

Abstract
For adoptive T cell therapy to be effective against solid tumors, tumor-specific T cells must be able to migrate to the tumor site. One requirement for efficient migration is that the effector cells express chemokine receptors that match the chemokines produced either by tumor or tumor-associated cells. In this study, we investigated whether the tumor trafficking of activated T cells (ATCs) bearing a chimeric antigen receptor specific for the tumor antigen GD2 (GD2-CAR) could be enhanced by forced co-expression of the chemokine receptor CCR2b, since this receptor directs migration towards CCL2, a chemokine produced by many tumors, including neuroblastoma. Neuroblastoma cell lines (SK-N-SH and SK-N-AS) and primary tumor cells isolated from six patients all secreted high levels of CCL2, but GD2-CAR transduced ATCs lacked expression of CCR2 (<5%) and migrated poorly to recombinant CCL2 or tumor supernatants. Following retroviral transduction, however, ATCs expressed high levels of CCR2b (>60%) and migrated well in vitro. We expressed firefly luciferase in CCR2b-expressing ATCs and observed improved homing (>10-fold) to CCL2-secreting neuroblastoma compared to CCR2 negative ATCs. As a result, ATCs co-modified with both CCR2b and GD2-CAR had greater anti-tumor activity in vivo.
Keywords: Cytotoxic T Lymphocyte, Chemokine Receptor, Chimeric Antigen Receptor, Immunotherapy, Adoptive T Cell Therapy
INTRODUCTION
Adoptive immunotherapy using T cells and their derivatives has shown promise as treatment for human malignancies(1–5), but to date the range of susceptible tumors has been limited. One of the constraints for successful treatment is the limited trafficking of lymphocytes, which requires effector T cells to express chemokine receptors matched with the chemokines produced by the tumor cells or their associated elements (6). Many human tumors produce low levels of chemokines, while others produce chemokines for which effector T cells lack receptors. As a consequence, adoptively transferred T cells may simply fail to “find” the tumors. Even if limited numbers arrive at the tumor, they may be overwhelmed by inhibitory elements of the immune system that have been recruited or expanded by tumor associated chemokines and cytokines. For example, we gave lymphocytes expressing a chimeric antigen receptor (CAR) specific for the GD2 tumor associated antigen to subjects with neuroblastoma (4). Tumor biopsies showed a scanty T cell infiltrate, and lacked a PCR signal derived from the transgenic GD2-CAR, even when such a signal was readily detectable in peripheral blood. Since 4/8 of the study group with measurable disease nonetheless had a tumor response or tumor necrosis, these data further support the concept that improved migration of infused T cells to tumor sites may increase clinical benefit.
Migration of adoptively transferred T cells to tumors is a multistep process that requires expression of adhesion and integrin receptors for binding to the vascular wall, as well as of chemokine receptors which detect local chemokines and initiate transmigration (7, 8). Kershaw and colleagues showed that deficiencies in T cell chemotaxis towards tumor cells could be overcome by transgenic expression of CXCR2, which recognizes tumor produced Gro-α (6). Similarly, Di Stasi et al demonstrated that overexpression of the chemokine receptor CCR4 by CD30-CAR expressing T cells enhanced migration in response to Hodgkin’s lymphoma secreted CCL17 (TARC; thymus- and activation-regulated chemokine) in murine xenograft experiments (9). We have now determined whether the chemokines produced by human neuroblastoma cells may be used to enhance trafficking of GD2-specific T cells to tumors using a transgenic chemokine receptor. We expressed transgenic CCR2b on in vitro expanded ATCs and show that ATCs engineered to co-express both GD2-CAR and CCR2b have improved tumor specific trafficking and significantly enhanced activity against neuroblastoma xenografts.
MATERIALS AND METHODS
Tumor cell lines
The neuroblastoma cell line LAN-1 was obtained from Dr. Seeger’s laboratory at the University of California Los Angeles. The cell lines JF, NB275, NB281, NB246, NB142, NB247 and NB175 were established in our laboratory. The cell lines IMR-32, SK-N-AS and SK-N-SH were obtained from the American Type Culture Collection (Rockville, MD). All neuroblastoma cell lines were maintained in DMEM supplemented with 10% fetal bovine serum (FBS; Hyclone, Logan, UT) and 2 mM L-glutamine (GlutaMAX-1; Invitrogen, Carlsbad, CA), hereafter referred to as NB media.
