

Abstract
DNA mismatch repair is central to the maintenance of genomic stability. It is initiated by the recognition of base–base mismatches and insertion/deletion loops by the family of MutS proteins. Subsequently, ATP induces a unique conformational change in the MutS–mismatch complex but not in the MutS–homoduplex complex that sets off the cascade of events that leads to repair. To gain insight into the mechanism by which MutS discriminates between mismatch and homoduplex DNA, we have examined the conformations of specific and nonspecific MutS–DNA complexes by using atomic force microscopy. Interestingly, MutS–DNA complexes exhibit a single population of conformations, in which the DNA is bent at homoduplex sites, but two populations of conformations, bent and unbent, at mismatch sites. These results suggest that the specific recognition complex is one in which the DNA is unbent. Combining our results with existing biochemical and crystallographic data leads us to propose that MutS: (i) binds to DNA nonspecifically and bends it in search of a mismatch; (ii) on specific recognition of a mismatch, undergoes a conformational change to an initial recognition complex in which the DNA is kinked, with interactions similar to those in the published crystal structures; and (iii) finally undergoes a further conformational change to the ultimate recognition complex in which the DNA is unbent. Our results provide a structural explanation for the long-standing question of how MutS achieves mismatch repair specificity.
DNA mismatch repair (MMR) is a highly conserved repair pathway targeting mismatched bases that arise through DNA replication errors and during homologous recombination (1–3). Inactivation of MMR genes results in a significant increase in the spontaneous mutation rate and, in humans, a predisposition to cancer (4). Escherichia coli provides the best-understood MMR system and serves as a prototype for the more complicated but homologous eukaryotic systems (5). In E. coli, the proteins MutS, MutL, and MutH are responsible for the initiation of MMR (6). MutS and MutL function as dimers and have intrinsic ATPase activities that are essential for MMR (7, 8). MMR is initiated by the binding of MutS to either a mismatch or a short insertion/deletion loop (IDL). Subsequently, ATP induces a conformational change in the MutS–mismatch complex and promotes its interaction with MutL. Assembly of the MutS–MutL–heteroduplex complex activates the endonuclease activity of MutH, which incises the newly synthesized (unmethylated) strand at a d(GATC) site. This incision confers strand specificity of MMR, directing repair exclusively to the newly synthesized strand containing the error. Excision repair completes the process.
Crystal structures of E. coli and Thermus aquaticus (Taq) MutS dimers complexed with a G/T base–base mismatch and a 1T–bulge, respectively, shed light on the structural components of mismatch recognition (9–11). Specific interactions include an aromatic ring stack of a conserved phenylalanine (Phe-39 in Taq or Phe-36 in E. coli) with the mismatched thymine and a hydrogen bond between a conserved glutamate (Glu-41 in Taq or Glu-38 in E. coli) and the mismatched thymine. Sequence-independent interactions include van der Waals interactions and hydrogen bonds between the DNA backbone and side chains of MutS. As a result of these protein–DNA interactions, the DNA is sharply kinked toward the major groove by 60° at the mismatch site (9, 10).
A fundamental problem in MMR is one of specificity, i.e., how recognition of a mismatch by MutS triggers repair. It has been demonstrated that MMR efficiency is not simply governed by the binding of MutS to a mismatch (12). Several models have been proposed to describe the initiation event in MMR (7). Although the exact mechanism of initiation has not been fully elucidated, there is general agreement that a key step is an ATP-induced conformational change in MutS that occurs only when MutS binds to a mismatch (11, 13–19). In this ATP-activated state, MutS appears to adopt a mobile clamp conformation that encircles the DNA. This ATP-induced mismatch-specific conformation is required for the subsequent interaction of MutS with MutL.
Insight into the mechanism of mismatch recognition and specificity can be gained by comparing the structures of MutS–DNA complexes at mismatch and homoduplex sites. In this study, we use atomic force microscopy (AFM) to characterize the conformations of specific and nonspecific MutS–DNA complexes. AFM is advantageous over bulk techniques, because it allows the conformations of MutS–DNA complexes at mismatches and those at homoduplex sites to be compared. Furthermore, the distribution of conformations within a complex population of molecules can be characterized (20). We demonstrate that MutS adopts a single population of conformations, in which the DNA is bent when bound to a homoduplex site, but adopts two populations of conformations, bent and unbent, when bound at a mismatch site. Our results suggest that DNA bending is important for initial mismatch recognition, whereas the ability of MutS–DNA complexes to adopt an unbent conformation governs, in part, repair specificity.
Methods
Protein and DNA Substrates. E. coli His6-tagged MutS and Taq MutS proteins were purified as described (18, 21). DNA substrates for AFM were created by ligating three DNA fragments, and those for electrophoresis were chemically synthesized, as described in Supporting Materials and Methods, which is published as supporting information on the PNAS web site, and in Fig. 1a.
Fig. 1.

