

Abstract
Tumor necrosis factor (TNF)-α, a homotrimeric, pleiotropic cytokine, is secreted in response to inflammatory stimuli in diseases such as rheumatoid arthritis and inflammatory bowel disease. TNF-α mediates both apoptosis and inflammation, stimulating an inflammatory cascade through the non-canonical pathway of NF-κB activation, leading to increased nuclear RelB and p52. In contrast, the common food additive carrageenan (CGN) stimulates inflammation through both the canonical and non-canonical pathways of NF-κB activation and utilizes the adaptor molecule BCL10 (B-cell leukemia/lymphoma 10). In a series of experiments, colonic epithelial cells and mouse embryonic fibroblasts were treated with TNF-α and carrageenan in order to simulate the possible effects of exposure to dietary CGN in the setting of a TNF-α-mediated inflammatory disease process. A marked increase in secretion of IL-8 occurred, attributable to synergistic effects on phosphorylated NF-κB-inducing kinase (NIK) in the non-canonical pathway. TNF-α induced the ubiquitination of TRAF2 (TNF receptor-associated factor 2), which interacts with NIK, and CGN induced phosphorylation of BCL10, leading to increased NIK phosphorylation. These results suggest that TNF-α and CGN in combination act to increase NIK phosphorylation, thereby increasing activation of the non-canonical pathway of NF-κB activation. In contrast, the apoptotic effects of TNF-α, including activation of caspase-8 and PARP-1 (poly(ADP-ribose) polymerase 1) fragmentation, were markedly reduced in the presence of CGN, and CGN caused reduced expression of Fas. These findings demonstrate that exposure to CGN drives TNF-α-stimulated cells toward inflammation rather than toward apoptotic cell death and suggest that CGN exposure may compromise the effectiveness of anti-TNF-α therapy.
Keywords: Apoptosis, Inflammation, NF-kappaB, Polysaccharide, TRAF, Tumor Necrosis Factor (TNF), Fas, Carrageenan, Phospho-NIK
Introduction
Carrageenan (CGN),2 a polysaccharide composed of sulfated galactose residues in α-1,3 and β-1,4 linkages, has been widely used for decades as a thickener, stabilizer, or emulsifying agent in many processed food products in the Western diet, including in dairy products (chocolate milk, ice cream, cottage cheese, yogurt, etc.), processed meats, soymilk, and infant formula. It is also widely used in a variety of non-food products, including cosmetics, toothpaste, room deodorizers, and pharmaceuticals (1–3). Current data suggest average consumption of 250 mg/day in the United States (5) (see the Encyclopaedia Britannica Online Web site). CGNs are derived by alkaline extraction from red seaweed (Rhodophycae) and structurally mimic the naturally occurring sulfated glycosaminoglycans (6). Multiple studies in mammals have demonstrated that CGN ingestion causes adverse effects on the intestine, including development of ulcerations, polyps, colitis, and colorectal tumors, and predictably induces inflammation in other sites in animal models, including in pleura, peritoneum, hind paw, and subcutaneous blebs (1–3, 7–10).
Investigation of CGN-related inflammatory processes in human colonic cell lines has demonstrated that CGN induces NF-κB activation and IL-8 secretion by three distinct cascades, involving both canonical (leading to nuclear translocation of RelA) and non-canonical (leading to nuclear translocation of RelB) pathways. Mediators and mechanisms of these pathways have been reported and include 1) a pathway mediated by reactive oxygen species, heat shock protein 27 (Hsp27), IKKβ (IκB kinase β), and phospho-IκBα, leading to nuclear translocation of RelA (p65) (11); 2) a TLR4 (Toll-like receptor 4), BCL10 (B-cell leukemia/lymphoma 10), IKKγ, and phospho-IκBα-mediated pathway of nuclear translocation of RelA (12, 13); and 3) a BCL10, phospho-NF-κB-inducing kinase (NIK), IKKα-mediated activation of the non-canonical pathway of NF-κB leading to nuclear translocation of RelB and p52 (NIK) (14). CGN also affects cell survival, leading to increased necrotic, non-apoptotic cell death (15). Thereby, the CGN-stimulated pathways of NF-κB involve the IKKα, IKKβ, and IKKγ (also called NEMO) components of the IKK signalosome.
Tumor necrosis factor-α (TNF-α), a potent proinflammatory cytokine, has well established, important roles in immunity, inflammation, differentiation, and apoptosis (16). TNF-α is involved in the pathogenesis of inflammatory diseases, such as Crohn disease, ulcerative colitis, and rheumatoid arthritis, and anti-TNF-α therapies have been widely used in the past decade in clinical practice (17–20). Considerable progress has been made in understanding the molecular mechanisms that mediate the different TNF-α-induced cellular responses. TNF-α exerts its effects through two distinct receptors, TNFR1 and TNFR2, and differences in therapeutic effectiveness and in cellular effects may be attributable in part to heterogeneity of the responses to these receptors (21, 22). Binding of the trimeric TNF-α induces receptor trimerization and recruitment of several signaling proteins to the cytoplasmic domains of the receptors. Variation in recruitment of specific proteins to the receptors affects the cellular response to TNFα, leading to either increased NF-κB and inflammation or increased activation of caspase-8 and apoptosis, as well as other downstream effects on ERKs, including responses of JNK, p38 MAPK, and p42/44 MAPK (23, 24).
The superfamily of TNF receptors contains a subfamily of death receptors (DRs), including TNFR1, TNFR2, Fas(CD95/APO-1), DR3, DR4, DR5, and DR6, each of which has an 80-amino acid-long death domain that is required for apoptosis (25). The death domains in the TNF receptors are composed of six α-helices in the cytoplasmic tail and can form oligomers, self-associating or forming combinations with caspase recruitment domain-containing proteins, death effector domains, or other interactions (26). TRADD (TNFR1-associated death domain protein) is recruited to TNFR1 and can serve as a platform to recruit other mediators, including FADD (Fas-associated death domain protein), TRAF2 (TNF-receptor associated factor 2), and RIP1 (receptor-interacting protein 1). The TRAF proteins undergo critical ubiquitinations, influencing either cell survival or cell death (27–29). The role of TRAFs as E3 ubiquitin ligases that can self-ubiquitinate adds further to the complexity of TNF-α-initiated processes (30). The mechanism by which TNF-α induces the activation of NF-κB involves crucial phosphorylations, including phosphorylations of TAK1 (transforming growth factor β-activating kinase 1) and NIK. The cascade of signaling leads to the phosphorylation of IKKα, the IKK signalosome component required for the non-canonical pathway of NF-κB activation (31–33).
Because CGN is a common ingredient in the diet as well as in other products to which exposure is common, this study was undertaken to determine if there were interactions between CGN- and TNF-α-initiated processes of inflammation and cell death, to define the loci of possible cross-talk between CGN- and TNF-α-initiated pathways, and to consider how these interactions might impact upon the effectiveness of anti-TNF-α therapy. In this report, we present the effects of combined CGN and TNF-α exposure on pathways of inflammation and apoptosis.
EXPERIMENTAL PROCEDURES
Cell Culture
The human colonic epithelial cell line NCM460, derived from normal adult human colon, was grown in M3:10 media (INCELL, San Antonio, TX) (34) and maintained at 37 °C in a humidified, 5% CO2 environment, as described previously (11–13).
Primary cultures of normal human colonic epithelial cells were initiated from deidentified colon specimens obtained at the time of colectomy, through an established protocol, approved by the Institutional Review Board of the University of Illinois at Chicago. Primary cultures were established as described previously (13).
Confluent NCM460 or primary colon cells grown in T-25 flasks (Costar) were harvested by trypsin/EDTA and were subcultured in multiwell tissue culture clusters (Costar). The cells were treated with different doses of TNF-α (Sigma) alone or in combination with λ-CGN (Sigma) (1 μg/ml) for 24 h. Spent media were collected from the control and treated wells and stored at −80 °C. Cells were harvested by scraping, lysed, and stored at −80 °C.