Chemokine ELISA
To measure the production of human CCL2, 1×106 tumor cells were plated in fresh NB media in 6-well plates. After 24 hours incubation, the supernatant was collected and centrifuged to remove remaining cells. We measured chemokine levels using an ELISA kit for CCL2 (R&D Systems Minneapolis, MN) per the manufacturer’s recommendations.
Retroviral vectors and supernatant
SFG.CCR2b was constructed by PCR cloning of CCR2 isoform b (CCR2b) from human monocytes and inserting the gene into the NcoI and MluI restriction enzyme sites of the SFG retroviral vector to make SFG.CCR2b. As a control chemokine receptor, CCR7 was cloned by PCR from human T cells and introduced into SFG (SFG.CCR7) at the same location. The GD2-specific CAR, incorporating the transmembrane signaling domains of CD28, OX40 and the T cell receptor ζ-chain (SFG.14g2a.CD28.OX40L.ζ; referred hereafter as CAR), was used as previously described (10). SFG.EGFPluciferase (EGFPluc) was constructed by insertion of the EGFP and firefly luciferase fusion gene (Promega, Madison, WI) into SFG at the NcoI and MluI restriction sites. To create retroviral supernatants, we first generated transient supernatant by co-transfecting 293T cells with the SFG vector plasmid, the Peg-Pam-e plasmid containing the sequence for MoMLV gag-pol and the RD114 plasmid encoding the RD114 envelope using GeneJuice (EMD Biosciences, Gibbstown, NJ) transfection reagent as recommended by the manufacturer. Supernatant containing the retrovirus was collected 48 hr and 72 hr after transfection.
Generation of OKT3 blasts
Following informed consent, peripheral blood mononuclear cells (PBMCs) were obtained from healthy donors using a Baylor College of Medicine IRB approved protocol. We generated OKT3 activated T cells and transduced them with the CAR using a protocol described previously(11). PBMC were resuspended T cell medium (TCM) in 45% RPMI 1640, 45% Click’s media (Irvine Scientific, Santa Ana, CA) supplemented with 10% FBS, 2 mM L-glutamine and 100 U/ml IL-2 (Proleukine; Chiron, Emeryville, CA). 5×106 PBMC were stimulated on non-tissue culture-treated 24-well plates coated with 1 μg/ml each of OKT3 (Ortho Biotech, Raritan, NJ) and anti-CD28 antibodies (BD Biosciences, San Diego, CA) in the presence of 100 U/ml IL-2. On day 3, activated T cells were harvested and transduced with retrovirus vectors or expanded in media supplementedwithIL-2 as described below.
Retroviral transduction
Non-tissue culture treated 24 well plates were coated with 7 μg/ml Retronectin(Takara Bio, Otsu, Shiga, Japan) overnight at 4°C. The wells were washed with phosphate-buffered saline then coated with retroviral supernatant. Subsequently, OKT3 blasts were plated at 5×105 cells per well in viral supernatant supplemented with 100 U/ml IL-2. After three days in culture, cells were harvested and expanded in tissue culture treated plates containing TCM plus 100 U/ml IL-2. For two or three-gene transductions, the protocol is identical to above except the wells were coated with equal amounts of each retroviral supernatant and OKT3 blasts were then plated into each well containing equal amounts of viral supernatant supplemented with 100U/ml IL-2.
Phenotyping
To assess the pattern of chemokine receptor expression on T cells, we used the following monoclonal antibodies conjugated to FITC, PE, PerCP or Cy5 (Becton Dickinson Biosciences, San Diego, CA): CD3, CD4, CD8, CCR4, CCR5, CCR7 and CXCR4. CCR2-PE antibody was purchased from R&D Systems (Minneapolis, MN). CAR expression was assessed using anti-F(ab′)2 (Jackson ImmunoResearch Laboratories, West Grove, PA). Cells were analyzed using a FACSCalibur flow cytometers (BD Biosciences) and FCSExpress software (De Novo Software, Los Angeles, CA).