Schematic view of the DNA substrates used in this AFM study and AFM images of free DNA and MutS–DNA complexes. (a) The name of the DNA fragment is shown below each diagram. The length of the fragments and the position of the mismatch in nucleotides from the nearest end are shown by the black arrows. The two flanking fragments (red and blue) are made from the DraIII- and BglI-digested PCR fragments amplified from M13mp18 and pUC18 plasmids, respectively. The middle fragments (black) with the mismatches are generated by annealing two synthesized oligos. (b Top) AFM images of free 783TBulge heteroduplex DNA (Left), E. coli MutS–982GT complexes (Center), and Taq MutS–783TBulge complexes (Right). The red arrows point to MutS–DNA complexes. (b Middle and Bottom) AFM surface plots of E. coli MutS bound to a G/T mismatch and Taq MutS bound to a 1T-bulge, respectively. MutS-induced DNA bend angles are shown on each image.
AFM. Protein–DNA complexes were formed by incubating a 1–5 nM E. coli MutS dimer or a 10–20 nM Taq MutS dimer with 1–2 nM hetero- or homoduplex DNA substrates for 1–5 min at room temperature or 65°C in binding buffer A (20 mM Hepes, pH 7.8/50 mM NaCl/5 mM MgCl2) in a total volume of 20 μl. The reaction was deposited onto freshly cleaved mica (Spruce Pine Mica Company, Spruce Pine, NC) at room temperature or 65°C. After a 1-min incubation, the mica surface was rinsed with HPLC-grade water, blotted dry, and then dried under a stream of nitrogen. The images were captured in air with a Nanoscope IIIa (Digital Instruments, Santa Barbara, CA) microscope in tapping mode. Pointprobe tapping mode silicon probes (Molecular Imaging, Tempe, AZ) with spring constants of ≈50 N·m–1 and resonance frequencies ≈170 KHz were used for all imaging. Images were collected at a speed of 3–4 Hz and a resolution of 512 × 512 pixels.
Image Analysis. DNA contour lengths and bend angles were measured by using either nanoscope IIIA software or a custom program written in matlab software (Mathworks, Natick, MA). The measurements from the two methods are equivalent. The angle, θ, at the intersection of the two DNA arms was measured. The DNA bend angle α is 180-θ. For measuring the intrinsic bending of DNA, circles of 10-nm radii, comparable to the size of MutS proteins in the AFM images, were put at the desired locations. Volume analysis was performed as described (22–24). For each type of DNA substrate, images from at least three independent experiments were analyzed and pooled. DNA molecules having MutS bound at more than one site were discarded from the statistical analysis of bend angles. The program kaleidagraph (Synergy Software, Reading, PA) was used to generate statistical plots.
Identification of Specific and Nonspecific Protein–DNA Complexes. The positions of the MutS-binding sites on the DNA templates were determined by measuring the length of the DNA from the intersection of the two extrapolated DNA arms to each end. The binding position was then defined as the ratio of the length of the shorter DNA tract divided by the total contour length. Complexes with centers within one standard deviation of the expected mismatch position were categorized as specific complexes. We did not end-label the DNA to unequivocally identify the DNA ends, which means that some nonspecific complexes will be counted as specific complexes, but not vice versa. Given the relative stabilities of the specific and nonspecific complexes, the data for the specific complexes should contain <5% nonspecific complexes.
Comparative Gel Electrophoresis. One hundred nanomole Taq MutS dimer was incubated with 1 nM 5′-32P-labeled DNA substrate in binding buffer A in the presence of 1 mM DTT and 0.1 mg·ml–1 BSA in a total volume of 10 μl. For each sample, 1 nM of plasmid was included to reduce nonspecific binding of MutS to the labeled DNA. Immediately before loading, Ficoll was added to samples to a final concentration of 3%. Samples were loaded under voltage onto a 6.5% polyacrylamide gel (29:1 acrylamide to bisacrylamide) containing Tris-glycine and 5 mM MgCl2. Buffers also contained Tris-glycine and 5 mM MgCl2 and were recirculated manually. After electrophoresis, gels were dried, exposed to phosphor screens, and analyzed on a Fuji BAS-1500 phosphorimager.
Results
MutS Bound to a Mismatch Exhibits Two Conformations: Bent and Unbent. In this study, we used three DNA substrates containing uniquely located mismatches (783GT, 783Tbulge, and 982GT; where the number indicates the length of the fragment, and the letters indicate the type of mismatch) and several different homoduplex DNAs (Fig. 1a and Table 1). The 783GT and 783Tbulge DNAs have a G/T mismatch and a single unpaired T, respectively, positioned 27% of the distance from one end, and the 982GT has aG/T mismatch 42% from one end (Fig. 1a). Representative AFM images of free DNA and DNA in the presence of E. coli and Taq MutS are shown in Fig. 1b. MutS can be clearly seen bound to the DNA in the deposition in the presence of protein. Because we know the positions of the mismatches on the DNA, we can discriminate between MutS bound at a mismatch (specific complex) and MutS bound at a homoduplex site (nonspecific complex) by measuring the distance of MutS from the ends of the DNA (see Methods). At room temperature, ≈60% of the MutS–DNA complexes are specific for both E. coli and Taq MutS proteins on the mismatch substrates. (If MutS was bound at multiple positions on the DNA or to the ends of the DNA, the complexes were not included in the analysis.) For all substrates that contain a mismatch, the distributions of positions of E. coli and Taq MutS are Gaussian centered at the positions of the mismatches, whereas on homoduplex DNA, the distributions of positions are random (Fig. 5, which is published as supporting information on the PNAS web site).
