

Abstract
The short chain fatty acid (SCFA) buyrate is a product of colonic fermentation of dietary fibers. It is the main source of energy for normal colonocytes, but cannot be metabolized by most tumor cells. Butyrate also functions as a histone deacetylase (HDAC) inhibitor to control cell proliferation and apoptosis. In consequence, butyrate and its derived drugs are used in cancer therapy. Here we show that aggressive tumor cells that retain the capacity of metabolizing butyrate are positively selected in their microenvironment. In the mouse xenograft model, butyrate-preselected human colon cancer cells gave rise to subcutaneous tumors that grew faster and were more angiogenic than those derived from untreated cells. Similarly, butyrate-preselected cells demonstrated a significant increase in rates of homing to the lung after intravenous injection. Our data showed that butyrate regulates the expression of VEGF and its receptor KDR at the transcriptional level potentially through FoxM1, resulting in the generation of a functional VEGF:KDR autocrine growth loop. Cells selected by chronic exposure to butyrate express higher levels of MMP2, MMP9, α2 and α3 integrins, and lower levels of E-cadherin, a marker for epithelial to mesenchymal transition. The orthotopic model of colon cancer showed that cells preselected by butyrate are able to colonize the animals locally and at distant organs, whereas control cells can only generate a local tumor in the cecum. Together our data shows that a butyrate-rich microenvironment may select for tumor cells that are able to metabolize butyrate, which are also phenotypically more aggressive.
Keywords: Colon Cancer, Fatty Acid Metabolism, Growth Factors, Tumor Metabolism, Tumor Metastases, Butyrate, VEGF:KDR Autocrine Loop, Tumor Microenvironment
Introduction
Short chain fatty acids are the final product of bacterial fermentation of dietary fibers (1). Butyrate, a 4-carbon fatty acid, is the main energy source for normal colonocytes (2, 3), which are found in high concentrations in the colonic lumen. It is also thought to be responsible for colorectal cancer prevention, as butyrate can also function as a histone deacetylase (HDAC)4 inhibitor to inhibit cell proliferation and induce apoptosis of cancer cells (4–6); probably because of this, cancer cells are normally unable to metabolize butyrate.
The anti-tumor effects of butyrate were described in studies using colorectal cancer cell lines, in which butyrate inhibits growth, and induces differentiation and apoptosis (7). In other studies butyrate was able to inhibit tumor growth in vivo in murine models (8, 9). Despite these findings, there is an unresolved paradox concerning the putative protective role of butyrate in colon cancer, colorectal cancers still develop and grow despite the high concentrations of butyrate in the colon. Furthermore, several studies have now shown that butyrate-resistant cells may be selected and give rise to more aggressive cancers (8, 10, 11). The mechanisms by which cancer cells develop resistance to butyrate and progress remain unknown.
Here, we demonstrate that chronic exposure of colon cancer cells to butyrate may result in the selection of more aggressive clones. Our data provides evidence showing that butyrate-resistant colon cancer cells acquire the ability to metabolize butyrate, resembling normal colonocytes. In butyrate-preselected cells we also found genes involved in EMT, cell proliferation, tumor angiogenesis, and metastasis to be up-regulated compared with the unselected cells.
EXPERIMENTAL PROCEDURES
Cell Culture
Human colon cancer cell lines (HCT15-CCL-225 and SW480-CCL-228, ATCC) were maintained in DMEM (Invitrogen 41965 with glucose 4.5 g/liter and l-glutamine) at 37 °C in a humidified 5% CO2 incubator. For butyric acid chronic exposure, cells were cultured in DMEM supplemented with 1 mm butyric acid (Sigma) for 5 days. For metabolic assays using NMR, cells grew in DMEM with 1 mm [U-13C]butyrate sodium salt (Sigma). For assaying if butyrate is directly incorporated into membranes or first transformed to acetate, HCT15 cells were grown in a total butyrate concentration of 1 mm: 20% [U-13C]butyrate and 80% butyric acid. For 13C NMR analysis of ethanol extracts HCT15 cells were grown overnight in DMEM with 10 mm [U-13C]butyrate sodium salt (Sigma).
Cell Extracts and NMR Spectroscopy
Control and butyrate-preselected HCT15 cells were collected and the lipidic fractions were extracted with 15 ml of chloroform/methanol/HCl 12 m (2:1:0.01, v/v/v) and 3.8 ml of KCl (66 mm). After centrifugation, the organic phase was evaporated under nitrogen, and lipids were suspended in chloroform d (deuterium) for NMR analysis. For analysis of the intracellular content, the cells were extracted with ice-cold ethanol (80%) and the supernatants were freeze-dried and suspended in D2O for NMR analyses.
Proton-decoupled 13C NMR spectra of cellular extracts were acquired in a Bruker AVANCE III 500 (Bruker, Rheinstetten, Germany) at 125.77 MHz, using a 5-mm 13C selective probe head. Proton decoupling was applied during the acquisition time only. Spectra were recorded at a temperature of 300 K. The chemical shifts in aqueous sample were referred to (trimethylsilyl) propanesulfonic acid, whereas the samples in chloroform d (deuterium) were referred to the solvent signal designated at 77.0 ppm. Assignments were made by comparison with chemical shifts found in the literature for metabolic intermediates. The 13C/13C-correlation spectrum (COSY) was acquired with a standard Bruker Pulse sequence using a 13C 90° flip angle under continuous 1H decoupling.
Glucose Quantification
The amount of glucose in culture supernatants was determined by using D_Glucose UV-method (Roche Applied Science/R-Biopharm).
Mice Models
Xenografted subcutaneous tumors were induced by inoculation of 5 × 106 HCT15 cells, butyric acid preselected and controls, into the subcutaneous region of 6-week-old male Balb/SCID mice. Animals were sacrificed at day 20. For the VEGF-KDR autocrine loop assay, the animals were injected intra-peritoneally every 3 days with 500 ng of IMC-11C1 (ImClone Systems, New York) or PBS. Tumors were fixed in 2% paraformaldehyde and embedded in gelatin. The antibodies used were anti-CD31 and anti-phospho-Histone3 (Table 1). Secondary antibodies were, respectively, anti-rat and rabbit FITC/phycoerythrin-coupled IgG (Alexa Fluor 488/594, Molecular Probes). Cells were examined in microscopy (Axioplan Microscope, Zeiss, Germany). Microvessel density was evaluated through CD31 immunoreactivity of the five most vascular areas (×200 field). Proliferation was evaluated through phospho-Histone3 immunoreactivity of the five most proliferative areas (×200 field).
