inhibitor. Parental SF295 cells and the drug-selected sublines all displayed similarly low levels of MRP1 expression. Finally, in the last column of Fig. 2C, ABCG2-overexpressing HEK293 cells readily transport the fluorescent compound pheophorbide A, shown by the difference in the histograms generated by incubating cells in pheophorbide alone (solid line) or with 10 μM FTC. Parental SF295 cells also express ABCG2, consistent with previous results (Robey et al., 2001a). In the drug-selected sublines, however, ABCG2 expression seems to be decreased compared with parental cells as evidenced by the smaller difference between the solid and dashed histgrams. Thus, the flow-cytometry results support the data obtained by immunoblot, suggesting that neither Pgp, MRP1, nor ABCG2 is up-regulated in the drug-selected sublines. These results suggested that the three major drug transporters associated with resistance to CPTs and hCPTs were not factors contributing to the resistance phenotype in the SF295/hCPT50 or SF295/BN50 sublines.
Reduced Formation of DNA-Protein Crosslinks Induced by CPT in Resistant SF295/BN50 cells
Because drug transporters were not implicated as the cause for drugresistance in the hCPT-selected cells, we next examined other possible mechanisms of resistance. We measured directly the formation of DNA-protein crosslinks (DPC) in parent SF295 and SF295/BN50 resistant cells after different concentrations of CPT treatment with the alkaline elution assay. The amounts of DPC in SF295/BN50 cells were approximately one third of those in SF295 cells after 1-h exposures to 0.1 to 1.0 μM CPT (Fig. 3). This finding demonstrated that drug resistance of the SF295/hCPT50 and SF295/BN50 cells was related to reduced formation of Top1 cleavage complexes, which is generally the mechanism of resistance to CPTs (Pommier et al., 1999).
Fig. 3.

DNA-protein crosslinks (DPCs) induced by CPT in human glioblastoma SF295 and BN80915-resistant SF295/BN50 cells. A, cells were prelabeled with [3H]thymidine and were then treated with 0.1, 0.3, and 1.0 μMCPT for 1 h at 37°C as described under Materials and Methods. DPCs were assayed by alkaline elution under nondeproteinizing conditions after ionizing radiation of cells with 3000 rads for reference (squares); or treatment with 0.1 μM CPT (circles); 0.3 μM CPT (triangles) or 1.0 μM CPT (diamonds) in SF295 (closed symbols) and SF295/BN50 (opened symbols) cells. B, DPCs were measured by alkaline elution assay and are expressed as rad equivalents at 12 h elution time in SF295 (◯) and SF295/BN50 (◻) cells. The average DPCs from two independent experiments are presented. Bars, S.E.M.
Reduced Expression of Top1 Protein and mRNA
Because we have previously observed decreased Top1 expression in CPT-selected cell lines (Fujimori et al., 1995), we next examined the hCPT and BN80915-selected lines for changes in Top1 expression. Top1 protein levels were clearly decreased in both SF295/hCPT50 and SF295/BN50 sublines (Fig. 4A). However, Top2 levels were not significantly increased (Fig. 4, B and C). We then asked whether the observed reduction of the Top1 protein was the result of reduced Top1 mRNA levels. Quantitative RT-PCR analysis of total RNA extracted from exponentially growing parent SF295, resistant SF295/hCPT50, and SF295/BN50 cells revealed consistently reduced (<50%) levels of Top1 mRNA in the resistant cells compared with SF295 cells (Fig. 4D). The mRNA results are consistent with the protein data, suggesting that the down-regulation of Top1 in the resistant cells occurs at thetranscriptional level.
Fig. 4.