Table 1.
Parameters from single and double Gaussian fits*
| DNA substrate or complex | Number of complexes* | Mean bend angle of first peak,°† | Mean bend angle of second peak,°‡ |
|---|---|---|---|
| Free DNA | |||
| 782Homo (at 27%) | 100 | 0 (35) | |
| 783TBulge (at 27%) | 100 | 0 (25) | |
| 783GT (at 27%) | 100 | 0 (35) | |
| 982GT (at 42%) | 100 | 0 (30) | |
| MutS—DNA complexes at specific sites | |||
| E. coli MutS + 783GT/982GT | 277 | 0 (12) | 74 (45) |
| Taq MutS + 783Tbulge (≈23°C) | 260 | 0 (11) | 42 (38) |
| Taq MutS + 783Tbulge (65°C) | 60 | 0 (10) | 32 (30) |
| MutS—DNA complexes at nonspecific sites | |||
| E. coli MutS + 783GT/982GT | 164 | 58 (52) | |
| Taq MutS + 783TBulge (≈23°C) | 159 | 39 (47) | |
| Taq MutS + 783TBulge (65°C) | 101 | 48 (48) | |
| E. coli MutS + pUC18§ | 93 | 47 (49) | |
| Taq MutS + pUC18 (≈23°C)§ | 306 | 58 (40) | |
| Taq MutS + 782Homo (≈23°C) | 287 | 52 (51) | |
| Taq MutS + 782Homo (65°C) | 235 | 62 (47) |
The parameters were used to generate the Gaussian fits of the distributions shown in Figs. 3 and 8.
Number of complexes or free DNA fragments analyzed for each distribution.
Mean bend angle determined from the single Gaussian curve fits or the mean bend angle of the first peak of the double Gaussian curve fits. The standard deviations are shown in parentheses.
Mean bend angle of the second peak of the double Gaussian curve fits. The standard deviations are shown in parentheses.
pUC18 plasmid was digested with DrdI, and the purified mixture of two linear fragments (817 and 1,869 bp) was used in this experiment.
To determine whether mismatched substrates have significant intrinsic bending, we measured DNA bend angles at the positions of the mismatches and at homoduplex sites in the absence of protein. Comparison of the bend angles at the mismatches vs. at homoduplex sites does not reveal any evidence of significant DNA bending at either G/T or 1T-bulge mismatches (Table 1). Specifically, bend angles measured at mismatches and at homoduplex sites all exhibit half-Gaussian distributions with similar breadths centered at 0° (Table 1). The lack of any significant intrinsic bending of DNA containing a 1T-bulge mismatch is supported by gel electrophoresis bending analysis, shown in Fig. 2b. These results are consistent with previous studies showing that one unpaired base or a G/T mismatch causes only a small bend in the DNA (<15°) (25–27) and suggest that MutS does not recognize a preexisting bend in the DNA.
Fig. 2.

Comparative gel electrophoresis of Taq MutS–DNA complexes. (a) A schematic demonstration of the principle of comparative gel electrophoresis. (b) Lanes 1–7, seven 90-bp DNA substrates with a single 1T-bulge at positions 15, 25, 35, 45, 55, 65, and 75 nt, respectively. Lanes 8–14, substrates in the same order as in lanes 1–7 complexed with 100 nM Taq MutS.
In contrast to the free DNA, we observe significant MutS-induced DNA bending at mismatches (Fig. 1b). This bending does not appear to involve any significant DNA looping, because the contour length of free and bound DNA is approximately the same (data not shown). Fig. 3 shows the distributions of bend angles for E. coli MutS bound to a G/T mismatch (Fig. 3a) and Taq MutS bound to a 1T-bulge at room temperature (Fig. 3b) and 65°C (Fig. 3c). Inspection of these data reveals that both E. coli and Taq MutS induce DNA bending at a mismatch, consistent with the x-ray crystallographic data (9, 10). Interestingly, however, in addition to the bent population, there is a population of complexes that is not bent. A binomial distribution analysis indicates that the peaks at 0° are significant, with P < 0.03 for E. coli MutS and P <10–7 for Taq MutS at both temperatures. In addition, these data do not fit well to a single Gaussian distribution, whereas they fit significantly better to a sum of two Gaussians, with peaks centered at 0° and 74° for E. coli MutS, at 0° and 42° for Taq MutS at room temperature, and at 0° and 32° for Taq MutS at 65°C (Table 1). Finally, two populations of complexes, bent and unbent, are also observed for E. coli MutS bound to a 1T-bulge and Taq MutS bound to a G/T mismatch (data not shown) and for E. coli MutS bound to a G/T mismatch in the presence of ADP (Fig. 6a, which is published as supporting information on the PNAS web site).
Fig. 3.