TABLE 1.
Primary antibodies used in immunofluorescense and Western blotting
| Antigen | Antibody | Dilution | Commercial | Techniquea |
|---|---|---|---|---|
| α2 Integrin | 611016 | 1:50 | BD Biosciences | IF |
| α3 Integrin | sc-7019 | 1:50 | Santa Cruz Biotechnology, Inc. | IF |
| α5 Integrin | sc-1354 | 1:50 | Santa Cruz Biotechnology, Inc. | IF |
| α6 Integrin | 1378 | 1:50 | Chemicon | IF |
| αV Integrin | 9969 | 1:50 | Santa Cruz Biotechnology, Inc. | IF |
| β3 Integrin | sc-20058 | 1:50 | Santa Cruz Biotechnology, Inc. | IF |
| KDR | V3003 | 1:25 | Sigma | IF |
| Cytochrome oxidase-2 | sc-65239 | 1:50 | Santa Cruz Biotechnology, Inc. | IF |
| E-Cadherin | 4A2C7 | 1:50 | Zymed | IF |
| CK-19 | RCK108 | 1:100 | DakoCytomation | IH |
| FoxO1 | C29H4 | 1:50 | Santa Cruz Biotechnology, Inc. | IF |
| FoxM1 | 263C2a | 1:50 | Santa Cruz Biotechnology, Inc. | IF |
| CD31 | 553370 | 1:100 | BD Pharmigen | IF |
| pH3 | 06–570 | 1:100 | Cell Signaling Solutions | IF |
| SCAD | ABVAP696Y | 1:1000 | Abnova | WB |
| MCAD | 100–1121 | 1:1000 | Novus Biologicals | WB |
| β-Actin | A5441 | 1:1000 | Sigma | WB |
a IF, immunofluorescense; IH, immunohistochemistry; WB, Western blotting.
“Metastasis” was induced by tail vein injection of 1 × 106 HCT15 cells, butyric acid-preselected and controls, in 6-week-old male Balb/SCID mice. Animals were sacrificed 4 weeks after injection and lung, liver, and spleen were collected, fixed in 10% formalin, and embedded in paraffin. Tissues were serially sectioned (3 μm), deparaffinized, and stained with hematoxylin and eosin (H&E).
Xenografted orthotopic tumors were induced by inoculation of 1 × 106 HCT15 cells, butyric acid selected and controls, into the visceral cecal wall of 6-week-old male Balb/SCID mice. Animals were sacrificed at day 60 and cecum, lung, liver, kidney, and spleen were collected, fixed in 10% formalin, and embedded in paraffin. Tissues were serially sectioned (3 μm), deparaffinized, and stained with hematoxylin and eosin (H&E), as well as used in immunohistochemistry for CK-19 detection and in immunofluorescense for CK-19 and E-cadherin detection. Antibodies used are presented in Table 1.
Immunofluorescence
Cells were fixed in 2% paraformaldehyde for 15 min at 4 °C. Antibodies used are presented in Table 1. Secondary antibodies used were anti-rat/goat/rabbit FITC/phycoerythrin-coupled IgG (Alexa Fluor 488/594, Molecular Probes). Cells were examined using Axioplan microscope (Zeiss) and confocal microscopy using a LSM510 META microscope (Zeiss).
SDS-PAGE and Western Blotting
Cell extracts were performed with RIPA buffer + 1 mm Na2VO3 + 1× proteinase inhibitors (Roche Applied Science), on ice for 30 min. Mitochondria extracts were performed according to the Mitochondria Isolation Kit (MITOISO1, Sigma).
Primary antibodies used are shown in Table 1. Secondary antibodies were conjugated with HRP (horseradish peroxidase) and reactivity was developed using ECL Western blotting detection kit (Amersham Biosciences). Bands were quantified using Image J software (rsb.info.nih.gov/ij).
Zymography
Media supernatants of HCT15 cells treated with 1 mm butyric acid were loaded in a 12% polyacrylamide gel (30% acryamide/bis solution, 29:1 (3.3% C), Bio-Rad) with 0.1% gelatin and electrophoresis was performed in 1× TGS buffer (Bio-Rad) during 6 h at 150 V. The gel was incubated in renaturating buffer (25% Triton X-100, v/v) in agitation at room temperature, after which the gel was incubated overnight at 37 °C in developing buffer (50 mm Tris base, 200 mm NaCl, 5 mm CaCl2, 0.02% Brij 35). Staining was performed with 0.5% (w/v) Coomassie Blue R-250 for 30 min and destaining with methanol/acetic acid/water (50:10:40). Bands were quantified using ImageJ software.
Transwell Migration Assay
HCT15 cells (1 × 105) were seeded in the upper well (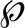 5 μm) in 100 μl of DMEM, 10% FBS and in the lower well in 500 μl of DMEM, 10% FBS, cells were added in the presence or absence of EGF (20 ng/μl) and/or 1 mm butyric acid. Each condition was performed in triplicate and after 12 h the number of cells in the lower well was counted.
5 μm) in 100 μl of DMEM, 10% FBS and in the lower well in 500 μl of DMEM, 10% FBS, cells were added in the presence or absence of EGF (20 ng/μl) and/or 1 mm butyric acid. Each condition was performed in triplicate and after 12 h the number of cells in the lower well was counted.
Enzyme-linked Immunosorbent Assay (ELISA)
Expression of VEGF was measured in medium from cultures of HCT15 control cells and HCT15 cells were treated with 1 mm butyric acid, using VEGF ELISA kit Human (Calbiochem) according to the manufacturer's instructions.
Reverse Transcription-Polymerase Chain Reaction (RT-PCR) and mRNA Arrays
RNA extraction (RNeasy mini kit, Qiagen), RT-PCR, and mRNA arrays (Oligo GEArray, SuperArray Bioscience corporation) were performed following standard protocols.