Top1 (A), Top2α (B), and Top2β (C) protein levels by Western blotting analysis, Top1 mRNA levels by real time quantitative-PCR analysis (D), and effect of decitabine (1 μM) and depsipeptide (2 ng/ml), alone or in combination, on mRNA expression of Top1 by semiquantitative RT-PCR analysis (E) in SF295, SF295/BN50, and SF295/hCPT50 human glioblastoma cells. For Western blotting assay, whole-cell lysates (50 μg) isolated from each cell line were probed with monoclonal mouse Top1 and polyclonal Top2 antibodies as described under Materials and Methods. β-actin was probed to show equal loading. For real-time quantitative-PCR analysis, total RNA was extracted, and real-time quantitative-PCR was performed as described under Materials and Methods. mRNA levels were normalized with 18S RNA and representative of at least two independent experiments. For semiquantitative RT-PCR analysis of Top1 in SF295 parental cells and the respective resistant sublines SF295/BN50 and SF295/hCPT50, cells were untreated or treated with either decitabine (1 μM) for 4 days, depsipeptide (2 ng/ml) for 1 day, or a combination of the two drugs. Representative results from three independent experiments are shown. The numbers indicates the -fold difference of Top1 relative to the untreated SF295 parental cells after normalization to GAPDH.
Low Top1 Gene Expression in the Resistant Cell Lines Could Not Be Rescued by Decitabine and Depsipeptide Treatment
To determine whether the reduction of Top1 protein and mRNA levels in both of the hCPT and BN80915-selected sublines might have been caused by epigenetic changes, such as histone methylation or deacetylation, in the promoter region of Top1 gene in these resistant cell lines (Fujimori et al., 1995), we explored the capacity of the DNA methyltransferase inhibitor 5-aza-2′-deoxycytidine (decitabine) and the histone deacetylase inhibitor depsipeptide, alone and in combination, to reactivate expression of the Top1 gene. Parental SF295 and drug-selected cells were treated with 1.0 μM decitabine alone for 4 days, 2 ng/ml depsipeptide alone for 1 day, or concomitantly with the two drugs, and RNA was subsequently isolated. Top1 levels were then determined by RT-PCR and normalized to GAPDH expression. Ratios of the amount of amplified DNA for the drugtreated samples compared with the control (parental SF295 levels set to a value of 10) were calculated to express the relative level of gene expression after drug treatment. Although decitabine treatment enhanced the Top1 transcript level by two thirds in parent SF295 cells, it failed to increase its level in either the resistant SF295/BN50 or SF295/hCPT50 cells (Fig. 4E). Treatment with depsipeptide alone also failed to increase expression of Top1, as did incubation with both depsipeptide and decitabine simultaneously (Fig. 4E). These results suggest that the decrease in Top1 expression is not associated with epigenetic modifications such as DNA methylation or histone deacetylation.
BrdU Incorporation and Cell Cycle Analysis
BrdU incorporation and cell cycle analyses of SF295, SF295/BN50, and SF295/hCPT50 cells were performed by FACS. Figure 5A shows a representative DNA histogram of BrdU-stained cells at different time points. Because BrdU is incorporated instead of thymidine into the DNA during DNA synthesis in proliferating cells, the percentage of BrdU-positive cells reflects the percentage of total S-phase cells. With cell cycle analysis, we detected the percentage of S-phase cells in the three cell lines at various times after the initial BrdU pulse. BrdU-positive cells in S-phase were normalized to the percentage of cells that were positive 3 h after the pulse (Fig. 5B). Compared with the parent SF295 cells, both the SF295/BN50- and the SF295/hCPT50-resistant cells showed slower S-phase progression. At 8.5 h after the BrdU pulse, the amount of cells that had stepped out the S phase was approximately 90, 40, and 20% for SF295, SF295/BN50, and SF295/hCPT50 cells, respectively. Thereafter we measured doubling times for the three cell lines. We found that doubling times for SF295, SF295/BN50, and SF295/hCPT50 were 31.5, 99, and 69 h, respectively.
Fig. 5.

BrdU incorporation and cell cycle analysis in SF295, SF295/BN50, and SF295/hCPT50 cells. A, cells were collected at indicated times after BrdU pulse, and stained with anti-BrdU antibody coupled to FITC and propidium iodide/RNase A. B, quantification of BrdU-positive cells that are still in the S-phase at indicated times after the BrdU pulse. The first time point (3 h after the BrdU pulse) is set up at 100%. The average percentages are presented from four independent experiments. Bars, S.E.M.