Histograms of DNA bend angles induced by E. coli and Taq MutS bound to mismatch (specific complexes, Upper) and homoduplex (nonspecific complexes, Lower) sites. DNA bend angles of specific complexes are shown for E. coli MutS bound at a G/T mismatch on the 783GT and 982GT DNA substrates (a) and for Taq MutS bound at a 1T-bulge on the 783TBulge substrate at room temperature (b) and at 65°C(c). DNA bend angles of nonspecific complexes are shown for E. coli MutS bound at homoduplex sites on 783GT and 982GT DNA substrates (d) and Taq MutS bound at homoduplex sites on 783Tbulge DNA substrate at room temperature (e) and at 65°C(f). The curves drawn in a–c are the double Gaussian fits to the data, and the curves drawn in d–f are the single Gaussian fits to the data. The parameters from these fits are shown in Table 1. A binomial analysis shows no peak at 0° for either E. coli or Taq MutS at homoduplex sites. In addition, fitting the curves of the bend angle distributions at homoduplex sites to a sum of two Gaussians does not significantly improve the fits. Because the data for MutS-induced DNA bending at homoduplex sites and at mismatch sites are obtained from the same depositions, the possibility that the differences seen at mismatch sites and homoduplex sites are artifacts of deposition is ruled out. See Figs. 6b and 8 for additional results on nonspecific complexes.
The breadths of the distribution are related to the flexibility of the protein–DNA complexes (28, 29). The breadths of distributions of angles for the bent states are greater than that of free DNA (≈40° vs. ≈30°), whereas they are narrower for the unbent states relative to free DNA (≈10° vs. ≈30°) (Table 1). These results indicate that the bent state of the protein–DNA complex is more flexible than free DNA, suggesting that the “bent state” represents a large ensemble of states that are in dynamic equilibrium. The conformational flexibility of the complex is consistent with the observation that the DNA-binding domains of Taq MutS are disordered in the crystal structure in the absence of DNA (9). In contrast to the bent state, the conformation of the unbent state appears to be relatively rigid (see Discussion).
For E. coli MutS, ≈10% of the specific complexes are in the unbent state, and for Taq MutS, ≈20% and ≈30% of the complexes are unbent at room temperature and 65°C, respectively. The ratio of the populations of the unbent state to the bent state is equal to the equilibrium constant between the two states [K(bent → unbent)]. This constant ranges from 0.1 (10%/90%) for E. coli MutS to 0.5 (30%/70%) for Taq MutS at 65°C, indicating that the unbent state is slightly less stable than the bent state (by 1.4–0.5 kcal/mol). Interestingly, increasing the temperature from room temperature to 65°C increases the population of the unbent state by 50% for Taq MutS, indicating that as the optimum temperature for Taq MutS is approached, an increasing proportion of the complexes are in the unbent state.
If the equilibrium between bent and unbent states is rapid relative to the rate of migration of the complex in a gel, a gel electrophoresis bending experiment would reveal a single population of complexes with an apparent bend angle that is the weighted average of the bent and unbent populations (30, 31). In contrast, if they are in a slow equilibrium, two bands would be expected: one corresponding to the bent and one to the unbent complex. Inspection of the gel electrophoresis data for Taq MutS in the presence of a series of seven 90-bp duplex DNA with a single 1T-bulge reveals a “frown” pattern that is characteristic of protein–DNA complexes in which the DNA is bent (Fig. 2) (32). Only a single population of complexes is observed, suggesting that the bent and unbent conformations are in “rapid” equilibrium with one another. Results from comparative gel electrophoresis of the yeast and human MutS homologs MSH2-MSH6 (MutSα) show similar results (Fig. 7, which is published as supporting information on the PNAS web site), suggesting that this idea may also hold for these eukaryotic homologs.
These AFM data indicate that MutS bound at a mismatch can impose two different conformations on the DNA, bent and unbent. Significantly, bimodal distributions have been observed in only a few of many microscopy studies of protein-induced DNA bending (33–35). Among these studies, human 8-oxoguanine DNA glycosylase has been found to induce bimodal distributions of DNA bending at nonspecific sites, but only a single bent population at specific sites (33). In the other two cases, two protein species or two different DNA-binding modes were present: one involving binding via the DNA ends and the other involving direct binding to a region of duplex DNA (34, 35).
Nonspecific MutS–DNA Complexes Exhibit Only One Conformation: Bent. One possible explanation for the unbent MutS–DNA complexes could be that they are intermediates in the formation of the bent state. Such a proposal has been put forth to explain the presence of unbent complexes at nonspecific sites for human 8-oxoguanine DNA glycosylase (33). To address the functional significance of the unbent complexes at mismatches, we have analyzed MutS-induced DNA bending at homoduplex sites on several different DNA substrates.
Inspection of the bend-angle data (Fig. 3 d–f, Table 1, Fig. 6b, and Fig. 8, which is published as supporting information on the PNAS web site) reveals that both E. coli and Taq MutS induce DNA bending at homoduplex sites, with average bend angles similar to those found for the bent population of the specific complexes (Table 1). In contrast to the specific sites, no significant population (<5%) of unbent complexes is observed at homoduplex sites (Figs. 3 d–f, 6b, and 8). These data indicate that in the absence of specific protein–DNA contacts, the most stable conformation of MutS–DNA complexes is one in which the DNA is bent. In contrast, on making specific interactions, MutS exists in an equilibrium between conformations in which the DNA is bent and in which it is unbent. These results strongly suggest that the unbent state is not a precursor to the bent state, but rather that the bent state is a precursor to the unbent state, with the unbent state resulting from interactions unique to the specific MutS–mismatch complex. If the unbent state were a precursor to the bent state, we would expect to observe the opposite result; that is, we would expect to see bent and unbent complexes at nonspecific sites and only bent complexes at specific sites, as was seen for human 8-oxoguanine DNA glycosylase (33).