Relative Quantifying PCR
RNA extraction (RNeasy mini kit, Qiagen) using RT-PCR was performed using SuperScript® II Reverse Transcriptase (Invitrogen) and RQ-PCR with SYBR® Green PCR Master Mix (Applied Biosystems) in a ABI PRISM® 7900HT (Applied Biosystems). The primers used to detect glucose transporters GLUT1 and SGLT1, monocarboxylates transporter 1 (MCT1), hexokinase II (HEXII), glucose-6-phosphate dehydrogenase, pyruvate kinase (PKLR), phosphoenolpyruvate carboxykinase (PEPCK), and SNAIL are presented in Table 2.
TABLE 2.
Primers used in relative quantifying PCR
| Gene | Primer | Sequence (5′–3′) |
|---|---|---|
| GLUT1 | FOR | CACGGCCTTCACTGTCGTG |
| REV | GGACATCCAGGGTAGCTGC | |
| SGLT1 | FOR | CACGGGTCTCTGGTCTGC |
| REV | CATGATGAACATGGGCATCAG | |
| MCT1 | FOR | GGACTCTTTGCACCTTTGGTGT |
| REV | CAACAAGGTCCATCAATGTTTCAA | |
| HEXII | FOR | GCATGAGTTTGACCAGGAG |
| REV | CAAAGAGCAGCTCCTCCTTG | |
| G6PD | FOR | GGCAACAGATACAAGAACGTGA |
| REV | GCAGAAGACGTCCAGGATGAG | |
| PKLR | FOR | AGGCGTGAAGAGGTTTGATGAG |
| REV | CCCCCGTGCCACCAT | |
| PEPCK | FOR | GCGGATCATGACGCGGATG |
| REV | GAGCGTCAGCTCCGGGTTG | |
| SNAIL | FOR | CTCTTTCCTCGTCAGGAAGC |
| REV | GGCTGCTGGAAGGTAAACTC |
pGL3basic Constructs and Luciferase Reporter Gene Assay
SCAD, MCAD, VEGF, and KDR promoters were analyzed for transcription factor binding site prediction using the TFBlast data base. Detection fragments corresponding to promoter regions were amplified by PCR using primers described in Table 3. For transient transfection assays, 2.5 × 105 HCT15 cells/well were seeded overnight in 24-well plates. Transfections were performed using Lipofectamine 2000 (Invitrogen), according to the standard protocol, with 0.5 μg of the Renilla luciferase expressing vector (transfection control) and 0.5 μg of the pGL3 construct of interest. After transfection, some cells were maintained in DMEM, 10% FBS and others in DMEM, 10% FBS, and 1 mm butyric acid, and all conditions were tested in triplicate. Total cell extracts were prepared after 48 h according to the Dual Luciferase reporter gene assay system protocol (Promega). The luciferase activity of test plasmids was expressed as -fold of induction compared with the activity of the corresponding empty vector (pGL3 basic, Promega), after correction for transfection efficiency with the levels of R. luciferase activity.
TABLE 3.
Primers used for SCAD, MCAD, VEGF, and KDR promoters constructs
| Promoter gene | Primer | Sequencea (5′–3′) |
|---|---|---|
| SCAD | FOR1 | ATGACGCGTCCTGGCCGCG AGCGCACCTC |
| SCAD | FOR2 | ATGACGCGTGGCCCTGATAGACAAGGCAGG |
| SCAD | REV | ATGAAGCTTCCAGGCTTCGCGACCTCCCG |
| MCAD | FOR1 | ATGACGCGTGTTGGCTACCCGGCGCCGGG |
| MCAD | FOR2 | ATGACGCGTGGGCCAGAGGTGGAAACGCAG |
| MCAD | FOR3 | ATGACGCGTCCCCACCGTTCAGCGCAACC |
| MCAD | REV | ATGAAGCTTGTTGGCTCCCGTTCCGGCCAC |
| VEGF | FOR1 | ATGACGCGTGGGTGCTAGAGGCGCAC |
| VEGF | FOR2 | ATGACGCGTCCAGATGAGGGCTCCAGTGG |
| VEGF | FOR3 | ATGACGCGTCCCCGCGGGCGCGTGTTC |
| VEGF | FOR4 | ATGACGCGTCCCGGCGGGGCGGAGCCA |
| VEGF | REV | ATGAAGCTTCCCCCAGCGCCACGACC |
| KDR | FOR1 | ATGACGCGTGGAGATCGCCGCCGGGTACC |
| KDR | FOR2 | ATGACGCGTCCAGCTTGCACCCGGCATAC |
| KDR | REV | ATGAAGCTTCGGGCGAAATGCCAGAACTCG |
a MluI and HindIII restriction sites (in bold) were included, respectively, in forward and reverse primers.
Statistical Analysis
Results are expressed as mean ± S.D. Data were analyzed using the unpaired two-tailed Student's t test or the Tukeys test one-way analysis of variance. p values of <0.05 were considered significant.
RESULTS
HCT15 Colon Cancer Cells Are Able to Fully Metabolize Butyrate
Under the culture conditions defined under “Experimental Procedures” (in the presence of 1 mm butyrate, for up to 5 days), the HCT15 colon cell line thrives and shows growth patterns similar to untreated cells, whereas SW480 grew much slower when exposed to butyrate (supplemental Fig. 1A) and the levels of cell death were 2-fold higher in the SW480 cell line compared with HCT15 after 5 days of exposure to butyrate (supplemental Fig. 1B). Therefore, we used these culture conditions to study the effects of butyrate in metabolic profiling and the acquisition of phenotypic changes by HCT15 cells.
The 13C NMR spectra of lipid extracts of butyrate-treated HCT15 cells showed no substantial alterations in lipid contents, compared with untreated cells (Fig. 1A). The major difference observed was an intensity increase of the resonances in the spectral region characteristic of the olephinic carbons (127 to 131 ppm), indicating an increase in lipid unsaturation. In the 13C NMR spectra of HCT15 cells treated with [U-13C]butyrate extracts, it was possible to observe the incorporation of the labeled carbons from [U-13C]butyrate in the lipid fraction (Fig. 1A). The peaks assigned to ω-1 (14 ppm), ω-2 (22 ppm), ω-3 (32 ppm), C1 (174 ppm), C2 (34 ppm), C3 (24–25 ppm), and the elongation carbons (27–30 ppm) of long chain fatty acids indicated the existence of carbons originating from the [U-13C]butyrate and from other sources. The incorporation of labeled carbons in glycerol was not detected (data not shown), indicating that in triacylglycerol molecules, carbons from butyrate were only incorporated in the fatty acid chain. For the SW480 cell line, there was no alteration in the spectra from the lipidic fraction with and without [U-13C]butyrate, suggesting that butyrate is not metabolized by these cells.