Mouse embryonic fibroblasts (MEF), including wild type (WT), IKKα−/−, and IKKβ−/−, were a generous gift from the laboratory of Dr. Michael Karin (University of California, San Diego). These cell lines were developed from embryos of transgenic homozygous mice in which either the IKKα or IKKβ gene was deleted (35). IKKα−/− and IKKβ−/− cells were grown in Dulbecco's modified Eagle's medium with 10% fetal bovine serum. The cells were maintained and treated with TNF-α, with similar procedures as in the NCM460 cells.
ELISA for IL-8
The secretion of IL-8 in the spent media of control and treated NCM460 cells and primary colon cells was measured by the DuoSet ELISA kit for human IL-8 (R&D Systems, Minneapolis, MN), as described previously (12–13). The wells of a microtiter plate were coated with specific anti-IL-8 monoclonal antibody, and the nonspecific sites were blocked by a blocking buffer with 1% bovine serum albumin (BSA). The secreted IL-8 molecules in the spent media were captured into the wells by this specific IL-8 antibody. Captured IL-8 molecules were then detected by biotin-conjugated secondary IL-8 antibody and streptavidin-horseradish peroxidase (HRP). Hydrogen peroxide-tetramethylbenzidine (TMB) chromogenic substrate was used to develop the color, and the intensity of color was measured at 450 nm in an ELISA plate reader (FLUOstar, BMG). The IL-8 concentrations were extrapolated from a standard curve, and sample values were normalized with total protein content (BCA Protein Assay Kit, Pierce (Rockford, IL)).
ELISA for KC
KC, the mouse homolog of IL-8, was measured in the spent medium of embryonic fibroblasts cells by a commercial DuoSet ELISA kit ELISA (R&D Systems) with specific KC antibodies (12, 13). The sample values were extrapolated from standard curves and normalized with the total cell protein concentrations. KC values were expressed as pg/mg protein.
FACE Assay for Total NIK and Phospho-NIK
Total and phospho-NIK in control and treated human cell samples were measured by a FACE assay (Active Motif) (36) using primary antibodies for NIK (sc-7211, Santa Cruz Biotechnology, Inc. (Santa Cruz, CA)) against the epitope 700–947 and for phospho-NIK (sc-12957, Santa Cruz Biotechnology, Inc.), which recognized Thr559 by appropriate secondary antibodies. Measurements of optical density were compared between control and treated samples and expressed as a percentage of untreated control.
Phospho-IκBα ELISA
Phospho-IκBα was determined by commercially available PathScan Sandwich ELISA (Cell Signaling Technology) (13). Phospho-IκBα in the samples was captured in microtiter wells that were coated with monoclonal antibody to phospho-IκBα. Immobilized phospho-IκBα molecules in the wells were detected by a specific rabbit phospho-IκBα antibody that detected Ser32 phosphorylation and was then recognized by an anti-rabbit IgG-HRP. The bound HRP activity was determined by adding hydrogen peroxide-TMB chromogenic substrate. The optical density for the developed color was measured in an ELISA reader at 450 nm, after stopping the reaction with 2 n sulfuric acid. The intensity of the developed color is proportionate to the quantity of phospho-IκBα in each sample. The sample values were normalized with the total cell protein and expressed as a percentage of control.
Silencing BCL10 Expression by siRNA
Silencing RNAs for BCL10 were obtained (Qiagen, Valencia, CA), and BCL10 expression was silenced, as described previously (13). Briefly, the cells were grown to 75% confluence in 12-well tissue culture clusters, and the medium of the growing cells was aspirated and replaced with 1.1 ml of fresh medium with serum. 0.3 μl of 20 μm siRNA (75 ng) was mixed with 100 μl of serum-free medium and 6 μl of HiPerfect transfection reagent (Qiagen). The mixture was incubated at room temperature for 10 min to allow the formation of transfection complexes and then added dropwise onto the cells. The plate was swirled gently, and treated cells were incubated at 37 °C in a humidified 5% CO2 environment. After 24 h, the spent medium was exchanged with fresh growth medium. The effectiveness of silencing of BCL10 expression was determined by ELISA assay and Western blot (12, 37).
Neutralization of TLR4 and TNF-α Receptors I and II by Blocking Antibodies
NCM460 cells were grown in 24-well tissue culture plates. At 60–70% confluence, the cells were treated with fresh media containing 20 μg/ml of either TLR4 antibody (BioLegend, San Diego, CA) or TNF-α receptor I (Zymed Laboratories Inc.) or II antibody (Abcam) for 2 h, prior to TNF-α and λ-CGN challenge for 24 h. At the end of treatment, the spent media were collected for the IL-8 assay. Cells from 12 wells were harvested, lysed, and used for other assays.
Detection of Nuclear NF-κB by Oligonucleotide-based ELISA
Nuclear extracts from treated and control NCM460 cells were prepared as described previously (14). Activated NF-κB in the samples was determined by oligonucleotide-based ELISA (Active Motif, Carlsbad, CA). The samples were added and incubated in the wells of the 96-well microtiter plate, precoated with an NF-κB consensus nucleotide sequence (5′-GGGACTTTCC-3′). The NF-κB proteins from the nuclear extract attached to the coated oligonucleotides of the microplate and the bound molecules were detected by specific antibodies to different NF-κBs (RelA, RelB, p50, and p52) and anti-rabbit-HRP-conjugated IgG. The specificity of the binding of NF-κB with the coated nucleotide sequence was determined by comparison with the binding when known quantities of free consensus nucleotide or mutated nucleotide were added in the reaction buffer. Colorimetric readout was performed with hydrogen peroxide-TMB chromogenic substrate. After the reaction was stopped with 2 n sulfuric acid, the optical density of the developed color was measured in an ELISA plate reader at 450 nm. The intensity of the developed color proportionately represents the quantity of NF-κB in each sample. The sample values were normalized with the total cell protein and expressed as percentage of control.
TRAF2 and TRAF6 Ubiquitination Assays
A 96-well ELISA plate was coated overnight at room temperature with specific antibody to ubiquitin (Santa Cruz Biotechnology, Inc.) at a concentration of 4 μg/ml. After coating, the wells were washed three times with wash buffer (PBS with 0.05% Tween 20) and blocked for 1 h at room temperature with blocking buffer (1% BSA in PBS). Control and treated NCM460 cell lysates were added to the coated wells and incubated for 2 h at room temperature to bind the ubiquitinated molecules, including TRAF2 and TRAF6, present in the cell lysates. Wells were washed three times to eliminate unbound molecules. The ubiquitinated TRAF2 or TRAF6 molecules were then identified by adding specific TRAF2 or TRAF6 antibody (2 μg/ml; Abcam), and the plate was incubated for 2 h at room temperature. Next, the ubiquitinated TRAF molecules were detected by adding anti-mouse IgG-HRP and incubated for 1 h at room temperature, and the bound HRP activity was determined by adding hydrogen peroxide-TMB chromogenic substrate. The optical density for the developed color was measured in an ELISA reader at 450 nm, after stopping the reaction with 2 n sulfuric acid. The intensity of the developed color was proportionate to the quantity of ubiquitinated TRAF2 or TRAF6 in each sample. The sample values were normalized with the total cell protein and expressed as a percentage of control. The specificity of the assay was determined by several control experiments using rabbit and mouse IgG to substitute for ubiquitin and the TRAF antibodies.
Western Blots of Phospho-NIK and PARP-1 Degradation Product
Proteins in the nuclear extract or in the whole cell lysate were separated by SDS-PAGE on a 10% gel and transferred to a PVDF membrane (Amersham Biosciences). Proteins were probed with phospho-NIK (Santa Cruz Biotechnology, Inc.) and cleaved PARP-1 (poly(ADP-ribose) polymerase) (89 kDa; Cell Signaling) primary antibodies and detected by donkey anti-goat HRP or goat anti-rabbit HRP secondary antibodies. β-Actin was used as a loading control. Immunoreactive bands were visualized using the ECL detection kit (Amersham Biosciences), and the ratios of the proteins of interest to actin were calculated and compared with the untreated control. The densitometry is the mean of three biological samples, and representative immunoblots are presented.