The observation that even at a mismatch, the unbent state is less stable than the bent state, is surprising, given that it is estimated to cost ≈3 kcal/mol to bend the DNA ≈60° (which correlates to an ≈200-fold effect on binding affinity) (29, 36). (This energy may be slightly less on mismatch DNA.) Consequently, on undergoing the transition from the bent to the unbent state, the complex would gain energy from the relaxation of the DNA. These results indicate that there is either a loss of protein–nucleic acid interactions or conformational rearrangements in the protein or both. Some of the energy stored in the bent DNA must be used to drive a conformational change in MutS such that it can accommodate DNA that is not bent, because the optimum conformation of MutS–DNA complexes in the absence of specific contacts is one in which the DNA is bent.
Conformations of MutS–DNA Complexes Are Independent of Oligomeric States of MutS. Both E. coli and Taq MutS proteins have been shown to exist in an equilibrium of dimers and tetramers (21, 37, 38). This observation raises the question of whether the bent and unbent populations correlate with the different oligomeric states of MutS proteins. To answer this question, we analyzed the oligomeric states of MutS–DNA complexes using a volume analysis method described previously (22–24). This analysis reveals that under the conditions of our experiments, both E. coli and Taq MutS exist primarily as dimers and tetramers, with ≈50% dimers for E. coli MutS and ≈75% dimers for Taq MutS, independent of whether they are bound to DNA (data not shown). These results are consistent with previous biochemical studies that indicate that the oligomeric states of MutS are independent of DNA binding (21, 37). Plots of AFM volumes of MutS vs. DNA bend angles (data not shown) show no correlation between DNA bend angles and oligomeric states of MutS–DNA complexes. These results indicate that the two populations of conformational states, bent and unbent, observed at mismatches do not result from different oligomeric states of MutS. Our simple explanation for this lack of correlation is that for the MutS tetramers, only one of the dimers is interacting with the DNA. Inspection of the AFM images of MutS–DNA complexes supports this idea. Although it is not always possible to distinguish the two dimers within a tetramer bound to DNA, in many of the images in which tetramers are bound to the DNA, it can be clearly seen that only one of them is contacting the DNA (Fig. 4).
Fig. 4.

Surface plot of a Taq MutS–783Tbulge DNA complex. White arrows point to free MutS dimers on the surface. Red and blue arrows point to two dimers in a MutS tetramer bound to the DNA. The horizontal distance between the peaks of those two dimers is 9 nm. The vertical distance between the peaks of those two dimers is 0.7 nm.
Discussion
In this study, we make the surprising observation that MutS–DNA complexes exhibit two populations of conformations at specific sites: one in which the DNA is bent and one in which the DNA is unbent, but only a single population in which the DNA is bent at nonspecific sites. These results indicate that the unbent state is the result of unique interactions between MutS and the mismatch base (or IDL) and suggest that the bent conformation is an intermediate in the formation of the unbent state. This suggestion is further supported by the observation of broad distributions of angles of bent complexes and narrow distributions of angles of the unbent complexes (Table 1 and Fig. 3), suggesting that the bent state represents a dynamic complex that is sampling a large number of conformations, whereas the unbent state is relatively rigid, with a narrow distribution of bend angles typical of specific protein–DNA complexes (29, 33).
These results, taken together with the published crystallographic data (9–11), suggest that MutS (i) binds to DNA nonspecifically and bends it (MutS:DNABENT,NS) in search of a mismatch; (ii) on specific recognition of a mismatch, undergoes a conformational change to an initial recognition complex (IRC) in which the DNA is kinked (MutS:DNAKINKED,SP), with interactions similar to those in the crystal structures (9–11); and (iii) undergoes a further conformational change to the ultimate recognition complex (URC) in which the DNA is unbent (MutS:DNAUNBENT,SP). In other words,
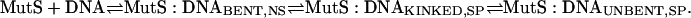 |
We propose that only the unbent URC is competent for ATP activation that leads to repair. Because ATP induces a further conformational change in the URC that is essentially irreversible on the time scale of repair, the unbent URC needs to be only modestly populated for repair to occur. In the discussion that follows, we provide additional support for this mechanism and discuss it in the context of the specificity of MMR.