Discussion
Most of the preclinical studies on drug resistance are carried out by using in vitro cellular models, including drugresistant cell lines selected from an originally sensitive counterpart. Resistance of cancer cells to CPT is multifactorial, involving reduced drug accumulation by overexpression of drug transporters (Bates et al., 2004), reduced expression of Top1 enzyme (Tan et al., 1989; Fujimori et al., 1996b), Top1 mutation (Pommier et al., 1999; Yanase et al., 2000; Urasaki et al., 2001), elevated DNA repair (Fujimori et al., 1996a; Pommier et al., 2006), and resistance to apoptosis (for review, see Pommier et al., 1999, 2006).
Besides our study, Eng et al. (1990) reported a camptothecin-resistant subline of P388 murine leukemia (P388/CPT) that was developed by repeated transplantation of P388 cells in mice treated with therapeutic doses of CPT and made hyper-resistant to CPT by passage in the presence of increasing concentrations of CPT. Both Top1 mRNA and the 100 kDa of Top1 enzyme levels were lower in these resistant cells. However, P388/CPT cells were not cross-resistant to other antineoplastic agents, including topoisomerase II inhibitors (Eng et al., 1990). It is noteworthy that the Top1-deficient leukemia P388/CPT45 cells were highly resistant to hCPT (Urasaki et al., 2000), which indicates that hCPT shares the same target Top1 with CPT. In P388/CPT45 cells, Top1 is not detectable by immunoblotting (Pourquier et al., 2000). In the present study, topotecan and CPT were much less effective, compared with hCPT and BN80915, in both SF295/hCPT- and SF295/BN50-resistant cells. These data are consistent with a previous report (Urasaki et al., 2000) that the antiproliferation activity of hCPT was greater than that of CPT in both parental and CPT-resistant cell lines. Taken together, these findings indicate that CPTresistant cells are cross-resistant to hCPT and the hCPT/BN80915-resistant cells show cross-resistance to CPTs, in both cases with reduced expression of common target Top1 at mRNA and protein level.
In the present study, both of the resistant SF295/BN50 and SF295/hCPT50 cell lines showed consistently reduced expression of Top1 protein and mRNA. The reduction of Top1 gene expression for drug resistance might be drug structure-specific, because no Top1 alteration was observed in a neuroblastoma model with in vivo-acquired resistance to irinotecan (Calvet et al., 2004). It is reasonable that the two resistant cell lines also show cross-resistance to other Top1 inhibitors because of decreased amount of their drug target, Top1. However, the cell lines resistant to hCPTs also showed increased sensitivity to Top2 inhibitors. These data suggest that Top2 inhibitors would be beneficial in patients with acquired resistance to hCPTs as a result of Top1 reduction after hCPTs treatment.
In addition to decreased Top1 levels, we noted a slower growth rate in our resistant cell lines. This phenomenon was also observed in a CPT-resistant subline of P388 leukemia cells with reduced Top1 content (Eng et al., 1990). In this study, slower growth rate did not seem to be a major mechanism of resistance to Top1 inhibitors because the BN- and hCPT-resistant cells are not resistant to Top2 inhibitors mitoxantrone and etoposide. Furthermore, drug treatment for cytotoxicity assays was performed by continuous drug exposure for 3 days. These extended drug exposure exceeded the cellular doubling times, which would minimize the impact of slower growth on the induction of DNA damage. Finally, DNA damage measured by alkaline elution showed reduced DNA-protein crosslinks in the resistant cells, which is most likely to contribute to drug resistance.