Cleaved PARP-1 ELISA
The cleaved PARP-1 in the cell lysates of control and treated NCM460 cells was measured by a commercial sandwich ELISA kit (Cell Signaling Technology, Danvers, MA). The 89-kDa fragment of PARP-1 is increased in apoptosis (38, 39). The wells of microtiter plates were coated with specific anti-cleaved PARP monoclonal antibody. The cleaved PARP molecules in the cell lysates were captured into the wells by this specific antibody. Captured cleaved PARP molecules were then sandwiched by a rabbit monoclonal cleaved PARP antibody and finally detected by anti-rabbit HRP secondary antibody. Hydrogen peroxide-TMB chromogenic substrate was used to develop the color, and the intensity of color was measured at 450 nm in an ELISA plate reader. The sample values were expressed as percentage of control.
Activated Caspase-8 Assay
The caspase-8 activity in the control and treated NCM460 cells was measured by a commercial Caspase-Glo® 8 Assay (Promega), a luminescence assay that measures caspase-8 activity. The assay provides a luminogenic caspase-8 substrate in a buffer system optimized for caspase activity, luciferase activity, and cell lysis. 100 μl of substrate solution was added to an equal volume of samples in a 96-well white plate (R & D). The plate was incubated for 1 h at room temperature. Activated caspase-8 in the samples cleaved the exogenous substrate and generated a luminescent signal, measured in a luminescence plate reader (FLUOstar, BMG). The signal generated was proportional to the amount of caspase-8 activity present.
Fas Assay
Fas (FasL receptor) was measured by FACE (Active Motif), using Fas antibody (Santa Cruz Biotechnology, Inc.). NCM460 cells were grown in 96-well plates and treated with TNF-α (0.1 ng/ml) or carrageenan (1 μg/ml), alone or in combination, for 24 h. Then the cells were washed and fixed with 4% formaldehyde, quenched for cellular peroxidase activity, and blocked with blocking buffer. The fixed cells were exposed to Fas antibody (2 μg/ml) to combine with Fas, and the captured Fas was detected by HRP-conjugated secondary antibody. Color was developed with hydrogen peroxide/TMB chromogenic substrate, and intensity of developed color was proportional to the Fas content. Optical densities were measured at 450 nm and compared between control and treated wells, and results are expressed as percentage of unexposed control.
MG-132 Inhibition of Proteasomal Processing
NCM460 cells were grown in 96-well plates and treated with the proteasomal inhibitor MG-132 (20 μm) for 2 h to inhibit proteasomal effects (40). A second plate was exposed to only the vehicle PBS and exposed to either TNF-α (0.1 ng/ml) or λCGN (1 μg/ml) alone or in combination for 24 h. At the end of this interval, the cells were harvested, cell lysates were prepared in lysis buffer, and PARP-1 was measured, as described previously. IL-8 in the spent media was measured by ELISA, as described above.
cDNA Microarray following Exposure to Carrageenan
Previously, we reported the cDNA microarray results from CGN treatment of NCM460 cells after exposure for 4 days (15). An Affymetrix microarray (human U133 2.0 gene chip) was also performed following CGN exposure (1 μg/ml) for 24 h. The significant results with corrected p value <0.05 are presented in the supplemental Table 1. Of the 54,613 transcripts present on the array, 438 transcripts had corrected p values of <0.05, and of these, 21 were up-regulated.
Statistics
Data presented are the mean ± S.D. of at least three independent experiments performed with technical replicates of each measurement. Statistical significance was determined either by one-way analysis of variance (ANOVA), followed by a post hoc Tukey-Kramer test for multiple comparisons or two-tailed, unpaired t test, using InStat3 software (GraphPad, La Jolla, CA). Under “Results” and in the figure legends, the unpaired t test, two-tailed, is used for all calculations of the significance of the difference between the calculated sum of the respective increases induced by exposure to CGN and to TNF-α versus the increase from combined exposure to CGN and TNF-α. A p value of ≤0.05 is considered statistically significant (***, p ≤ 0.001; **, p ≤ 0.01; *, p ≤ 0.05).
RESULTS
IL-8 Secretion from Human Colonic Epithelial Cells Was Synergistically Increased by the Combination of TNF-α and CGN
NCM460 cells in culture were treated with different concentrations of TNF-α (0.01–5.0 ng/ml), either alone or in combination with CGN (1 μg/ml) for 24 h (Table 1 and Fig. 1A). TNF-α treatment increased the IL-8 secretion in a dose-dependent manner, and the IL-8 secretion increased synergistically when the cells were treated with TNF-α and CGN in combination. The difference between the increase in secreted IL-8 from the calculated sum of TNF-α alone and CGN alone versus the result of TNF-α and CGN in combination was statistically significant at all concentrations tested (0.0002 ≤ p ≤ 0.0044, unpaired t test, two-tailed). Base-line IL-8 secretion was 0.623 ± 0.06 ng/mg protein in the NCM460 cells.
TABLE 1.
IL-8 (ng/mg protein) following CGN, TNF-α, and CGN plus TNF-α combined in NCM460 cells
Values are expressed as ng/mg cell protein ± S.D. (shown in parentheses). IL-8 results from combined exposure to CGN and TNF are in boldface type. Differences between additive value and combined exposure value are in italic type. Base-line level of IL-8 in the spent medium of untreated control samples after 24 h is 0.623 (0.061) ng/mg protein.
| Exposure to TNFα (24 h) | Exposure to CGN (24 h) | CGN-induced increase in IL-8 from base line | TNF-α−induced increase in IL-8 from base line | Sum of increases from CGN and from TNF-α | CGN and TNF-α in combination-induced increase in IL-8 from base line | Difference in IL-8 between sum of increases from CGN and from TNF-α versus CGN and TNF-α in combination | p value for differencea |
|---|---|---|---|---|---|---|---|
| ng/ml | μg/ml | ng/mg protein | ng/mg protein | ng/mg protein | ng/mg protein | ng/mg protein | |
| 0.01 | 1 | 0.73 (0.04) | 0.320 (0.02) | 1.05 (0.07) | 1.92 (0.10) | 0.87 (0.05) | 0.0002 |
| 0.05 | 1 | 0.73 (0.04) | 1.75 (0.03) | 2.48 (0.07) | 3.43 (0.16) | 0.95 (0.22) | 0.0007 |
| 0.1 | 1 | 0.73 (0.04) | 3.36 (0.22) | 4.09 (0.30) | 5.40 (0.22) | 1.31 (0.05) | 0.0037 |
| 1 | 1 | 0.73 (0.04) | 10.94 (0.31) | 11.67 (0.38) | 13.91 (0.10) | 2.24 (0.34) | 0.0006 |
| 2 | 1 | 0.73 (0.04) | 14.30 (0.85) | 15.03 (0.92) | 18.33 (0.36) | 3.30 (0.40) | 0.0044 |
| 5 | 1 | 0.73 (0.04) | 16.94 (0.54) | 17.67 (0.61) | 21.23 (0.77) | 3.56 (0.11) | 0.0033 |
a Differences in the IL-8 increase between the sum of increases from CGN and from TNF-α versus CGN and TNF-α in combination are compared by unpaired t test, two-tailed.
FIGURE 1.

TNF-α and CGN produced synergistic increases in IL-8 or KC secretion. A, IL-8 secretion was measured by ELISA in the spent media of NCM460 cells following stimulation by either TNF-α (at concentrations ranging from 0.01 to 5 ng/ml) or CGN (1 μg/ml) or their combination for 24 h. The increase from the base-line value (base line = 0.623 ± 0.06 ng/mg protein) is represented on the y axis. At all of the concentrations of TNF-α tested, exposure to the combination of TNF-α and CGN stimulated the IL-8 secretion to a significantly greater value than the sum of the increases of the individual exposures (all p values <0.01, unpaired t test, two-tailed). B, similarly, the increase in IL-8 secretion in primary human colonic epithelial cells was significantly greater following the combined exposure to CGN (1 μg/ml) and TNF-α (0.01 ng/ml) for 24 h (1.386 ± 0.024 ng/mg protein; p < 0.0001, unpaired t test, two-tailed) than the sum (0.686 ± 0.034 ng/mg protein) of the increases from exposure to CGN and TNF-α individually. C, in the MEF, KC, the mouse homolog of IL-8, increased synergistically in response to TNF-α and CGN in the WT and IKKβ−/− cells (p = 0.0004 and p = 0.0006, respectively; unpaired t test, two-tailed) but not in the IKKα−/− cells. **, p ≤ 0.01; ***, p ≤ 0.001. Error bars, S.D.