FIGURE 1.

HCT15 colon cancer cells are able to fully metabolize butyrate. A, 13C NMR spectra of the lipid fraction of HCT15 and SW480 control cells, treated with butyric acid, and sodium [U-13C]butyrate showed that in HCT15 the presence of butyrate induces a slight change in lipid profile and its carbons are incorporated in all positions of triacylglycerides; B, 13C NMR spectra of ethanolic extracts of HCT15 control cells and treated with sodium [U-13C]butyrate showed that carbons from butyrate are incorporated in carbons 4 and 5 of glutamate and carbon 5 of praline; and C, 13C-13C NMR COSY spectra of the lipid extract of HCT15 cells treated with 20% [U-13C]butyrate and 80% of butyric acid, showed only cross-peaks between two 13C indicating that butyrate is metabolized into acetate prior to incorporation.
In the 13C-13C COSY spectra of lipid extracts from HCT15 cells grown in 20% [U-13C]butyrate and 80% of butyric acid, each signal shows a single COSY correlation, i.e. ω-2 correlated only with ω-1 and not with ω-3, which in turn presented a correlation with an elongation carbon (Fig. 1C), meaning that carbon is incorporated in two atom blocks. Hence, 13C-13C COSY spectra showed that butyrate enters in β-oxidation and is catabolized to acetate prior to incorporation into fatty acids.
The 13C NMR spectra of ethanol extracts of HCT15 cells treated overnight with 10 mm [U-13C]butyrate showed that the labeled carbons were incorporated in position 5 of proline and in position 4 of glutamate (Fig. 1B). The J13C-13C coupling constant present in carbon 4 of glutamate indicated that carbon 5 is also labeled. These results suggest that carbons from butyrate enter the oxidative pathway of the Kreb's cycle as acetyl-CoA, being converted to glutamate through oxaloacetate. These data are in agreement with increased expression of SCAD and MCAD (Fig. 2A), acyldehydrogenases that can catalyze the first step of butyrate β-oxidation. Luciferase reporter gene assays showed that the SCAD and MCAD promoters were activated in HCT15 cells previously exposed to butyrate (Fig. 2B). We also found that butyrate-preselected cells express more resistin (RETN) (Fig. 3), which is a hormone that represses the uptake of glucose. This indicates that like normal colonocytes, these cells preferred to metabolize butyrate rather than glucose. Taken together, these data showed that at least a subset of HCT15 cells can fully metabolize butyrate.
FIGURE 2.

The expression of enzymes and transporters are altered after butyrate exposure. A, Western blotting for MCAD and SCAD showed that these oxidation enzymes are increased in cells preselected by butyrate (p < 0.05); B, luciferase reporter gene assay showed that both SCAD promoter sequences −119 to −1 and −399 to −1 are significantly stimulated in cells preselected by butyric acid (p = 0.0278 for pSCAD1 and p = 0.037 for pSCAD3), the same results were observe for the MCAD promoter sequences −201 to −1 and −421 to −1 (p = 0.0213 for pMCAD1 and p = 0.0158 for pMCAD3); C, RQ-PCR showed, in HCT15, a decreased expression of mRNAs from glucose transporters GLUT1 and SGLT1, whereas MCT1 is increased in HCT15 cells preselected by butyrate; the mRNAs encoding the HEXII from glycolysis and glucose-6-phosphate dehydrogenase (G6PD) from the pentose-phosphate pathway are decreased, whereas pyruvate kinase (PKLR) from glycolysis and phosphoenolpyruvate carboxykinase (PEPCK) from gluconeogenesis are increased; in SW480, mRNA related to GLUT1, MCT1, and G6PD are increased; and E, immunoreactivity for cytochrome oxidase 2 showed mitochondria aggregation in butyrate-preselected HCT15 cells, whereas in control cells they are disperse as well as in SW480 control and butyrate-selected cells. D, HCT15 cells preselected by butyrate consume 20% less glucose titan control cells.
FIGURE 3.

HCT15 cells preselected with butyrate express more FoxM1 and membrane and metastases-associated proteins. SuperArray analysis for cell membrane and metastasis markers revealed that HCT15 cells preselected with butyric acid express higher levels of genes related to inhibition of glucose uptake, low differentiation, cell cycle and proliferation, and a more invasive (metastatic) behavior. These data were obtained from three independent experiments, with consistent results.
RQ-PCR showed decreased mRNA expression of glucose transporters GLUT1 and SGLT1, whereas MCT1 was increased in the butyrate-preselected HCT15 cells, indicating that HCT15 cells uptake butyrate rather than glucose, because MCT1 is a transporter for butyrate (Fig. 2C). Despite the fact that HEXII is not the unique hexokinase encoding gene, its decreased expression is in agreement with the lower rates of glucose uptake observed in HCT15 cells cultured in butyrate-supplemented medium, which is about 20% less than control cells (Fig. 2D). The expression of PKLR was increased, indicative of glycolysis. The decreased expression of glucose-6-phosphate dehydrogenase indicates that the pentose-phosphate pathway was partially affected by the lack of glucose, however, the increased expression of PEPCK indicates that gluconeogenesis was activated (Fig. 2C). In SW480, the expression of GLUT1 was slightly increased in butyrate-preselected cells, whereas SGLT1 was expressed at the same levels as in control cells. MCT1 expression was also increased in SW480 butyrate-preselected cells, indicating the uptake of butyrate. The expression levels of HEXII, PKLR, and PEPCK were similar expression levels in SW480 control and butyrate-preselected cells, whereas glucose-6-phosphate dehydrogenase is up-regulated in butyrate-preselected cells. In addition, butyrate-preselected cells formed mitochondria aggregates in their cytoplasm, whereas the mitochondria were dispersed in the controls as well as the SW480 control and butyrate-exposed cells (Fig. 2E).