In mammalian cells, expression of ABC transporters such as Pgp (MDR1) and ABCG2 confers resistance to CPT and its derivatives (Hoki et al., 1997; Brangi et al., 1999; Rajendra et al., 2003). Although Pgp is not implicated in resistance to hCPTs (Lavergne et al., 2000a; Larsen et al., 2001), hCPTs have been shown to be subject to transport by MRP1 (Chauvier et al., 2002) and ABCG2 (Bates et al., 2004). In addition, selection with CPTs usually results in overexpression of ABCG2 (Maliepaard et al., 1999; Kawabata et al., 2001). In light of these previous findings, it is surprising that selection with hCPTs did not result in overexpression of ABC transporters. Thus, drug transporters may be not a major resistance challenge to hCPTs, and especially to BN80915, in clinical use.
Acknowledgments
We thank Dr. Paola Principe (Beaufour-Ipsen Pharma, Paris, France) for providing homocamptothecin and BN80915.
This work was supported by the Intramural Program of the National Cancer Institute, Center for Cancer Research.
Glossary
ABBREVIATIONS
- CPT
camptothecin
- hCPT
homocamptothecin
- Top1
DNA topoisomerase I
- BN80915
diflomotecan (5-ethyl 9,10-difluoro-4,5,dihydro-5-hydroxy-1H-oxepino[3′,4′,6,7]indolizino [1,2-b]quinoline-3,15[13H]-dione)
- NSC724998
MJ-III-65 (NSC 706744), 6-[3-(2-hydroxyethyl)amino-1-propyl]-5,6-dihydro-2,3-dimethoxy-8,9-methylenedioxy-5,11-dioxo-11H-indeno[1,2-c]isoquinoline
- ABCG2
ATP-binding cassette, subfamily G (WHITE), member 2
- MRP1
multidrug resistance-associated protein 1
- DPC
DNA-protein cross-link(s)
- PAGE
polyacrylamide gel electrophoresis
- PBS-T
phosphate-buffered saline-Tween 20
- PCR
polymerase chain reaction
- RT
reverse transcription
- ABC
ATP-binding cassette
- Pgp
P-glycoprotein
- MRP1
multidrug resistance-associated protein 1
- MK571
3-[[3-[2-(7-chloroquinolin-2-yl)vinyl]phenyl]-(2-dimethylcarbamoylethylsulfanyl)methylsulfanyl] propionic acid
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
References
- Bailly C, Lansiaux A, Dassonneville L, Demarquay D, Coulomb H, Huchet M, Lavergne O, Bigg DC. Homocamptothecin, an E-ring-modified camptothecin analogue, generates new topoisomerase I-mediated DNA breaks. Biochemistry. 1999;38:15556–15563. doi: 10.1021/bi990947h. [DOI] [PubMed] [Google Scholar]
- Bates SE, Medina-Perez WY, Kohlhagen G, Antony S, Nadjem T, Robey RW, Pommier Y. ABCG2 mediates differential resistance to SN-38 (7-ethyl-10-hydroxycamptothecin) and homocamptothecins. J Pharmacol Exp Ther. 2004;310:836–842. doi: 10.1124/jpet.103.063149. [DOI] [PubMed] [Google Scholar]
- Brangi M, Litman T, Ciotti M, Nishiyama K, Kohlhagen G, Takimoto C, Robey R, Pommier Y, Fojo T, Bates SE. Camptothecin resistance: role of the ATP-binding cassette (ABC), mitoxantrone-resistance half-transporter (MXR), and potential for glucuronidation in MXR-expressing cells. Cancer Res. 1999;59:5938–5946. [PubMed] [Google Scholar]