Smooth Bending vs. Kinking Governs IRC Formation. In this mechanism, there are two steps that govern mismatch recognition. The first step is the formation of a MutS–mismatch complex in which the DNA is kinked at the mismatch with specific interactions between the mismatch base and MutS, as seen in the crystal structures (9, 10). If this bent IRC is not formed, then the unbent URC cannot form; therefore, the formation of this complex plays a central role in governing MMR. Inspection of the crystal structures of MutS–mismatch complexes (9, 10) reveals that the majority of nonspecific interactions involve van der Waals interactions and hydrogen bonds between MutS and the sugar phosphate backbone of DNA in regions flanking the mismatch but not immediately adjacent to it. Assuming that these same interactions are used when MutS binds to homoduplex DNA, it is likely to be more favorable to spread the bend over several base pairs of the homoduplex DNA than to kink it locally. The presence of a mismatch or an IDL, however, would facilitate kinking locally, because there is already local destabilization at the mismatch (39). This idea is supported by studies showing that small-molecule intercalators preferentially bind to mismatches, indicating greater local flexibility at a mismatch than at homoduplex sites (40). This tendency to kink locally at a mismatch would provide the opportunity for phenylalanine to interact specifically with the mismatch base and permit the formation of the bent IRC, whereas, in the absence of a mismatch, the DNA would be smoothly bent, reducing the chance for the phenylalanine to interact with a correctly paired base. The preference for kinking the DNA locally at a mismatch (or IDL) vs. bending it smoothly at a homoduplex site provides a signal for MutS to check for specific interactions. In this way, MutS can rapidly scan the DNA checking for specific contacts only where MutS can induce local kinking (or where the DNA has a tendency to be kinked or both), thereby facilitating the initial identification of a mismatch. If a mismatch is present, the additional interactions that are gained by the interaction of the mismatch base with phenylalanine will further stabilize this kinked complex at the mismatch and permit the formation of the IRC. Consequently, the local flexibility around a mismatch is key to the formation of the kinked MutS–DNA conformation, as has been proposed (26, 40). A similar role for bending in specific site recognition has been proposed for λ cro protein (29).
The Relative Stability of the Bent and Unbent States Governs, in Part, MMR. Once the bent IRC is formed, it can undergo a conformational change to the unbent URC, which we propose is the complex that signals repair. The relative stability of the bent IRC and the unbent URC will determine whether a mismatch in the DNA will form an URC with MutS. If the bent IRC is significantly more stable than the unbent URC, the specific unbent recognition state will not be significantly populated; and if the unbent state must be populated to signal downstream events, no repair will ensue. Consequently, mismatches that stabilize the bent IRC relative to the unbent URC are expected to be less efficiently repaired.
Studies of T4 endonuclease VII activity on DNAs containing mismatches provide an estimate of the extent to which different DNA mismatches stabilize kinked DNA (41). T4 endonuclease VII catalyzes the cleavage of Holliday junctions and of DNA that can be easily kinked. It exhibits low activity on all G containing mismatches; intermediate activity on A/A, A/C, and T/T mismatches and single-base IDLs; and high activity on C/C mismatches and large IDLs (>4 bases), suggesting that G containing mismatches are not easily kinked, but that C/C mismatches and large IDLs are easily kinked.
Comparing these results with the efficiencies of repair of different mismatches by MMR indicates that those that are easily kinked are not efficiently repaired, whereas those that are not easily kinked are efficiently repaired. Specifically, in E. coli, purine–pyrimidine, purine–purine, and small IDLs are repaired significantly better than pyrimidine–pyrimidine mismatches or large IDLs, with C/C mismatches being refractory to repair and G/T mismatches being one of the most efficiently repaired (12). These results demonstrate an inverse correlation between the facility of DNA bending (or kinking) and the efficiency of repair, supporting our assertion that the unbent state is the URC that signals repair. In addition, the observation that mismatches are more efficiently repaired in GC-than AT-rich DNA is consistent with this idea (42), because in general, AT-rich DNA is more easily bent than GC-rich DNA (43). Similarly, it is consistent with the observation that MMR is less efficient on mismatch bases that are looped out and extrahelical (44), because such mismatches should be easier to bend. Finally, this idea explains the observation that yeast MSH2–MSH6 (yMutSα) binds tightly to palindromic mispairs, but a ternary complex including MSH2–MSH6 and MLH1–PMS1 (yMutLα) does not form in the presence of a palindromic mispair in vitro, and palindromic mispairs are refractory to MMR in vivo (45–47). Specifically, palindromic mispairs are statically bent (48), providing a good substrate for binding; however, they would be difficult to unbend, which is required to form the URC that, in turn, would make them refractory to repair. Our observation of an unbent state that is unique to complexes containing mismatches provides an answer to the long-standing question of how MutS achieves repair specificity and can explain why binding of E. coli MutS to a mismatch does not necessarily lead to its correction (12). MutS presents an unusual case, however, in that the unbent URC is slightly less stable than the bent IRC at the specific site. Based on the above discussion, whether the downstream events that lead to repair will occur is expected to be determined by the ability of MutS to adopt the unbent URC. Consequently, if the unbent state were significantly more stable than the bent state, MutS–homoduplex complexes could adopt the unbent state, and futile cycles of repair would occur. In addition, by evolving such that the unbent URC is only modestly stable relative to the bent IRC, MutS can recognize the mismatches that occur more often during DNA replication, such as G/T mismatches, more efficiently than the mismatches that occur less often, such as C/C mismatches (39, 49).
The requirement for the formation of the bent IRC prevents homoduplex DNA from being futilely repaired, and the relative stability of the bent IRC and unbent URC can explain the general trend in MMR efficiencies. The questions remain of why an unbent conformation would be more stable at a mismatch than at a homoduplex site, and how the unbent state provides specific mismatch recognition. One plausible explanation for these questions is that in the unbent URC, one of the mismatch base partners is flipped out and interacting with MutS. In this way, MutS could achieve specific recognition of a mismatched base, and the unbent state with a base flipped out would be more stable at a mismatch than at a correctly paired base (50). Such mechanisms have been found for other DNA repair enzymes, such as DNA glycosylases and T4 endonuclease V (51).