Transwell migration assay
To determine if T cells expressing CCR2b had improved migration in response to CCL2, we used transwell migration assays in which CCR2b-modified and control ATCs (non-transduced or modified with SFG.CCR7) were labeled with 50 μCi Chromium51 (Cr51; MP Biomedicals, Solon, OH) and 1.5×105 cells were placed into the upper chamber of 24-well 6.5 mm diameter, 5 μm pore size transwell chambers (Costar Transwell, Corning, NY). Media alone, media containing 50 ng/ml recombinant CCL2 (R&D Systems) or supernatants from tumor cell lines were placed in the bottom chamber. Plates were then incubated for 3 hours at 37°C. Following incubation, the cells in the bottom well were collected and analyzed using a γ-counter (Cobra Quantum; Perkin Elmer, Shelton, CT). Specific migration was calculated using the following equation: Specific migration (%) = (experimental − spontaneous [media alone])/(maximum [1.5×105 cells] − spontaneous) × 100.
Cytotoxicity and co-culture assays
We used a standard 4-hour 51Cr-release assay to confirm the ability of CAR ATCs to kill GD2 expressing neuroblastoma cells. Briefly, the GD2 positive neuroblastoma cell lines LAN-1 and SK-N-AS were labeled with 51Cr and co-cultured with non-transduced T cells or with cells modified with CAR alone, CCR2b alone, or following transduction with both CAR and CCR2b. Specific lysis was calculated using the equation (experimental lysis -spontaneous lysis)/(maximum lysis -spontaneous lysis) × 100. To examine the ability of CAR and CAR-CCR2b transduced T cells to control tumor growth in vitro, 1×105 GFP transduced tumor cells were plated with increasing numbers of gene-modified allogeneic ATCs at a 10:1 T cell to tumor cell ratio. T cell expansion and tumor cell frequency measured after 7 days by FACS analysis using CD3 PerCP and GFP, respectively.
SCID xenograft animal model
To measure enhanced homing of CCR2b gene-modified T cells and determine if CCR2b expression was associated with increased in vivo tumor killing by GD2-specific T cells, we used a human neuroblastoma xenograft into severe combined immune deficient mice (SCID [strain NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ]; Jackson Laboratory, Bar Harbor, Maine). All mouse experiments accorded with Baylor College of Medicine Animal Husbandry and Institutional Animal Care and Use Committee (IACUC) guidelines.
In vivo migration assay following CCR2b gene modification
To examine improved homing to CCL2-secreting neuroblastoma tumors, SCID mice were injected subcutaneously (s.c.) in the right flank with 1×106 SK-N-SH tumor cells resuspended in Matrigel (BD Biosciences, San Diego, CA). After establishment of the tumor (7–10 days), two groups of mice were injected via the tail vein (intravenous; i.v.) with either 1×107 ATCs transduced with EGFPluc (control; EGFPluc) or with both CCR2b and EGFPluc (experimental; CCR2b-EGFPluc). Following T cell infusion, mice received 1000 U/mouse recombinant human IL-2 intraperitoneally (i.p) every 2–3 days. Biodistribution of T cells was examined by i.p. injection 150 mg/kg D-luciferin (Xenogen, Alameda, CA) at time points of 1, 24, 48 and 72 hours and imaging using the IVIS imaging system (Xenogen). Photon emission was analyzed by constant region-of-interest (ROI) drawn over the tumor region and the signal measured as total photon/sec/cm2/steridian (p/s/cm2/sr) as previously validated (12).
In vivo proliferation assay following CCR2b gene modification
To examine the effects of the CAR on ATC proliferation in vivo, SCID mice were injected s.c. in the right flank with 1×106 SK-N-AS tumor cells resuspended in Matrigel. After establishment of the tumor (7–10 days), two groups of mice were injected i.v. with either 1×107 ATCs transduced with the CAR and EGFPluc (control; CAR-EGFPluc) or with the CAR, CCR2b, and EGFPluc (experimental; CAR-CCR2b-EGFPluc). Following T cell infusion, mice received 1000 U/mouse recombinant human IL-2 i.p. every 2–3 days. Biodistribution and expansion of T cells was examined by i.p. injection of D-luciferin on days 1, 7, and 14 using the procedure described above.