Colon epithelial cells in primary culture showed a similar response to TNF-α and CGN in combination as the NCM460 cells. TNF-α (0.01 ng/ml) and CGN (1 μg/ml) stimulated the IL-8 secretion from the base-line value of 0.411 ± 0.0006 ng/mg protein to 0.666 ± 0.014 and 0.843 ± 0.034 ng/mg protein, respectively (Fig. 1B). The combination of TNF-α and CGN increased the IL-8 secretion to 1.80 ± 0.024 ng/mg protein, an increase above base line that was significantly higher than the sum of the individual increases (p < 0.0001, unpaired t test, two-tailed).
TNF-α and CGN synergistically stimulated KC secretion from IKK WT and IKKβ−/− cells but not from IKKα−/− cells (Fig. 1C). The increases in secreted KC were significantly greater in the WT and IKKβ−/− MEF when exposed to TNF-α (0.01 ng/ml) and CGN (1 μg/ml) in combination than the sum of the increases following the individual exposures. In contrast, in the IKKα−/− cells, KC did not increase following TNF-α, and no synergistic response occurred following the combined exposure. This result demonstrated that the enhanced effect of the combination of CGN and TNF-α required IKKα and was attributable to the non-canonical pathway of NF-κB activation.
TNF-α and CGN Increased Nuclear p52 and RelB Synergistically in NCM460 Cells and in IKK WT and IKKβ−/− MEF but Not in IKKα−/− Cells
Nuclear content of NF-κB components in NCM460 cells and MEF was measured by an oligonucleotide ELISA in which the wells of a microplate were coated with the NF-κB consensus sequence. TNF-α (0.1 ng/ml for 24 h) stimulated the activation of p52 and RelB but not p65 (RelA) or p50 in the NCM460 cells (Fig. 2A), consistent with a unique effect on the non-canonical pathway. In contrast, CGN induced the activation of p50, p52, RelA, and RelB in the NCM460 cells, indicating activation of both canonical and non-canonical pathways. In these cells, the combined exposure to CGN and TNF-α synergistically and significantly increased the p52 and RelB beyond the sum of their increases (p = 0.019 and p = 0.0005, respectively; unpaired t test, two-tailed) but had no increased effect on p65 or p50.
FIGURE 2.
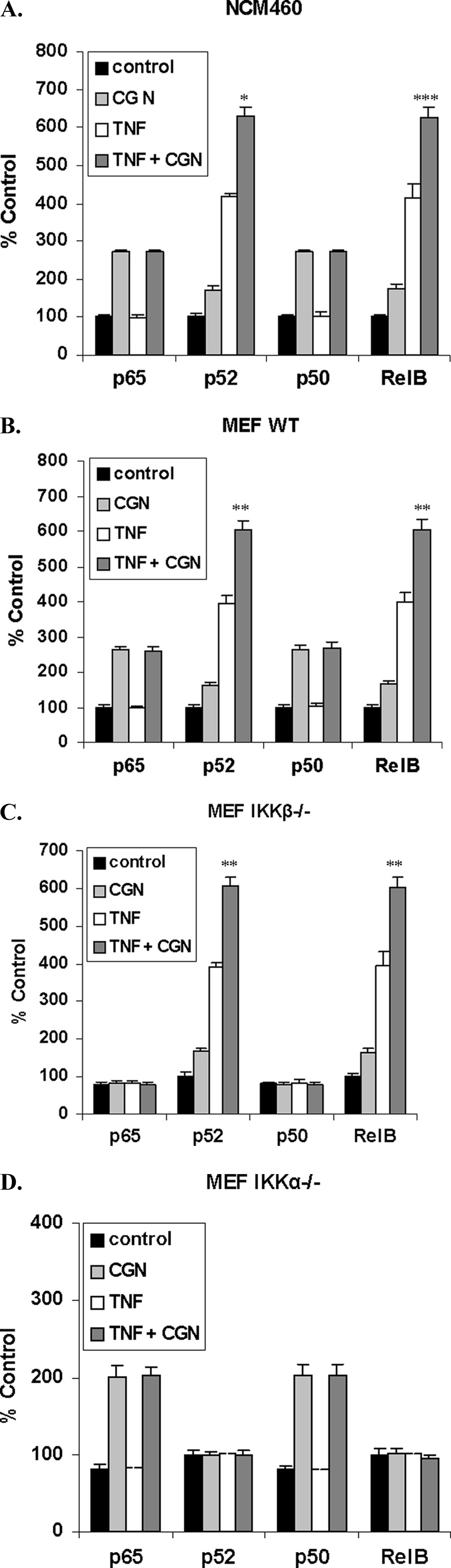
TNF-α and CGN in combination increased p52 and RelB beyond an additive effect, but p65 (RelA) and p50 did not increase. A, nuclear content of NF-κB components was measured by oligonucleotide ELISA. In the NCM460 nuclear extracts, combined λCGN (1 μg/ml) and TNF-α (0.1 ng/ml) exposure for 24 h significantly increased the nuclear p52 and RelB (p = 0.019 and p = 0.0005, respectively; unpaired t test, two-tailed). TNF-α exposure produced no increases in either p65 or p50 over the control, and no synergistic increases occurred in p65 or p50 with the combination of CGN and TNF-α. B, similarly, in the WT MEF, CGN and TNF-α in combination increased the nuclear p52 and RelB significantly beyond the sum of the increases induced by CGN and TNF-α individually (p = 0.0016 and 0.0041, unpaired t test, two-tailed). C, in the IKKβ−/− MEF, CGN and TNF-α in combination significantly increased the nuclear p52 and RelB, beyond the sum of the CGN-induced and TNF-α-induced increases (p = 0.008 and p = 0.0013, unpaired t test, two-tailed), as in the NCM460 and the WT MEF. D, in contrast, in the IKKα−/− cells, the combination of CGN and TNF-α produced no further increases in NF-κB components. *, p ≤ 0.05; **, p ≤ 0.01; ***, p ≤ 0.001. Error bars, S.D.
Combined treatment with CGN and TNF-α in the WT (Fig. 2B) and IKKβ−/− (Fig. 2C) MEF cells produced increases in nuclear p52 and RelB that exceeded the sum of the individual increases produced by CGN and TNF-α. Increases were statistically significant for p52 (p = 0.0016 (WT) and p = 0.008 (IKKβ−/−)) and for RelB (p = 0.0041 (WT) and p = 0.0013 (IKKβ−/−)). However, in the IKKα−/− cells, no synergistic effects were evident, reflecting that the interaction between CGN and TNF-α requires IKKα and the non-canonical pathway.
TNF-α and CGN Synergistically Increased Phospho-NIK
Neither TNF-α nor CGN exposure in the NCM460 cells or the MEF increased total NIK (Fig. 3, A–E). However, both TNF-α and CGN increased phospho(Thr559)-NIK in NCM460 cells, as evident by ELISA (Fig. 3A) and Western blot (Fig. 3B) and in the WT (Fig. 3C), IKKβ−/− (Fig. 3D), and IKKα−/− (Fig. 3E) MEF. The combined treatment of TNF-α (0.1 ng/ml) and CGN (1 μg/ml) for 24 h, in both NCM460 cells and MEF cells, had a synergistic effect on phospho-NIK, producing increases significantly greater than the sum of the individual increases by CGN and by TNF-α (p ≤ 0.001, unpaired t test, two-tailed, for each cell type). Densitometric determinations of phospho-NIK in the NCM460 cells indicated that the ratio of phospho-NIK to β-actin increased to 1.93 ± 0.14 times the control value following CGN, to 4.03 ± 0.47 times following TNF-α, and to 7.22 times following CGN and TNF-α in combination (n = 3).
FIGURE 3.