Conclusion
MutS appears to have evolved an unusual mechanism of specific site recognition, in that it bends the DNA in search of its specific site and, on specific site recognition, uses the energy stored in the bent DNA to help drive the conformational change in MutS that leads to the formation of the specific unbent recognition complex. As such, MutS represents an example of a protein in which the formation of a bent complex precedes the formation of an unbent (straight) complex and not vice versa. These results demonstrate the dynamic nature of MutS–DNA complexes.
Supplementary Material
Acknowledgments
We are grateful for helpful discussions with Drs. T. Kunkel, M. Guthold, S. Walker IV, and G. Pielak. We thank Drs. T. Selmane (National Institute of Diabetes and Digestive and Kidney Diseases) for purifying protein and G. Bassi, A. Wilson, S. Nayak, and K. Yoshioka for assistance. The AFM studies of E. coli and Taq MutS were done by H.W. and Y.Y., respectively. This work was supported by the National Institutes of Health and the American Cancer Society.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: MMR, mismatch repair; AFM, atomic force microscopy; IDL, insertion/deletion loop; Taq, Thermus aquaticus; IRC, initial recognition complex; URC, ultimate recognition complex.
References
- 1.Buermeyer, A. B., Deschenes, S. M., Baker, S. M. & Liskay, R. M. (1999) Annu. Rev. Genet. 33, 533–564. [DOI] [PubMed] [Google Scholar]
- 2.Kolodner, R. D. & Marsischky, G. T. (1999) Curr. Opin. Genet. Dev. 9, 89–96. [DOI] [PubMed] [Google Scholar]
- 3.Harfe, B. D. & Jinks-Robertson, S. (2000) Mutat. Res. 451, 151–167. [DOI] [PubMed] [Google Scholar]
- 4.Jiricny, J. & Marra, G. (2003) Curr. Opin. Genet. Dev. 13, 61–69. [DOI] [PubMed] [Google Scholar]
- 5.Modrich, P. & Lahue, R. (1996) Annu. Rev. Biochem. 65, 101–133. [DOI] [PubMed] [Google Scholar]
- 6.Au, K. G., Welsh, K. & Modrich, P. (1992) J. Biol. Chem. 267, 12142–12148. [PubMed] [Google Scholar]
- 7.Hsieh, P. (2001) Mutat. Res. 486, 71–87. [DOI] [PubMed] [Google Scholar]
- 8.Yang, W. (2000) Mutat. Res. 460, 245–256. [DOI] [PubMed] [Google Scholar]
- 9.Obmolova, G., Ban, C., Hsieh, P. & Yang, W. (2000) Nature 407, 703–710. [DOI] [PubMed] [Google Scholar]
- 10.Lamers, M. H., Perrakis, A., Enzlin, J. H., Winterwerp, H. H., de Wind, N. & Sixma, T. K. (2000) Nature 407, 711–717. [DOI] [PubMed] [Google Scholar]
- 11.Junop, M. S., Obmolova, G., Rausch, K., Hsieh, P. & Yang, W. (2001) Mol. Cell 7, 1–12. [DOI] [PubMed] [Google Scholar]
- 12.Su, S. S., Lahue, R. S., Au, K. G. & Modrich, P. (1988) J. Biol. Chem. 263, 6829–6835. [PubMed] [Google Scholar]
- 13.Allen, D. J., Makhov, A., Grilley, M., Taylor, J., Thresher, R., Modrich, P. & Griffith, J. D. (1997) EMBO J. 16, 4467–4476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Blackwell, L. J., Martik, D., Bjornson, K. P., Bjornson, E. S. & Modrich, P. (1998) J. Biol. Chem. 273, 32055–32062. [DOI] [PubMed] [Google Scholar]
- 15.Gradia, S., Acharya, S. & Fishel, R. (1997) Cell 91, 995–1005. [DOI] [PubMed] [Google Scholar]
- 16.Gradia, S., Subramanian, D., Wilson, T., Acharya, S., Makhov, A., Griffith, J. & Fishel, R. (1999) Mol. Cell 3, 255–261. [DOI] [PubMed] [Google Scholar]
- 17.Sixma, T. K. (2001) Curr. Opin. Struct. Biol. 11, 47–52. [DOI] [PubMed] [Google Scholar]
- 18.Schofield, M. J., Nayak, S., Scott, T. H., Du, C. & Hsieh, P. (2001) J. Biol. Chem. 276, 28291–28299. [DOI] [PubMed] [Google Scholar]
- 19.Joshi, A. & Rao, B. J. (2002) Biochemistry 41, 3654–3666. [DOI] [PubMed] [Google Scholar]
- 20.Bustamante, C. & Rivetti, C. (1996) Annu. Rev. Biophys. Biomol. Struct. 25, 395–429. [DOI] [PubMed] [Google Scholar]
- 21.Biswas, I., Ban, C., Fleming, K. G., Qin, J., Lary, J. W., Yphantis, D. A., Yang, W. & Hsieh, P. (1999) J. Biol. Chem. 274, 23673–23678. [DOI] [PubMed] [Google Scholar]