Butyrate-preselected Cells Are More Effective in Tumor Growth, Angiogenesis, and Metastasis in Vivo
Having shown that HCT15 butyrate-preselected cells fully metabolize butyrate, we reasoned that these cells might exhibit different cell phenotypes and behavior in vivo and in vitro compared with control HCT15 cells. Accordingly, butyrate pre-treatment of HCT15 cells prior to subcutaneous inoculation into Balb/SCID mice generated tumors that were significantly more aggressive than those established from control cells. In particular, the tumor sizes (Fig. 4A) and proliferation rates (Fig. 4B) of butyrate-preselected tumors were significantly higher than those established from control cells. Moreover, the microvessel density of butyrate-preselected tumors was also increased (Fig. 4C). In addition to the significant effects in primary tumor growth, butyrate treatment also promoted the formation of lung homing spots. Butyrate-preselected HCT15 cells formed a significant number of lung metastases shortly after inoculation, whereas control cells formed significantly fewer metastases (Fig. 4D). In both groups of animals the liver demonstrated no alterations or metastasis (data not shown). Taken together, these data show that butyrate-preselected cells have increased tumorigenic and metastatic potential.
FIGURE 4.

HCT15 cells preselected with butyrate, in vivo (Balb/SCID mice), induce more vascularized and proliferative tumors, generating of a functional VEGF:KDR autocrine loop, and exhibited a higher capacity of homing in lungs. A, subcutaneous inoculation of HCT15 cells treated with butyric acid gave rise to significantly bigger tumors (p = 0.0097) than non-treated HCT15 cells, treatment of mice, inoculated with butyrate-preselected HCT15 cells, with IMC-1C11 blocks the augmented tumor growth; B, immunoreactivity for phospho-Histone3 showed that tumors developed from HCT15 cells treated with butyric acid are significantly more proliferative (p = 0.02) than tumors from non-treated HCT15 cells, treatment with IMC-1C11 also decreases proliferation; C, immunoreactivity for CD31 in subcutaneous tumors showed that tumors developed from HCT15 cells preselected with butyric acid are significantly more vascularized (p < 0.0001) than tumors from non-treated HCT15 cells, treatment with IMC-1C11 also decreases vascularization; D, H&E from lung sections (quantified in the plot) showed that mice injected with HCT15 cells preselected with butyric acid developed significantly more lung homing spots than control cells. These data were obtained from three independent experiments (6 mice/experiment/experimental condition), with consistent results. *, statistical significance.
These cancer cells were also implanted orthotopically into the cecal wall. Sixty days after implantation, mice injected with control HCT15 cells showed gross lesions only in the cecum, whereas those injected with butyrate-preselected HCT15 cells showed gross lesions in the peritoneum, liver, pancreas, kidney, lung, and mediastinum (Fig. 5A). Histological examination showed that tumors derived from the control HCT15 cells were multifocal to diffuse, with severe infiltration of the muscle layer, submucosa, and lamina propria. The tumor patterns were mainly nested, with areas of solid growth and moderate lymphatic invasion (Fig. 5B). Primary tumors from butyrate-preselected HCT15 cells were multifocal, with much smaller foci confined to the submucosa. These tumors were rarely detected in the muscle layer, with no infiltration of the lamina propria; but lymphatic invasion was severe for these tumors (Fig. 5B). Notably, metastasis to the liver and lung occurred through vascular dissemination but not by diffusion/infiltration from the serosal surfaces, as observed in the spleen, kidney, and pancreas (Fig. 5C).
FIGURE 5.

Xenografted orthotopic models in Balb/SCID mice showed that butyrate-preselected HCT15 cells are more invasive and metastastic than control cells. A, macroscopic observation showed that mice inoculated with control cells had only a tumor in the cecum, whereas mice inoculated with butyrate-preselected HCT15 cells had a local tumor and distant metastasis in the liver, kidney, and mediastinum; B, histological analysis showed that mice inoculated with control cells had a bigger tumor mass in the point of injection, in lamina propria and forming gland-like structures, whereas mice inoculated with butyrate-preselected HCT15 cells had a smaller tumor mass and an extended angio invasion; and C, mice inoculated with butyrate-preselected HCT15 cells presented liver, lung, and kidney metastasis and serous invasion of the spleen.
Butyrate-preselected Cells Showed Increased MMPs and Migration
Next, we explored the molecular basis for the phenotypic features seen in tumors from butyrate-preselected HCT15 cells. First, as determined by zymography, butyrate-preselected HCT15 cells secreted significantly higher levels of active MMP9 and MMP2 (Fig. 6A) compared with control cells, suggestive of increased invasive behavior. Moreover, butyrate-preselected HCT15 cells showed increased migratory capacity to EGF gradient (Fig. 6B), and increased VEGF production into the culture supernatants (Fig. 7A), consistent with the increased angiogenesis seen in vivo (Fig. 4C), and the increased KDR expression (Fig. 7B).
FIGURE 6.

HCT15 cells preselected with butyrate exhibit a more invasive and motile phenotype. A, analysis of cell culture supernatants by gelatinolytic zymography showed that HCT15 cells after 120 h of butyric acid selection produce more matrix metalloproteinases 9 and 2 (MMP9 and MMP2) than those produced by control cells; B, transwell assay showed that HCT15 cells preselected with butyric acid migrate more in response to EGF (p < 0.04); C, butyrate-preselected HCT15 cells have reduced E-cadherin expression by immunofluorescense and Western blotting; D, RQ-PCR showed increased levels of mRNA from SNAIL in butyrate-preselected HCT15 cells; E, as shown by immunofluorescence butyrate-preselected HCT15 cells have increased expression of integrins related to invasion and migration (α2, α3, αv, and β3), and decreases the expression of integrins related to the maintenance of epithelium architecture (α5 and α6). These data were obtained from three independent experiments, with consistent results.
FIGURE 7.