In vivo efficacy assay following CCR2b gene modification
To determine if CCR2b gene modification improved the anti-neuroblastoma efficacy of CAR T cells, SCID mice were injected s.c. in the right flank with 1×106 SK-N-AS-EGFPluc (GD2 positive, CCL2 secreting neuroblastoma modified to express EGFPluc) tumor cells resuspended in Matrigel. Three days post-tumor inoculation, two groups of mice were injected i.v. with either 1×107 ATCs genetically modified to express the CAR (control; CAR) or both the CAR and CCR2b (experimental; CAR-CCR2b). Gene-modified ATCs were then injected weekly on days 7 and 14 for a total of three ATC doses per mouse. As described above, mice received 1000 U/mouse recombinant human IL-2 i.p. every 2–3 days post T cell infusion. Tumor growth was monitored by bioluminescence imaging on days 0, 8, 18 and 22 using the procedure described above.
Statistical analyses
Data are expressed as means ± standard deviation. Student’s t test was used where appropriate. Where indicated by *, P ≤ .05 was accepted as statistically significant.
RESULTS
ATCs do not express the receptor for the CCL2 chemokine secreted by NB
We first confirmed that CCL2 was secreted from neuroblastoma cell lines and primary tumor cells obtained from biopsies. MYCN non-amplified tumor cell lines and primary tumors (n=6) consistently secreted high concentrations of CCL2, while MYCN amplified tumors did not (Table 1). We then measured chemokine receptor expression on CAR-modified T cells generated from PBMCs from six subjects with neuroblastoma. CD4+ and CD8+ ATCs expressed both CCR5 and CXCR4, the receptors for the CCL5 and CXCL12 chemokines respectively, but lacked CCR2 expression (Figure 1). Hence, CCR2 is not amongst the chemokine receptors expressed by ex vivo expanded and gene-modified T cells.
Table 1.
Neuroblastoma cell line characteristics.
| Cell Line | CCL2 (pg/ml) | GD2%/MFI | Myc-N Status |
|---|---|---|---|
| A. Established cell lines | |||
| IMR-32 | 36 ± 3 | 79%/75 | Amplified |
| JF | 40 ± 1 | 82%/89.8 | Amplified |
| LA-N-1 | 79 ± 5 | 85%/97.3 | Amplified |
| SK-N-AS | > 2000 | 47%/7.5 | Non-amplified |
| SK-N-SH | > 2000 | 6%/0.1 | Non-amplified |
| B. Primary cell lines | |||
| P142 | 1393 | 60%/29.2 | Non-amplified |
| P175 | 457 | 39%/4.9 | Mixed |
| P246 | 1600 | n.d. | n.d. |
| P247 | 932 | 34 | n.d. |
| P275 | 5929 | n.d. | Non-amplified |
| P283 | 1530 | 8%/0.1 | Non-amplified |
n.d., not determined
Figure 1. ATCs lack expression of CCR2.

A) ATCs generated for adoptive cell therapy from six patients were examined for chemokine receptor expression using a panel of antibodies to CCR2, CCR4, CCR5, CCR7 and CXCR4. ATCs were examined for chemokine receptor expression for both CD4+ (left panel) and CD8+ T cells(right panel). Mean receptor expression is shown.
CCR2b gene-modified ATCs migrate in response to rhCCL2 and neuroblastoma supernatants
To determine if transgenic expression of the CCR2 receptor by CAR expressing patient T cells could modify their ability to migrate in response to CCL2, we transduced ATCs generated from healthy donor PBMC with retrovirus encoding either CCR2b or a control chemokine receptor, CCR7. Analysis showed that the SFG.CCR2b, as well as SFG.CCR7, efficiently transduced both CD4+ and CD8+ ATCs (Figures 2a and 2b). CAR ATCs lacking CCR2 migrated poorly in response to CCL2, but following transduction with CCR2b, migration was significantly enhanced (Figure 2c). This chemotactic response was receptor-ligand specific since ATCs transduced with CCR2 failed to migrate in response to CCL21, whereas control cells expressing CCR7 migrated on exposure to their ligand CCL21, but not to CCL2 (Figure 2c). The migration of CCR2b-transduced ATCs was also increased in response to supernatants from tumors that produced CCL2 (Table 1), so that CCR2b transduced ATCs increased their chemotaxis in response to SK-N-SH supernatants, but did not respond to supernatants from the lines IMR-32 and JF, which are low CCL2 producers (Figure 2d). Importantly, migration of CCR2b ATCs was significantly increased following exposure to supernatants from primary neuroblastoma cells obtained from three patients (Figure 2e). Therefore, transgenic CCR2b is functional when expressed in patient T cells, enhancing migration in response to tumor derived CCL2 chemokine.