TNF-α and CGN increased NIK phosphorylation in NCM460 and MEF cells. A, combined TNF-α and CGN increased NIK phosphorylation in the NCM460 cells by ∼9.2 times the base-line value. TNF-α alone increased phospho-NIK by ∼4.3 times the base line, and CGN alone increased phospho-NIK by ∼2.2 times the base line (p < 0.001, unpaired t test, two-tailed). Total NIK content did not change. B, representative Western blot demonstrates a marked increase in phospho-NIK following exposure to TNF-α and CGN in combination. Densitometric measurements of the ratio of phospho-NIK to β-actin were averaged and indicated relative values of 1.0 for control, 4.03 ± 0.47 for TNF-α, 1.93 ± 0.14 for CGN, and 7.22 ± 0.52 for CGN and TNF-α in combination, corresponding to the increases obtained by the cell-based ELISA. C–E, both TNF-α and CGN independently induced the phosphorylation of NIK in the WT MEF (C), and the combined treatment was also synergistic in the IKKβ−/− (D) and in the IKKα−/− (E) MEF (p ≤ 0.001, unpaired t test, two-tailed). Total NIK did not change in the MEF. ***, p ≤ 0.001. Error bars, S.D.
In contrast to the effect on phospho-NIK and consistent with the interaction in the non-canonical pathway of NF-κB activation, combined treatment with CGN and TNF-α had no impact on the CGN-induced increase in phospho-IκBα (Fig. 4A). In the presence of TNFR1 antibody or TLR4 blocking antibody, the TNF-α- or the CGN-induced increase in IL-8, respectively, was inhibited and demonstrated no cross-reactivity in response at the receptor level (Fig. 4B). These findings further suggest that the synergism between CGN and TNF-α does not involve the canonical pathway and is downstream of the receptors at the cell membrane.
FIGURE 4.

Additional evidence localizing synergistic effect on inflammation. A, in contrast to the TNF-α-induced increase in phospho-NIK, TNF-α exposure did not increase phospho-IκBα in the NCM460 cells, and there was no synergistic effect between CGN (1 μg/ml) and TNF-α (0.01 ng/ml) for 24 h, consistent with restriction of the synergistic effect of CGN and TNF-α to the non-canonical pathway. B, inhibition by TLR4 blocking antibody of the TLR4 receptor nullified the CGN-induced increase in IL-8 but had no effect on the TNF-α (0.1 ng/ml for 24 h)-induced IL-8 response. Inversely, blocking of TNFR1 receptor by antibody against sTNFR1 abrogated the TNF-α-induced IL-8 response but not the CGN-induced IL-8 response. Error bars, S.D.
BCL10 Silencing Had No Effect on TNF-α-induced Increases in IL-8 or Phospho-NIK
Silencing BCL10 with specific siRNA had no impact upon the TNF-α-induced increase in IL-8 (Fig. 5A). Similarly, BCL10 silencing had no effect on the TNF-α-induced increases in phospho(Thr559)-NIK in the NCM460 cells (Fig. 5B) or in the WT MEF cells (Fig. 5C), in contrast to reduction of the CGN-induced increases. In the IKKβ−/− and the IKKα−/− MEF, findings were similar and indicated no impact of BCL10 silencing on the TNF-α induced increase in phospho-NIK (data not shown).
FIGURE 5.

BCL10 silencing had no impact on TNF-α induced effects. A, BCL10 silencing had no effect on TNF-α (0.1 ng/ml for 24 h)-induced IL-8 secretion in the NCM460 cells. B and C, BCL10 silencing also had no effect on TNF-α-induced increases in phospho-NIK in the NCM460 cells or in the MEF cells. Similarly, in the IKKα−/− and IKKβ−/−cells, BCL10 silencing also had no effect on the TNF-α-induced increase in phospho-NIK (data not shown). control si, control siRNA; BCL10 si, BCL10 siRNA. Error bars, S.D.
TNF-α but Not CGN Induced TRAF2 Ubiquitination
Treatment of NCM460 cells with TNF-α (0.1 ng/ml for 24 h) induced the ubiquitination of TRAF2 (Fig. 6A). This effect was inhibited by pretreatment of the cells with TNFR1 antibody. CGN exposure (1 μg/ml) had no impact upon TRAF2 ubiquitination (Fig. 6A). BCL10 silencing did not inhibit the TNF-α-induced increase in TRAF2 ubiquitination, and CGN in combination with TNF-α had no effect on TRAF2 ubiquitination (Fig. 6B). In contrast, TRAF6 ubiquitination was increased following exposure to CGN (1 μg/ml for 24 h) and reduced by BCL10 silencing (p < 0.001, one-way ANOVA with Tukey-Kramer post-test) but was unaffected by exposure to TNF-α and not modified by TNF-α in combination with CGN (Fig. 6C).
FIGURE 6.

TNF-α, but not CGN, induced the ubiquitination of TRAF2. A, ubiquitination of TRAF2 was induced by TNF-α (0.1 ng/ml for 24 h) but not by CGN (1 μg/ml for 24 h) in the NCM460 cells. Soluble TNFR1 antibody blocked the TNF-α-induced ubiquitination of TRAF2. B, BCL10 silencing had no impact on the TNF-α-induced TRAF2 ubiquitination, and CGN in combination with TNF-α did not modify the TNF-α-induced increase. C, in contrast, TRAF6 ubiquitination was increased following exposure to CGN (1 μg/ml for 24 h) and reduced by BCL10 silencing (p < 0.001, one-way ANOVA with Tukey-Kramer post-test). TNF-α had no impact on the CGN-induced TRAF6 ubiquitination. ***, p ≤ 0.001. Error bars, S.D.
CGN Reduced the TNF-α-induced Increases in Activated Caspase-8 and in PARP-1 89-kDa Fragment
Proapoptotic effects of TNF-α were evaluated by the extent of the increase in measurements of activated caspase-8 and of PARP-1 fragmentation. When NCM460 cells were treated with both CGN (1 μg/ml) and TNF-α (0.1 ng/ml) for 24 h, the increases in activated caspase-8 and PARP-1 89-kDa fragment induced by TNF-α declined significantly (p < 0.001, one-way ANOVA with Tukey-Kramer post-test). Activated caspase-8 following TNF-α alone increased to 314.5 ± 19.9% of the base-line value and declined to 145.6 ± 3.2% of base line following CGN and TNF-α (Fig. 7A). The concentration of the 89-kDa fragment of PARP-1, which is increased in apoptosis, declined from 270.3 ± 4.8% of the base-line value that followed exposure to TNF-α to 159.5 ± 5.6% of base line when cells were exposed to both CGN and TNF-α (Fig. 7B). Representative immunoblot demonstrated the prominent PARP-1 fragment (89 kDa) following exposure to TNF-α, which was absent following exposure to CGN and very faint following exposure to combined CGN and TNF-α (Fig. 7C). BCL10 silencing had no effect on the TNF-α-induced increase in the PARP-1 fragment (Fig. 7D), consistent with no effect of BCL10 on apoptosis. These responses demonstrate that CGN co-exposure reduced the apoptotic effect of TNF-α, in contrast to its impact on increasing the TNF-α-associated inflammation by the non-canonical pathway.
FIGURE 7.

TNF-α-induced activation of caspase-8 and fragmentation of PARP-1 were reduced by combined exposure with CGN. A, the TNF-α-induced increase in activated caspase-8 was significantly reduced by combined CGN and TNF-α exposure. B, PARP-1 fragmentation (89 kDa) was increased following exposure to TNF-α, but this increase was inhibited following exposure to CGN and TNF-α in combination. C, Western blot of nuclear PARP-1 fragment indicated near absence of the PARP-1 89-kDa fragment following CGN and the combination of CGN and TNF-α. D, BCL10 silencing had no impact on PARP-1 fragmentation. ***, p ≤ 0.001. Error bars, S.D.
Additional Antiapoptotic Effects of Carrageenan
cDNA microarray data of apoptosis-associated genes affected by λCGN (1 μg/ml for 24 h) are summarized in Table 2. (Complete microarray data at 24 h are presented in the supplemental Table 1.) Findings included down-regulation of Fas, BAX, BCL2L1, DEDD, DAPK1, MCL1, and TP53, all consistent with an antiapoptotic effect of CGN. The decline in Fas was confirmed by a FACE assay that demonstrated marked reduction in Fas (p < 0.001, one-way ANOVA with Tukey-Kramer post-test) following CGN exposure (Fig. 8), in contrast to no effect of TNF-α on Fas.
TABLE 2.