HCT15 cells preselected with butyrate have a more angiogenic phenotype. A, as determined by ELISA, butyrate-preselected HCT15 cells express more VEGF than control cells (p = 0.03); B, as determined by immunofluorescence, butyrate-preselected HCT15 cells express more KDR than control cells. These data were obtained from three independent experiments, with consistent results; C, luciferase reporter gene assay showed that the VEGF promoter sequence −131 to −1 is significantly more active (p < 0.02) than the other tested sequences in HCT15 cells, and deletion constructs pVEGF1, pVEGF2, and pVEGF4 are significantly stimulated in cells preselected by butyric acid (p = 0.041 for pVEGF1, p = 0.0073 for pVEGF2 and p = 0.0098 for pVEGF4); D, luciferase reporter gene assay showed that the KDR promoter sequence −446 to −1 is significantly more active (p = 0.046) than the sequence −796 to −1 in HCT15 cells, and both deletion constructs of KDR promoter are significantly stimulated in cells preselected by butyric acid (p = 0.0205 for pKDR1 and p = 0.0477 for pKDR2; E, as shown by immunofluorescence, butyrate-preselected HCT15 cells show low levels of FoxO1 and increased expression of FoxM1 transcription factor.
Butyrate-preselected Cells Showed Higher VEGF and KDR Transcriptional Levels
We exploited further the regulation of VEGF and KDR expression in butyrate-preselected HCT15 cells. As shown using luciferase reporter gene assays, both VEGF and KDR promoters exhibited higher activities in butyrate-preselected cells (Fig. 7, C and D). The regulatory elements responsible for the induction of promoter activity in the butyrate-preselected cells appeared to locate between −131 and −66 upstream of the ATG translational start site, whereas for the KDR promoter the responsive region was mapped between −446 and the ATG. We next analyzed the involvement of the forkhead Fox transcription factors (previously shown to regulate VEGF expression (12, 13)) in the establishment of butyrate-preselected HCT15 cells. As shown in Fig. 7E, there were no detectable differences in FoxO1 expression between the control and butyrate-preselected cells, whereas FoxM1 expression was increased in the butyrate-preselected cells compared with the controls. Taken together, these data strongly suggest that butyrate-preselected cells have higher levels of activated VEGF and KDR, and this induction is mediated at the transcriptional level; in the case of VEGF, it may involve the transcription factor FoxM1 (14, 15).
Butyrate-preselected Tumors Showed VEGF:KDR Autocrine Loop
Because butyrate-selected HCT15 cells showed increased expression of VEGF and KDR, we reasoned it might result in the generation of a functional VEGF:KDR autocrine loop, as shown in other tumor models (16–19), including colon cancer cells (20). Indeed, as shown in Fig. 4A, treatment of mice harboring butyrate-preselected HCT15 tumors with a human KDR neutralizing antibody, IMC-1C11, inhibited the increment in tumor growth induced by butyrate. Accordingly, the proliferation rate (Fig. 4B), and the tumor microvessel densities (Fig. 4C), of preselected butyrate tumors that received IMC-1C11 were identical to the control tumors; possibly due to decreased tumor growth. These data strongly suggests that preselected butyrate cells exhibit a functional VEGF:KDR autocrine loop that promotes tumor growth.
Butyrate-preselected HCT15 Cells Showed Lower Expression of Membrane Proteins, Namely E-cadherin, Suggestive of Selection of EMT Positive Cells
The expression of integrins by HCT15 was altered in cells preselected by butyrate (Fig. 6E). HCT15 cells preselected with butyrate expressed higher levels of α2, α3, αv, and β3 integrins and lower levels of α5 and α6 integrins compared with control cells. Butyrate exposure also resulted in the selection of E-cadherin negative cells (Fig. 6C), which has been reported to be evidence for EMT (21, 22). The repression of E-cadherin expression is mediated through Snail whose expression is also increased in butyrate-preselected cells (Fig. 6D).
We also characterized a more global gene expression profile of butyrate-preselected versus control cells, focusing on differentiation, cell cycle, and metastasis-related genes. As shown in Fig. 3, the expression of poorly differentiation markers dipeptidyl phosphatase 4 (DPP4) and cytokeratin (KRT17) were increased in the butyrate-preselected cells. Similarly, genes related to increased proliferation, namely cytokeratin (KRT18), myosin 9 (MYH9), and cyclin-dependent kinase 4 (CDK4) were also induced in the butyrate-preselected cells. Moreover, several genes indicative of invasive and metastatic behavior were increased upon butyrate selection, including dysadherin (FXYD5), GTPase KRas (KRAS), α2 and α3 integrins (ITGA2 and -3), and Rho-related GTP-binding protein RhoC (RHOC), among others. Collectively, these data show that butyrate treatment results in the selection of HCT15 cells that have a more metastatic phenotype, including EMT.
DISCUSSION
In this study, we describe in detail the role of colonic butyrate in the selection of aggressive colon cancer cells that retain the capacity of normal cells to metabolize butyrate. Our data shows biochemical indicators of metabolic adaptation by HCT15 colon cancer cells preselected by butyrate.
As shown by NMR spectroscopy, HCT15 butyrate-resistant cells, like normal colonocytes, are able to fully metabolize butyrate. We show that butyrate is degraded into 2 carbon molecules (acetyl-ACP), which is in agreement with the increased expression of SCAD and MCAD, the only acyldehydrogenases that can catalyze the first step of butyrate β-oxidation. The 13C atoms from butyrate were incorporated in long carbon chains that probably constitute long chain fatty acids, because the acetyl-CoA formed from acetyl groups resulting from butyrate degradation are used as blocks in the synthesis of long chains fatty acids. It was also observed that glutamate was synthesized from butyrate 13C atoms, indicating that acetyl-CoA from butyrate degradation is also entering Kreb's cycle. RETN is a hormone that represses the uptake of glucose by cells and has been implicated in insulin resistance (23), and its overexpression (as shown here) indicates that butyrate-preselected HCT15 cells prefer to metabolize butyrate rather than glucose. RETN has also been described in throphoblastic cells as being responsible for GLUT1 down-regulation (24) as we saw in HCT15 butyrate-preselected cells. A gut-derived resistin-like protein has recently been pointed out as a down-regulator of SGLT1 (25). The low rate of glucose uptake corroborates with the down-regulation of HEXII and glucose-6-phosphate dehydrogenase genes that encode hexokinase type 2 from glycolysis and glucose-6-phosphate dehydrogenase from the pentose-phosphate pathway, respectively. Nevertheless, there are other genes encoding for hexokinases whose expression was not evaluated. The expression of the PEPCK gene is up-regulated in HCT15 butyrate-preselected cells, indicating that gluconeogenesis is activated. On the other hand the increased expression of PKLR can be due to the fact that cells are producing glucose to be degraded in glycolysis. The SW480 cells import glucose and butyrate, as the expression of GLUT1 and MCT1 are increased in butyrate-preselected cells. The butyrate uptake by SW480 cells is in agreement with the apoptotic levels observed in SW480 following exposure to butyrate, because these cells are not able to metabolize butyrate, which can function as an HDAC inhibitor.