- Calvet L, Santos A, Valent A, Terrier-Lacombe MJ, Opolon P, Merlin JL, Aubert G, Morizet J, Schellens JH, Benard J, et al. No topoisomerase I alteration in a neuroblastoma model with in vivo acquired resistance to irinotecan. Br J Cancer. 2004;91:1205–1212. doi: 10.1038/sj.bjc.6602079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamberlain MC. Salvage chemotherapy with CPT-11 for recurrent glioblastoma multiforme. J Neurooncol. 2002;56:183–188. doi: 10.1023/a:1014532202188. [DOI] [PubMed] [Google Scholar]
- Chamberlain MC. Treatment options for glioblastoma. Neurosurg Focus. 2006;20:E2. doi: 10.3171/foc.2006.20.4.12. [DOI] [PubMed] [Google Scholar]
- Chauvier D, Morjani H, Manfait M. Homocamptothecin-daunorubicin association overcomes multidrug-resistance in breast cancer MCF7 cells. Breast Cancer Res Treat. 2002;73:113–125. doi: 10.1023/a:1015244604336. [DOI] [PubMed] [Google Scholar]
- Chen AY, Yu C, Potmesil M, Wall ME, Wani MC, Liu LF. Camptothecin overcomes MDR1-mediated resistance in human KB carcinoma cells. Cancer Res. 1991;51:6039–6044. [PubMed] [Google Scholar]
- Chen ZS, Furukawa T, Sumizawa T, Ono K, Ueda K, Seto K, Akiyama SI. ATP-Dependent efflux of CPT-11 and SN-38 by the multidrug resistance protein (MRP) and its inhibition by PAK-104P. Mol Pharmacol. 1999;55:921–928. [PubMed] [Google Scholar]
- Cloughesy TF, Filka E, Kuhn J, Nelson G, Kabbinavar F, Friedman H, Miller LL, Elfring GL. Two studies evaluating irinotecan treatment for recurrent malignant glioma using an every-3-week regimen. Cancer. 2003;97(9 Suppl):2381–2386. doi: 10.1002/cncr.11306. [DOI] [PubMed] [Google Scholar]
- Eng WK, McCabe FL, Tan KB, Mattern MR, Hofmann GA, Woessner RD, Hertzberg RP, Johnson RK. Development of a stable camptothecin-resistant subline of P388 leukemia with reduced topoisomerase I content. Mol Pharmacol. 1990;38:471–480. [PubMed] [Google Scholar]
- Friedman HS, Petros WP, Friedman AH, Schaaf LJ, Kerby T, Lawyer J, Parry M, Houghton PJ, Lovell S, Rasheed K, et al. Irinotecan therapy in adults with recurrent or progressive malignant glioma. J Clin Oncol. 1999;17:1516–1525. doi: 10.1200/JCO.1999.17.5.1516. [DOI] [PubMed] [Google Scholar]
- Fujimori A, Gupta M, Hoki Y, Pommier Y. Acquired camptothecin resistance of human breast cancer MCF-7/C4 cells with normal topoisomerase I and elevated DNA repair. Mol Pharmacol. 1996a;50:1472–1478. [PubMed] [Google Scholar]
- Fujimori A, Harker WG, Kohlhagen G, Hoki Y, Pommier Y. Mutation at the catalytic site of topoisomerase I in CEM/C2, a human leukemia cell line resistant to camptothecin. Cancer Res. 1995;55:1339–1346. [PubMed] [Google Scholar]
- Fujimori A, Hoki Y, Popescu NC, Pommier Y. Silencing and selective methylation of the normal topoisomerase I gene in camptothecin-resistant CEM/C2 human leukemia cells. Oncol Res. 1996b;8:295–301. [PubMed] [Google Scholar]
- Gelderblom H, Salazar R, Verweij J, Pentheroudakis G, de Jonge MJ, Devlin M, van Hooije C, Seguy F, Obach R, Prunonosa J, et al. Phase I pharmacological and bioavailability study of oral diflomotecan (BN80915), a novel E-ring-modified camptothecin analogue in adults with solid tumors. Clin Cancer Res. 2003;9:4101–4107. [PubMed] [Google Scholar]
- Hoki Y, Fujimori A, Pommier Y. Differential cytotoxicity of clinically important camptothecin derivatives in P-glycoprotein-overexpressing cell lines. Cancer Chemother Pharmacol. 1997;40:433–438. doi: 10.1007/s002800050682. [DOI] [PubMed] [Google Scholar]