Apoptosis and ubiquitin-associated genes down-regulated following exposure to λ-CGN at 24 and 96 h
| Gene symbol | Gene description | Change | Corrected p value |
|---|---|---|---|
| -fold | |||
| 24 h | |||
| Fas | TNF receptor superfamily, member 6 | −1.67 | 0.033 |
| −1.73 | 0.0031 | ||
| BAX | BCL2-associated X protein | −2.44 | 0.023 |
| BCL2L1 | BCL2-like 1 | −3.67 | 8.07 × 10−5 |
| −4.21 | 2.21 × 10−6 | ||
| DAPK1 | Death-associated protein kinase 1 | −2.41 | 0.009 |
| DEDD | Death effector domain-containing | −2.28 | 0.036 |
| MCL1 | Myeloid cell leukemia seq 1 (BCL2-related) | −2.94 | 0.00076 |
| TP53 | Tumor protein p53 | −3.75 | 8.11 × 10−6 |
| USP28 | Ubiquitin-specific peptidase 28 | −2.51 | 0.022 |
| USP34 | Ubiquitin-specific peptidase 34 | −3.16 | 6.38 × 10−6 |
| UBE1L2 | Ubiquitin-activating enzyme E1-like 2 | −2.35 | 0.00030 |
| UEV3 | Ubiquitin-conjugating enzyme E2-like | −2.85 | 0.0024 |
| UBE2M | Ubiquitin-conjugating enzyme E2 M (UBC12 homolog, yeast) | −2.56 | 0.026 |
| 96 h | |||
| USP38 | Ubiquitin-specific protease 38 | 1.43 | 0.00079a |
| USP53 | Ubiquitin-specific protease 53 | 1.97 | 0.0012 |
| 1.94 | 0.0060 | ||
| UBE2J1 | Ubiquitin-conjugating enzyme E2, J1 (UBC6 homolog, yeast) | 1.46 | 0.00019a |
| 1.49 | 0.00043a | ||
| 1.48 | 0.000030a | ||
a Uncorrected.
FIGURE 8.

CGN reduced Fas in NCM460 cells. NCM460 cells were exposed to λ-CGN (1 μg/ml for 24 h), and Fas was measured by FACE. CGN exposure markedly reduced Fas, to ∼69.4 ± 1.8% of the base-line value. ***, p ≤ 0.001. Error bars, S.D.
In addition to direct effects on down-regulation of several apoptosis-associated genes at 24 h, CGN had significant effects on several ubiquitination-associated genes. At 24 h, expressions of two ubiquitin-specific peptidases (USP28 and USP34), the ubiquitin-activating enzyme E1-like 2, and two ubiquitin-conjugating enzymes (UEV3 and UBE2M) were significantly reduced. In contrast, the ubiquitin-conjugating enzyme UBE2J1 was increased as well as two ubiquitin-specific proteases (USP38 and USP53).
Impact of Proteasomal Inhibition by MG-132
Prior exposure to the proteasomal inhibitor MG-132 (20 mm for 2 h) completely inhibited the CGN-, TNF-α-, and CGN in combination with TNF-α-induced increases in IL-8 secretion (Fig. 9A), consistent with the requirement for ubiquitination in the inflammatory pathways. By inhibition of ubiquitinations (e.g. of TRAF2) in the TNF-α-induced pathway and (e.g. of TRAF6, phospho-IκBα, and IKKγ) in the CGN-induced pathways, MG-132 completely suppressed the IL-8 response.
FIGURE 9.
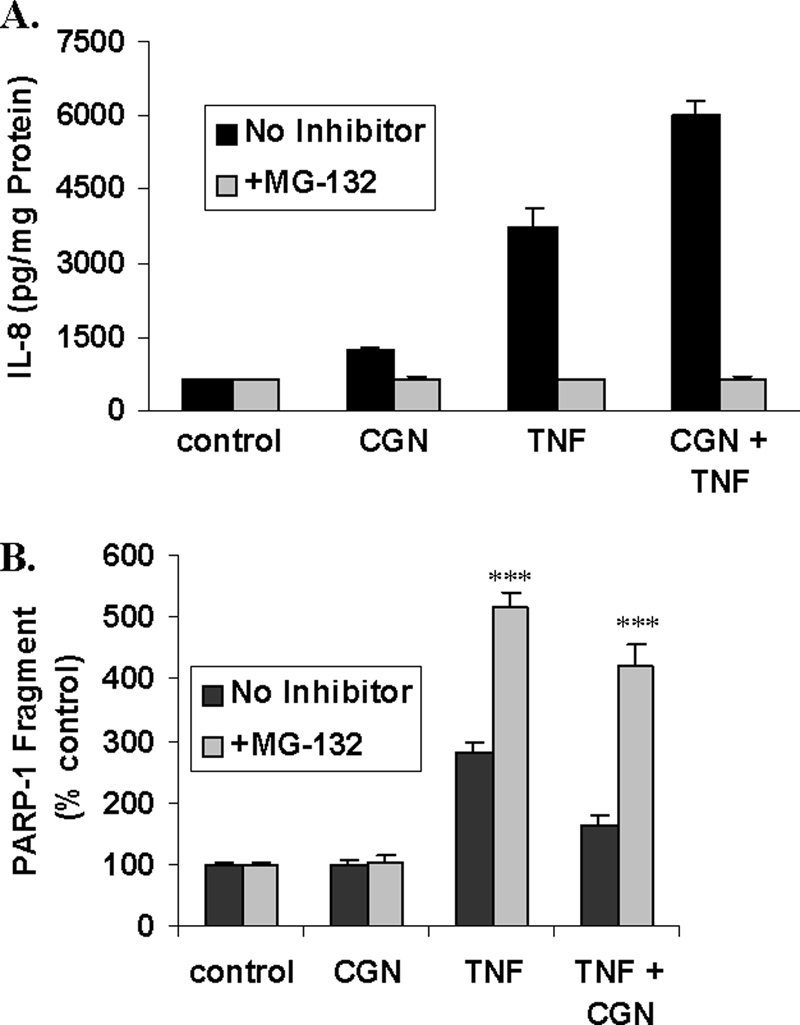
Effects of proteasomal inhibitor MG-132. A, when cells were exposed to MG-132 (20 mm x 2 h) prior to exposure to CGN, TNF-α, or the combination of CGN and TNF-α, proteasomal inhibition completely reversed the increases in IL-8. B, in contrast, the increases in the PARP-1 89-kDa fragment that followed TNF-α or combined TNF-α + CGN exposures persisted when proteasomal processing was inhibited (p < 0.001, one-way ANOVA with Tukey-Kramer post-test). ***, p ≤ 0.001. Error bars, S.D.
In contrast, the TNF-α- and the TNF-α plus CGN-induced increases in the PARP-1 89-kDa fragment were enhanced by treatment of the cells with MG-132 (Fig. 9B). These findings demonstrate that inhibition of proteasomal degradation augmented the apoptotic response to TNF-α. In the presence of MG-132, CGN (1 μg/ml) in combination with TNF-α (0.1 ng/ml) reduced the TNF-α-induced PARP-1 degradation, suggesting that CGN effects on reducing the apoptotic response to TNF-α were upstream of the proteasome and consistent with the CGN-induced decline in Fas.
DISCUSSION
The overall effects of combined exposure to CGN and TNF-α on inflammation and apoptosis are presented in Fig. 10. Together, they increase the activation of the non-canonical pathway of NF-κB, leading to increased nuclear translocation of RelB and p52. In contrast, the combination of CGN and TNF-α reduced the TNF-α-induced apoptotic effects, including activation of caspase-8 and increased PARP-1 fragmentation. Hence, exposure to CGN influenced the TNF-α-mediated cellular responses toward inflammation and away from apoptosis. Due to the high daily exposure to CGN in the human diet and environment, these effects may be clinically relevant. Hundreds of milligrams of CGN may be consumed daily, in contrast to digoxin, a glycosidic pharmaceutical agent that is prescribed in less than 1-mg quantities, or heparin (0.002 mg/unit), which is administered in far smaller quantities. In human cells, CGN exposure appears to activate a pathway of innate immunity mediated by BCL10 (6, 12). This effect may be attributable, at least in part, to the unusual α-1,3-galactosidic linkage in the CGN disaccharide structural unit because this bond is a known immune epitope in humans, who lack the α-1,3-galactosyltransferase required to form this bond (6, 41, 42).
FIGURE 10.