The mitochondria aggregation observed in butyrate-preselected HCT15 cells is a phenomenon that has previously been described during embryonic development (26, 27), in which mitochondria tend to group when cell energy demand increases, to support nucleus activity. In our study this aggregation can be related to an increased rate of β-oxidation. Taken together, the evidence for metabolic adaptation (selection) of HCT15 colon cancer cells exposed to butyrate strongly suggested that this might occur to the selection for cells with a more aggressive phenotype. In agreement, we show in vivo that HCT15 cells preselected by butyrate generated more proliferative, more angiogenic, and more metastatic tumors than control cells.
Subcutaneous tumors established from butyrate-preselected HCT15 cells have a higher proliferation rate and are more angiogenic than tumors established from untreated cells. Although the increased proliferation may be a direct consequence of the fact that they are metabolically adapted, the increased angiogenesis observed in butyrate-preselected HCT15 cells results from transcriptional modulation of VEGF, which may involve FoxM1 and FoxO1 transcription factors as previously suggested (13–15). Very recently, Behren et al. (26) published that FoxM1 is an essential factor for migration and invasion, and it has been identified as a key downstream target of Ras. In our study we also observed an up-regulation of KRAS as mentioned below. In addition, butyrate-preselected cells also express higher levels of KDR, resulting in establishment of a functional VEGF:KDR autocrine loop. Accordingly, in vivo treatment of mice inoculated with butyrate-preselected HCT15 cells and treated with IMC-1C11, a human KDR blocking antibody, reverted tumor growth to control levels. Our previous work showed the VEGF:KDR autocrine loops are essential for the growth of subsets of hematologic malignancies (16–19), whereas others have suggested their involvement in solid tumor growth as well (16–20). In prostate cancer the VEGF autocrine loop is involved in EMT (27). Taken together, we demonstrate for the first time a link between metabolic adaptation (to butyrate) and the selection of tumor cells with increased tumorigenic potential, which includes the generation of a functional angiogenic growth factor autocrine loop.
The increased in vivo metastasis and tumor spread of butyrate-preselected HCT15 cells is in agreement with the in vitro reduced expression of α5 and α6 integrins, which bind components of the basal lamina and adhesion molecules such as E-cadherin (19, 22), and also with the reduced expression of E-cadherin, a strong indication of EMT (21, 22). In turn, butyrate selection is accompanied by an increase in α2, α3, αv, and β3 integrins, whose expression may be associated with increased metastatic behavior because they bind to extracellular matrix proteins and endothelial selectins. Butyrate-preselected cells also produce more active MMP2 and MMP9, well known for their role in metastasis formation in numerous models (28, 29). Other known metastases regulators also modulated by butyrate include dysadherin (FXYD5), which down-regulates E-cadherin (30) and reduces cell adhesion and promoting metastasis; KRAS whose overactivity is related to the disruption of β-catenin and E-cadherin binding and is implicated in tumor metastases formation including colon cancer (31); RHOC, which regulates a signal transduction pathway, promoting the assembly of focal adhesion molecules and actin stress fibers formation and has been associated with metastases formation in the lung (32); FSCN1, an actin-binding protein that is involved in cell membrane stabilization playing a role in stress fibers formation and consequent cell migration, and is also associated with the metastatic process (32, 33). Taken together, butyrate exposure can result in the selection of colon cancer cells that show evidence for EMT; and express genes related to increased metastatic behavior.
Cancer cells that are capable of metabolizing butyrate are protected against its role as a HDACs inhibitor, because butyrate does not accumulate in cytoplasm and is not transferred into the nucleous. Our data suggests that the colonic, butyrate-rich, microenvironment may play a role in the selection of more aggressive colon cancer cells that maintain the ability to use butyrate as a carbon and energy source.
Supplementary Material
Acknowledgments
We acknowledge the Serviço de Anatomia Patológica, Instituto Português de Oncologia de Francisco Gentil, Centro de Lisboa, EPE (CIPM/IPOLFG) for the support in histological analysis. We thank Prof. Helena Santos (ITQB) for the scientific support in NMR experiments. We acknowledge the use of the NMR spectrometers at CERMAX, which are part of the National NMR Network and were acquired with funds from FCT and FEDER.
This work was supported in part by grants from the Fundação Luso Americana para o Desenvolvimento (FLAD) and Liga Portuguesa Contra o Cancro (LPCC), Núcleo Regional Sul.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Fig. S1.
- HDAC
- histone deacetylase
- PEPCK
- phosphoenolpyruvate carboxykinase
- PKLR
- pyruvate kinase
- MCT1
- monocarboxylates transporter 1
- EMT
- epithelial to mesenchymal transition
- RQ
- relative quantifying
- MMP
- matrix metalloproteinases
- HEXII
- hexokinase II.