- Kawabata S, Oka M, Shiozawa K, Tsukamoto K, Nakatomi K, Soda H, Fukuda M, Ikegami Y, Sugahara K, Yamada Y, et al. Breast cancer resistance protein directly confers SN-38 resistance of lung cancer cells. Biochem Biophys Res Commun. 2001;280:1216–1223. doi: 10.1006/bbrc.2001.4267. [DOI] [PubMed] [Google Scholar]
- Kohn KW. DNA filter elution: a window on DNA damage in mammalian cells. Bioessays. 1996;18:505–513. doi: 10.1002/bies.950180613. [DOI] [PubMed] [Google Scholar]
- Larsen AK, Gilbert C, Chyzak G, Plisov SY, Naguibneva I, Lavergne O, Lesueur-Ginot L, Bigg DC. Unusual potency of BN 80915, a novel fluorinated E-ring modified camptothecin, toward human colon carcinoma cells. Cancer Res. 2001;61:2961–2967. [PubMed] [Google Scholar]
- Lavergne O, Demarquay D, Bailly C, Lanco C, Rolland A, Huchet M, Coulomb H, Muller N, Baroggi N, Camara J, et al. Topoisomerase I-mediated antiproliferative activity of enantiomerically pure fluorinated homocamptothecins. J Med Chem. 2000a;43:2285–2289. doi: 10.1021/jm000129j. [DOI] [PubMed] [Google Scholar]
- Lavergne O, Demarquay D, Kasprzyk PG, Bigg DC. Homocamptothecins: E-ring modified CPT analogues. Ann N Y Acad Sci. 2000b;922:100–111. doi: 10.1111/j.1749-6632.2000.tb07029.x. [DOI] [PubMed] [Google Scholar]
- Maliepaard M, van Gastelen MA, de Jong LA, Pluim D, van Waardenburg RC, Ruevekamp-Helmers MC, Floot BG, Schellens JH. Overexpression of the BCRP/MXR/ABCP gene in a topotecan-selected ovarian tumor cell line. Cancer Res. 1999;59:4559–4563. [PubMed] [Google Scholar]
- Parney IF, Chang SM. Current chemotherapy for glioblastoma. Cancer J. 2003;9:149–156. doi: 10.1097/00130404-200305000-00003. [DOI] [PubMed] [Google Scholar]
- Pommier Y. Topoisomerase I inhibitors: camptothecins and beyond. Nat Rev Cancer. 2006;6:789–802. doi: 10.1038/nrc1977. [DOI] [PubMed] [Google Scholar]
- Pommier Y, Barcelo JM, Rao VA, Sordet O, Jobson AG, Thibaut L, Miao ZH, Seiler JA, Zhang H, Marchand C, et al. Repair of topoisomerase I-mediated DNA damage. Prog Nucleic Acid Res Mol Biol. 2006;81:179–229. doi: 10.1016/S0079-6603(06)81005-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pommier Y, Pourquier P, Urasaki Y, Wu J, Laco GS. Topoisomerase I inhibitors: selectivity and cellular resistance. Drug Resist Updat. 1999;2:307–318. doi: 10.1054/drup.1999.0102. [DOI] [PubMed] [Google Scholar]
- Pourquier P, Takebayashi Y, Urasaki Y, Gioffre C, Kohlhagen G, Pommier Y. Induction of topoisomerase I cleavage complexes by 1-β-D-arabinofuranosylcytosine (ara-C) in vitro and in ara-C-treated cells. Proc Natl Acad Sci U S A. 2000;97:1885–1890. doi: 10.1073/pnas.97.4.1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajendra R, Gounder MK, Saleem A, Schellens JH, Ross DD, Bates SE, Sinko P, Rubin EH. Differential effects of the breast cancer resistance protein on the cellular accumulation and cytotoxicity of 9-aminocamptothecin and 9-nitrocamptothecin. Cancer Res. 2003;63:3228–3233. [PubMed] [Google Scholar]