Schematic representation of CGN and TNF-α pathways. The CGN and TNF-α inflammatory pathways intersect to increase phospho-NIK and nuclear RelB and p52, consistent with a synergistic impact on the IKKα-mediated, non-canonical NF-κB pathway of inflammation. In contrast, the effect of CGN and TNF-α in combination reduced caspase-8 and PARP-1 fragmentation, indicating inhibition of TNF-α-induced apoptosis. This antiapoptotic effect may be attributable to CGN inhibition of the extrinsic pathway of apoptosis by reduction of Fas.
TNF-α is a pleiotropic inflammatory cytokine secreted from epithelial as well as myeloid cells, and its inhibition has evolved as an important treatment modality in inflammatory disorders. TNF-α has a unique and pivotal role in regulating the cellular choice between proapoptotic and antiapoptotic inflammatory signaling pathways. TNF-α can trigger apoptosis by a cascade involving TRADD, FADD, and activation of caspase-8 (43). The recruitment of specific proteins in the receptor complex may determine the activation of the inflammatory pathway versus the apoptotic pathway. Binding of TRADD, TRAF2, and RIP1 to TNFR1 leads to stimulation of the inflammatory pathway and activation of NF-κB and inhibition of the apoptotic pathway (42). The recruitment of FADD to TNFR1 triggers the apoptotic pathway, with activation of caspases and inhibition of the inflammatory pathway (44). The NF-κB pathway might also inhibit the apoptotic pathway through the induction of cellular inhibitors of apoptosis, which function as specific caspase inhibitors (45).
In this study, we have demonstrated that TNF-α and CGN exposures cooperated to increase NIK phosphorylation. This occurred by TNF-α increasing TRAF2 ubiquitination and reducing its restraint on NIK phosphorylation and by CGN increasing BCL10-mediated phosphorylation of NIK. Subsequently, phospho-NIK interacts with IKKα to activate the non-canonical pathway of NF-κB involving p52 and RelB. After translocation to the nucleus, RelB induces the expression of NF-κB dependent genes, including IL-8 and BCL10. Because there appears to be an NF-κB binding site in the BCL10 promoter (34, 46), the initial CGN exposure and increase in NF-κB may lead to a constitutive activation of BCL10, thus propagating the inflammatory effect of CGN and continuing to amplify the inflammatory effect of TNF-α, either by transcriptional effects of NF-κB or at the level of phospho-NIK interaction.
BCL10 is a caspase recruitment domain (CARD)-containing protein and may interact with other proteins with CARDs. BCL10 was reported to be proapoptotic in MCF-7 cells, attributable to interaction with caspase-9 at its C-terminal and the presence of a CARD at its N terminus (47). However, the findings in this report suggest a greater role for BCL10 in inflammation than in apoptosis. Future work that clarifies specific CARD-CARD interactions involving BCL10 are needed to better understand its influence.
The receptor-interacting protein kinases, including RIP1, -2, and -3 have been associated with TNF-α pathways, and overexpression of RIP1 in mammalian cells was found to be a key effector in the activation of NF-κB by TNF-α (48). Overexpression of RIP2 and RIP3 in mammalian cells elicited NF-κB activation and cell death, although their precise role in TNF-α signaling and interaction with TRAFs remains to be established (49, 50).
Our findings of IL-8 decline and PARP-1 fragment increase in response to the proteasomal inhibitor MG-132 following TNF-α stimulation demonstrate the critical importance of the proteasome and ubiquitinations in the determination of cell fate between inflammation and apoptotic cell death. The complex effects of ubiquitination involve multiple mediators of NF-κB activation (51) and lead the TNF-α response to inflammation over apoptosis in the presence of CGN. The protein A20 (TNF-α-induced protein 3; TNFAIP3) has been identified as an NF-κB-responsive protein that has both anti-inflammatory and antiapoptotic effects, attributable to its complex ubiquitin-associated functions (52). A20 interacts with TRAF1 and TRAF2, inhibiting TRAF2-mediated NF-κB activation (53).
Interestingly, in contrast to the A20 anti-inflammatory and antiapoptotic effects and the TNF-α proinflammatory and proapoptotic effects, CGN appears to have antiapoptotic and proinflammatory effects. In the cDNA microarray, CGN increased the expression of UBE2J1. UBE2J1 interacts with cellular inhibitor of apoptosis-1 (BIRC2), an E3 ubiquitin ligase that affects TRAF2, TRAF1, and RIP1 and inhibits apoptosis (54). This suggests a potential impact of CGN exposure on critical ubiquitin-associated processes. Also, CGN exposure was associated with significant increases in transcription of the ubiquitin protease USP53 that has been linked with TRAF2 in a yeast hybrid assay (55). Because carrageenan is so highly sulfated and thioester reactions involving sulfonyl groups are required for the sequence of ubiquitination reactions (4, 56), the increase in ubiquitination reactions that favor inflammation post-CGN exposure may be attributable to the increased availability of sulfate groups following exposure to the highly sulfated CGN.
CGN-induced inflammatory responses in tissues can lead to the activation of resident or recruited macrophages, dendritic cells, lymphocytes, and other myeloid cells, responding to IL-8 or other cytokine/chemokine stimuli. In turn, cells may produce TNF-α, as well as other cytokines, potentiating the inflammatory response. The use of anti-TNF-α therapy has unanticipated adverse effects on host defense, such as reactivation of latent tuberculosis (18, 19). Increased recognition of specific signaling selection mechanisms by TNF-α and of the impact of CGN exposure on these cellular responses, favoring either apoptosis or inflammation, may lead to improvements in effectiveness of immune modulation therapy and in better understanding of the relationships between inflammation, apoptosis, and neoplasia.
Supplementary Material
This work was supported by Veterans Affairs Merit funds (to J. K. T.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Table 1.
- CGN
- carrageenan
- FACE
- fast activated cell-based ELISA
- MEF
- mouse embryo fibroblast(s)
- TMB
- tetramethylbenzidine
- NIK
- NF-κB-inducing kinase
- CARD
- caspase recruitment domain
- ANOVA
- analysis of variance.