REFERENCES
- 1.Bugaut M. (1987) Comp. Biochem. Physiol. B 86, 439–472 [DOI] [PubMed] [Google Scholar]
- 2.Roediger W. E. (1982) Gastroenterology 83, 424–429 [PubMed] [Google Scholar]
- 3.Wong J. M., de Souza R., Kendall C. W., Emam A., Jenkins D. J. (2006) J. Clin. Gastroenterol. 40, 235–243 [DOI] [PubMed] [Google Scholar]
- 4.Leng S. L., Leeding K. S., Gibson P. R., Bach L. A. (2001) Carcinogenesis 22, 1625–1631 [DOI] [PubMed] [Google Scholar]
- 5.Hinnebusch B. F., Meng S., Wu J. T., Archer S. Y., Hodin R. A. (2002) J. Nutr. 132, 1012–1017 [DOI] [PubMed] [Google Scholar]
- 6.Shi S. L., Wang Y. Y., Liang Y., Li Q. F. (2006) World J. Gastroenterol. 12, 1694–1698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Heerdt B. G., Houston M. A., Augenlicht L. H. (1994) Cancer Res. 54, 3288–3293 [PubMed] [Google Scholar]
- 8.Mariadason J. M., Velcich A., Wilson A. J., Augenlicht L. H., Gibson P. R. (2001) Gastroenterology 120, 889–899 [DOI] [PubMed] [Google Scholar]
- 9.Medina V., Afonso J. J., Alvarez-Arguelles H., Hernández C., González F. (1998) J. Parenter. Enteral. Nutr. 22, 14–17 [DOI] [PubMed] [Google Scholar]
- 10.Chai F., Evdokiou A., Young G. P., Zalewski P. D. (2000) Carcinogenesis 21, 7–14 [DOI] [PubMed] [Google Scholar]
- 11.López de Silanes I., Olmo N., Turnay J., González de Buitrago G., Pérez-Ramos P., Guzmán-Aránguez A., García-Díez M., Lecona E., Gorospe M., Lizarbe M. A. (2004) Cancer Res. 64, 4593–4600 [DOI] [PubMed] [Google Scholar]
- 12.Wang Z., Banerjee S., Kong D., Li Y., Sarkar F. H. (2007) Cancer Res. 67, 8293–8300 [DOI] [PubMed] [Google Scholar]
- 13.Furuyama T., Kitayama K., Shimoda Y., Ogawa M., Sone K., Yoshida-Araki K., Hisatsune H., Nishikawa S., Nakayama K., Nakayama K., Ikeda K., Motoyama N., Mori N. (2004) J. Biol. Chem. 279, 34741–34749 [DOI] [PubMed] [Google Scholar]
- 14.Zhang Y., Zhang N., Dai B., Liu M., Sawaya R., Xie K., Huang S. (2008) Cancer Res. 68, 8733–8742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dormond O., Madsen J. C., Briscoe D. M. (2007) J. Biol. Chem. 282, 23679–23686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Santos S. C., Dias S. (2004) Blood 103, 3883–3889 [DOI] [PubMed] [Google Scholar]
- 17.Dias S., Hattori K., Heissig B., Zhu Z., Wu Y., Witte L., Hicklin D. J., Tateno M., Bohlen P., Moore M. A., Rafii S. (2001) Proc. Natl. Acad. Sci. U.S.A. 98, 10857–10862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weigand M., Hantel P., Kreienberg R., Waltenberger J. (2005) Angiogenesis 8, 197–204 [DOI] [PubMed] [Google Scholar]
- 19.Sher I., Adham S. A., Petrik J., Coomber B. L. (2009) Int. J. Cancer 124, 553–561 [DOI] [PubMed] [Google Scholar]
- 20.Calvani M., Trisciuoglio D., Bergamaschi C., Shoemaker R. H., Melillo G. (2008) Cancer Res. 68, 285–291 [DOI] [PubMed] [Google Scholar]
- 21.Contreras H. R., Ledezma R. A., Vergara J., Cifuentes F., Barra C., Cabello P., Gallegos I., Morales B., Huidobro C., Castellón E. A. (2010) Urologic Oncology 28, 534–540 [DOI] [PubMed] [Google Scholar]
- 22.von Burstin J., Eser S., Paul M. C., Seidler B., Brandl M., Messer M., von Werder A., Schmidt A., Mages J., Pagel P., Schnieke A., Schmid R. M., Schneider G., Saur D. (2009) Gastroenterology 137, 361–371 [DOI] [PubMed] [Google Scholar]
- 23.Park S., Hong S. M., Sung S. R., Jung H. K. (2008) Endocrinology 149, 445–454 [DOI] [PubMed] [Google Scholar]
- 24.Di Simone N., Di Nicuolo F., Marzioni D., Castellucci M., Sanguinetti M., D'lppolito S., Caruso A. (2009) J. Cell. Mol. Med. 13, 388–397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Krimi R. B., Letteron P., Chedid P., Nazaret C., Ducroc R., Marie J. C. (2009) Diabetes 58, 2032–2038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mitchell M., Schulz S. L., Armstrong D. T., Lane M. (2009) Biol. Reprod. 80, 622–630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barnett D. K., Kimura J., Bavister B. D. (1996) Dev. Dyn. 205, 64–72 [DOI] [PubMed] [Google Scholar]
- 28.Gentner B., Wein A., Croner R. S., Zeittraeger I., Wirtz R. M., Roedel F., Dimmler A., Dorlaque L., Hohenberger W., Hahn E. G., Brueckl W. M. (2009) Anticancer Res. 29, 67–74 [PubMed] [Google Scholar]
- 29.Struckmann K., Mertz K., Steu S., Storz M., Staller P., Krek W., Schraml P., Moch H. (2008) J. Pathol. 214, 464–471 [DOI] [PubMed] [Google Scholar]
- 30.Izumi T., Oda Y., Hasegawa T., Nakanishi Y., Kawai A., Sonobe H., Takahira T., Kobayashi C., Yamamoto H., Tamiya S., Hirohashi S., Iwamoto Y., Tsuneyoshi M. (2007) Am. J. Surg. Pathol. 31, 85–94 [DOI] [PubMed] [Google Scholar]
- 31.Miranda E., Destro A., Malesci A., Balladore E., Bianchi P., Baryshnikova E., Franchi G., Morenghi E., Laghi L., Gennari L., Roncalli M. (2006) Br. J. Cancer 95, 1101–1107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vignjevic D., Schoumacher M., Gavert N., Janssen K. P., Jih G., Laé M., Louvard D., Ben-Ze'ev A., Robine S. (2007) Cancer Res. 67, 6844–6853 [DOI] [PubMed] [Google Scholar]
- 33.Lee T. K., Poon R. T., Man K., Guan X. Y., Ma S., Liu X. B., Myers J. N., Yuen A. P. (2007) Cancer Lett. 254, 308–315 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.