- Robey RW, Fetsch PA, Polgar O, Dean M, Bates SE. The livestock photosensitizer, phytoporphyrin (phylloerythrin), is a substrate of the ATP-binding cassette transporter ABCG2. Res Vet Sci. 2006;81:345–349. doi: 10.1016/j.rvsc.2006.04.003. [DOI] [PubMed] [Google Scholar]
- Robey RW, Honjo Y, van de Laar A, Miyake K, Regis JT, Litman T, Bates SE. A functional assay for detection of the mitoxantrone resistance protein, MXR (ABCG2) Biochim Biophys Acta. 2001a;1512:171–182. doi: 10.1016/s0005-2736(01)00308-x. [DOI] [PubMed] [Google Scholar]
- Robey RW, Medina-Perez WY, Nishiyama K, Lahusen T, Miyake K, Litman T, Senderowicz AM, Ross DD, Bates SE. Overexpression of the ATP-binding cassette half-transporter, ABCG2 (Mxr/BCrp/ABCP1), in flavopiridol-resistant human breast cancer cells. Clin Cancer Res. 2001b;7:145–152. [PubMed] [Google Scholar]
- Robey RW, Steadman K, Polgar O, Morisaki K, Blayney M, Mistry P, Bates SE. Pheophorbide a is a specific probe for ABCG2 function and inhibition. Cancer Res. 2004;64:1242–1246. doi: 10.1158/0008-5472.can-03-3298. [DOI] [PubMed] [Google Scholar]
- Schneider E, Horton JK, Yang CH, Nakagawa M, Cowan KH. Multidrug resistance-associated protein gene overexpression and reduced drug sensitivity of topoisomerase II in a human breast carcinoma MCF7 cell line selected for etoposide resistance. Cancer Res. 1994;54:152–158. [PubMed] [Google Scholar]
- Tan KB, Mattern MR, Eng WK, McCabe FL, Johnson RK. Nonproductive rearrangement of DNA topoisomerase I and II genes: correlation with resistance to topoisomerase inhibitors. J Natl Cancer Inst. 1989;81:1732–1735. doi: 10.1093/jnci/81.22.1732. [DOI] [PubMed] [Google Scholar]
- Troconiz IF, Garrido MJ, Segura C, Cendros JM, Principe P, Peraire C, Obach R. Phase I dose-finding study and a pharmacokinetic/pharmacodynamic analysis of the neutropenic response of intravenous diflomotecan in patients with advanced malignant tumours. Cancer Chemother Pharmacol. 2006;57:727–735. doi: 10.1007/s00280-005-0112-6. [DOI] [PubMed] [Google Scholar]
- Urasaki Y, Laco GS, Pourquier P, Takebayashi Y, Kohlhagen G, Gioffre C, Zhang H, Chatterjee D, Pantazis P, Pommier Y. Characterization of a novel topoisomerase I mutation from a camptothecin-resistant human prostate cancer cell line. Cancer Res. 2001;61:1964–1969. [PubMed] [Google Scholar]
- Urasaki Y, Takebayashi Y, Pommier Y. Activity of a novel camptothecin analogue, homocamptothecin, in camptothecin-resistant cell lines with topoisom-erase I alterations. Cancer Res. 2000;60:6577–6580. [PubMed] [Google Scholar]
- Vassal G, Doz F, Frappaz D, Imadalou K, Sicard E, Santos A, O'Quigley J, Germa C, Risse ML, Mignard D, et al. A phase I study of irinotecan as a 3-week schedule in children with refractory or recurrent solid tumors. J Clin Oncol. 2003;21:3844–3852. doi: 10.1200/JCO.2003.08.175. [DOI] [PubMed] [Google Scholar]
- Yanase K, Sugimoto Y, Tsukahara S, Oh-Hara T, Andoh T, Tsuruo T. Identification and characterization of a deletion mutant of DNA topoisomerase I mRNA in a camptothecin-resistant subline of human colon carcinoma. Jpn J Cancer Res. 2000;91:551–559. doi: 10.1111/j.1349-7006.2000.tb00980.x. [DOI] [PMC free article] [PubMed] [Google Scholar]