- 22.Ratcliff, G. C. & Erie, D. A. (2001) J. Am. Chem. Soc. 123, 5632–5635. [DOI] [PubMed] [Google Scholar]
- 23.Xue, Y., Ratcliff, G. C., Wang, H., Davis-Searles, P. R., Gray, M. D., Erie, D. A. & Redinbo, M. R. (2002) Biochemistry 41, 2901–2912. [DOI] [PubMed] [Google Scholar]
- 24.Yang, Y., Wang, H. & Erie, D. A. (2003) Methods 29, 175–187. [DOI] [PubMed] [Google Scholar]
- 25.Woodson, S. A. & Crothers, D. M. (1988) Biochemistry 27, 3130–3141. [DOI] [PubMed] [Google Scholar]
- 26.Isaacs, R. J., Rayens, W. S. & Spielmann, H. P. (2002) J. Mol. Biol. 319, 191–207. [DOI] [PubMed] [Google Scholar]
- 27.Lilley, D. M. (1995) Proc. Natl. Acad. Sci. USA 92, 7140–7142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.van Noort, J., Orsini, F., Eker, A., Wyman, C., de Grooth, B. & Greve, J. (1999) Nucleic Acids Res. 27, 3875–3880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Erie, D. A., Yang, G., Schultz, H. C. & Bustamante, C. (1994) Science 266, 1562–1566. [DOI] [PubMed] [Google Scholar]
- 30.Miick, S. M., Fee, R. S., Millar, D. P. & Chazin, W. J. (1997) Proc. Natl. Acad. Sci. USA 94, 9080–9084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shubsda, M., Goodisman, J. & Dabrowiak, J. C. (1999) Biophys. Chem. 76, 95–115. [DOI] [PubMed] [Google Scholar]
- 32.Crothers, D. M., Gartenberg, M. R. & Shrader, T. E. (1991) Methods Enzymol. 208, 118–146. [DOI] [PubMed] [Google Scholar]
- 33.Chen, L., Haushalter, K. A., Lieber, C. M. & Verdine, G. L. (2002) Chem. Biol. 9, 345–350. [DOI] [PubMed] [Google Scholar]
- 34.Rippe, K., Guthold, M., von Hippel, P. H. & Bustamante, C. (1997) J. Mol. Biol. 270, 125–138. [DOI] [PubMed] [Google Scholar]
- 35.Valle, M., Valpuesta, J. M., Carrascosa, J. L., Tamayo, J. & Garcia, R. (1996) J. Struct. Biol. 116, 390–398. [DOI] [PubMed] [Google Scholar]
- 36.Kahn, J. D. & Crothers, D. M. (1992) Proc. Natl. Acad. Sci. USA 89, 6343–6347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bjornson, K. P., Allen, D. J. & Modrich, P. (2000) Biochemistry 39, 3176–3183. [DOI] [PubMed] [Google Scholar]
- 38.Bjornson, K. P., Blackwell, L. J., Sage, H., Baitinger, C., Allen, D. & Modrich, P. (2003) J. Biol. Chem. 278, 34667–34673. [DOI] [PubMed] [Google Scholar]
- 39.Marra, G. & Schar, P. (1999) Biochem. J. 338, 1–13. [PMC free article] [PubMed] [Google Scholar]
- 40.Rajski, S. R., Jackson, B. A. & Barton, J. K. (2000) Mutat. Res. 447, 49–72. [DOI] [PubMed] [Google Scholar]
- 41.Solaro, P. C., Birkenkamp, K., Pfeiffer, P. & Kemper, B. (1993) J. Mol. Biol. 230, 868–877. [DOI] [PubMed] [Google Scholar]
- 42.Jones, M., Wagner, R. & Radman, M. (1987) Genetics 115, 605–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hogan, M., LeGrange, J. & Austin, B. (1983) Nature 304, 752–754. [DOI] [PubMed] [Google Scholar]
- 44.Fazakerley, G. V., Quignard, E., Woisard, A., Guschlbauer, W., van der Marel, G. A., van Boom, J. H., Jones, M. & Radman, M. (1986) EMBO J. 5, 3697–3703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bowers, J., Tran, P. T., Liskay, R. M. & Alani, E. (2000) J. Mol. Biol. 302, 327–338. [DOI] [PubMed] [Google Scholar]
- 46.Detloff, P., White, M. A. & Petes, T. D. (1992) Genetics 132, 113–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nag, D. K. & Petes, T. D. (1991) Genetics 129, 669–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hagerman, P. J. (1984) Proc. Natl. Acad. Sci. USA 81, 4632–4636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kunkel, T. A. & Bebenek, K. (2000) Annu. Rev. Biochem. 69, 497–529. [DOI] [PubMed] [Google Scholar]
- 50.O'Gara, M., Horton, J. R., Roberts, R. J. & Cheng, X. (1998) Nat. Struct. Biol. 5, 872–877. [DOI] [PubMed] [Google Scholar]
- 51.Roberts, R. J. & Cheng, X. (1998) Annu. Rev. Biochem. 67, 181–198. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.