REFERENCES
- 1.Tobacman J. K. (2003) in Reviews in Food and Nutrition Toxicity, Vol. 1 (Preedy V. R., Watson R. R. eds) pp. 204–229, Taylor & Francis, New York [Google Scholar]
- 2.Tobacman J. K. (2001) Environ. Health Perspect. 109, 983–994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Joint FAO/WHO Expert Committee on Food Additives (2007) Summary and Conclusions of the Sixty-eighth Meeting, United Nations, Geneva [Google Scholar]
- 4.Huang D. T., Hunt H. W., Zhuang M., Ohi M. D., Holton J. M., Schulman B. A. (2007) Nature 445, 394–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.West J., Miller K. A. (2001) Agarophytes and Carrageenophytes. California's Living Marine Resources: A Status Report, p. 286, California Department of Fish and Game, Sacramento, CA [Google Scholar]
- 6.Bhattacharyya S., Liu H., Zhang Z., Jam M., Dudeja P. K., Michel G., Linhardt R. J., Tobacman J. K. (2010) J. Nutr. Biochem. 21, 906–913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.IARC Working Group on the Evaluation of the Carcinogenic Risk of Chemicals to Humans (1983) IARC Monogr. Eval. Carcinog. Risk Hum. 31, 79–94 [PubMed] [Google Scholar]
- 8.Ceccarelli M., Bani D., Cinci L., Nistri S., Uliva C., Ragazzo E., Vannacci A., Manoni M., Gori A. M., Abbate R., Gensini G. F., Masini E. (2009) J. Cell. Mol. Med. 13, 2704–2712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Benitz K. F., Golberg L., Coulston F. (1973) Food Cosmet. Toxicol. 11, 565–575 [DOI] [PubMed] [Google Scholar]
- 10.Marcus R., Watt J. (1969) Lancet 2, 489–490 [DOI] [PubMed] [Google Scholar]
- 11.Bhattacharyya S., Dudeja P. K., Tobacman J. K. (2008) Biochim. Biophys. Acta. 1780, 973–982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bhattacharyya S., Gill R., Chen M. L., Zhang F., Linhardt R. J., Dudeja P. K., Tobacman J. K. (2008) J. Biol. Chem. 283, 10550–10558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Borthakur A., Bhattacharyya S., Dudeja P. K., Tobacman J. K. (2007) Am. J. Physiol. Gastrointest. Liver Physiol. 292, G829–G838 [DOI] [PubMed] [Google Scholar]
- 14.Bhattacharyya S., Borthakur A., Tyagi S., Gill R., Chen M. L., Dudeja P. K., Tobacman J. K. (2010) J. Biol. Chem. 285, 522–530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bhattacharyya S., Borthakur A., Dudeja P. K., Tobacman J. K. (2008) J. Nutr. 138, 469–475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Baud V., Karin M. (2001) Trends Cell Biol. 11, 372–377 [DOI] [PubMed] [Google Scholar]
- 17.Podolsky D. K. (2002) N. Engl. J. Med. 347, 417–429 [DOI] [PubMed] [Google Scholar]
- 18.Sfikakis P. P. (2010) Curr. Dir. Autoimmun. 11, 180–210 [DOI] [PubMed] [Google Scholar]
- 19.Lin J., Ziring D., Desai S., Kim S., Wong M., Korin Y., Braun J., Reed E., Gjertson D., Singh R. R. (2008) Clin. Immunol. 126, 13–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Behm B. W., Bickston S. J. (2008) Tumor Necrosis Factor-Alpha Antibody for Maintenance of Remission in Crohn's Disease, Cochrane Database of Systematic Reviews, Issue 1, Art. No.: CD006893. DOI:10.1002/14651858.CD006893 [DOI] [PubMed] [Google Scholar]
- 21.Kollias G., Kontoyiannis D. (2002) Cytokine Growth Factor Rev. 13, 315–321 [DOI] [PubMed] [Google Scholar]
- 22.McFarlane S. M., Pashmi G., Connell M. C., Littlejohn A. F., Tucker S. J., Vandenabeele P., MacEwan D. J. (2002) FEBS Lett. 515, 119–126 [DOI] [PubMed] [Google Scholar]
- 23.Mohamed A. A., Jupp O. J., Anderson H. M., Littlejohn A. F., Vandenabeele P., MacEwan D. J. (2002) Biochem. J. 366, 145–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Karin M., Gallagher E. (2009) Immunol. Rev. 228, 225–240 [DOI] [PubMed] [Google Scholar]
- 25.Weinlich R., Brunner T., Amarante-Mendes G. P. (2010) Cell. Mol. Life Sci. 67, 1631–1642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Weber C. H., Vincenz C. (2001) Trends Biochem. Sci. 26, 475–481 [DOI] [PubMed] [Google Scholar]
- 27.Ha H., Han D., Choi Y. (2009) Curr. Protoc. Immunol., Chapter 11, Unit 11.9D [DOI] [PubMed] [Google Scholar]
- 28.Chung J. Y., Lu M., Yin Q., Wu H. (2007) Adv. Exp. Med. Biol. 597, 93–113 [DOI] [PubMed] [Google Scholar]
- 29.Chung J. Y., Park Y. C., Ye H., Wu H. (2002) J. Cell Sci. 115, 679–688 [DOI] [PubMed] [Google Scholar]
- 30.Xu Y., Huang S., Liu Z. G., Han J. (2006) J. Biol. Chem. 281, 8788–8795 [DOI] [PubMed] [Google Scholar]
- 31.Takeuchi M., Rothe M., Goeddel D. V. (1996) J. Biol. Chem. 271, 19935–19942 [DOI] [PubMed] [Google Scholar]
- 32.Stanger B. Z., Leder P., Lee T. H., Kim E., Seed B. (1995) Cell 81, 513–523 [DOI] [PubMed] [Google Scholar]
- 33.Ting A. T., Pimentel-Muiños F. X., Seed B. (1996) EMBO J. 15, 6189–6196 [PMC free article] [PubMed] [Google Scholar]
- 34.Moyer M. P., Manzano L. A., Merriman R. L., Stauffer J. S., Tanzer L. R. (1996) In Vitro Cell. Dev. Biol. Anim. 32, 315–317 [DOI] [PubMed] [Google Scholar]
- 35.Hu Y., Baud V., Delhase M., Zhang P., Deerinck T., Ellisman M., Johnson R., Karin M. (1999) Science 284, 316–320 [DOI] [PubMed] [Google Scholar]
- 36.Bhattacharyya S., Borthakur A., Dudeja P. K., Tobacman J. K. (2010) Exp. Cell Res. 316, 3317–3327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bhattacharyya S., Pant N., Dudeja P. K., Tobacman J. K. (2007) J. Immunoassay Immunochem. 28, 173–188 [DOI] [PubMed] [Google Scholar]
- 38.Shah G. M., Shah R. G., Poirier G. G. (1996) Biochem. Biophys. Res. Commun. 229, 838–844 [DOI] [PubMed] [Google Scholar]
- 39.Kotova E., Jarnik M., Tulin A. V. (2010) Proc. Natl. Acad. Sci. U.S.A. 107, 6406–6411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Steinhilb M. L., Turner R. S., Gaut J. R. (2001) J. Biol. Chem. 276, 4476–4484 [DOI] [PubMed] [Google Scholar]
- 41.Galili U. (2005) Immunol. Cell Biol. 83, 674–686 [DOI] [PubMed] [Google Scholar]
- 42.Tanemura M., Yin D., Chong A. S., Galili U. (2000) J. Clin. Invest. 105, 301–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hsu H., Shu H. B., Pan M. G., Goeddel D. V. (1996) Cell 84, 299–308 [DOI] [PubMed] [Google Scholar]
- 44.Chinnaiyan A. M., O'Rourke K., Tewari M., Dixit V. M. (1995) Cell 81, 505–512 [DOI] [PubMed] [Google Scholar]
- 45.Deveraux Q. L., Reed J. C. (1999) Genes Dev. 13, 239–252 [DOI] [PubMed] [Google Scholar]
- 46.Borthakur A., Bhattacharyya S., Alrefai W. A., Tobacman J. K., Ramaswamy K., Dudeja P. K. (2010) Inflamm. Bowel Dis. 16, 593–603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yan M., Lee J., Schilbach S., Goddard A., Dixit V. (1999) J. Biol. Chem. 274, 10287–10292 [DOI] [PubMed] [Google Scholar]
- 48.Kelliher M. A., Grimm S., Ishida Y., Kuo F., Stanger B. Z., Leder P. (1998) Immunity 8, 297–303 [DOI] [PubMed] [Google Scholar]
- 49.McCarthy J. V., Ni J., Dixit V. M. (1998) J. Biol. Chem. 273, 16968–16975 [DOI] [PubMed] [Google Scholar]
- 50.Yu P. W., Huang B. C., Shen M., Quast J., Chan E., Xu X., Nolan G. P., Payan D. G., Luo Y. (1999) Curr. Biol. 9, 539–542 [DOI] [PubMed] [Google Scholar]
- 51.Wertz I. E., Dixit V. M. (2010) Cold Spring Harb. Perspect. Biol. 2, a003350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Coornaert B., Carpentier I., Beyaert R. (2009) J. Biol. Chem. 284, 8217–8221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shembade N., Ma A., Harhaj E. W. (2010) Science 327, 1135–1139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Varfolomeev E., Goncharov T., Fedorova A. V., Dynek J. N., Zobel K., Deshayes K., Fairbrother W. J., Vucic D. (2008) J. Biol. Chem. 283, 24295–24299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rual J. F., Venkatesan K., Hao T., Hirozane-Kishikawa T., Dricot A., Li N., Berriz G. F., Gibbons F. D., Dreze M., Ayivi-Guedehoussou N., Klitgord N., Simon C., Boxem M., Milstein S., Rosenberg J., Goldberg D. S., Zhang L. V., Wong S. L., Franklin G., Li S., Albala J. S., Lim J., Fraughton C., Llamosas E., Cevik S., Bex C., Lamesch P., Sikorski R. S., Vandenhaute J., Zoghbi H. Y., Smolyar A., Bosak S., Sequerra R., Doucette-Stamm L., Cusick M. E., Hill D. E., Roth F. P., Vidal M. (2005) Nature 437, 1173–1178 [DOI] [PubMed] [Google Scholar]
- 56.Scheffner M., Nuber U., Huibregtse J. M. (1995) Nature 373, 81–83 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.