Figure 2. Transduction of ATCs with CCR2b or CCR7 augments migration to chemokines.

ATCs were transduced with SFG.CCR2b (A) or SFG.CCR7 (B) retrovirus and analyzed for expression of the transgene by flow cytometry using CCR2 or CCR7 antibodies. C) CCR2b or CCR7 transduced ATCs showed enhanced migration to CCL2 or CCL21, respectively, in transwell migration assays compared to ATCs transduced with the mismatched receptor. D) CCR2b transduced ATCs show improved migration to SK-N-SH (CCL2+) tumor supernatant, but not to IMR-32 or JF which do not secrete CCL2. E) ATCs transduced with CCR2b show enhanced transwell migration compared to non-transduced (NT) ATCs when cultured with tumor supernatants taken from neuroblastoma cell lines derived from primary patient tissue.
Increased in vivo migration of CCR2b-transduced ATCs in a neuroblastoma xenograft
We established s.c. CCL2-secreting SK-N-SH tumor xenografts in SCID mice and subsequently injected ATCs transduced either with EGFPluc (control; EGFPluc) or with both CCR2b and EGFPluc (experimental; CCR2b-EGFPluc) intravenously. We then measured the biodistribution of EGFPluc and CCR2b-EGFPluc ATCs over 72 hours using bioluminescence imaging. Consistent with our in vitro observations, accumulation of CCR2b-EGFPluc ATCs progressively increased over the initial 1–3 days of the experiment, while no signal from ATCs expressing EGFPluc alone were detected at the tumor site (Figures 3a and 3b). These data suggest that the increased in vitro migratory function of CCR2b-transduced ATCs to CCL2 is associated with similar in vivo behavior.
Figure 3. CCR2b transduced ATCs exhibit enhanced tumor trafficking in vivo.
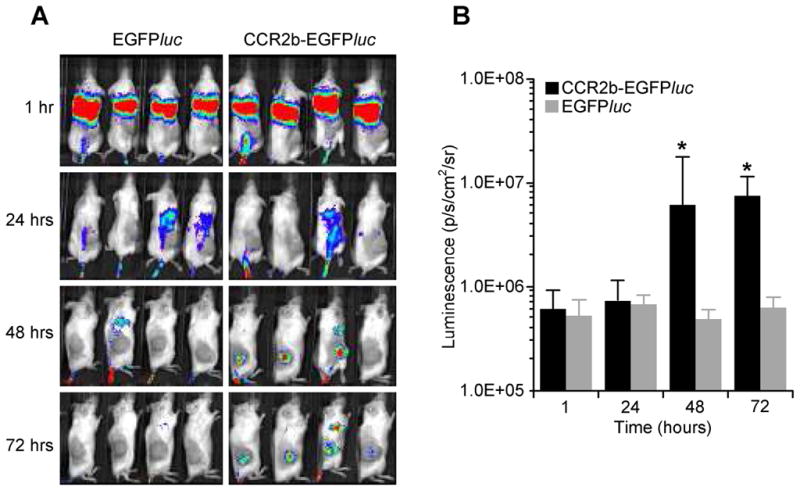
A) SCID mice received a s.c. injection in the right flank with SK-N-SH (CCL2+) tumor cells followed i.v. injection of ATCs transduced with EGFPluc alone, or with EGFPluc and CCR2b. Following injection, ATCs were monitored by bioluminescence imaging for 72 hours. B) Bioluminescence was calculated by region of interest (ROI) analysis at the tumor site.
CAR-T cells that co-express CCR2b migrate to CCL2 in vitro and kill GD2+CCL2+ neuroblastomas
We modified ATCs to express both CAR and CCR2b and determined if they were able to migrate to and kill GD2+ and CCL2 secreting SK-N-AS neuroblasts in vitro (Table 1). For this experiment, the SK-N-AS cell line was used due to their known expression of GD2 and secretion of CCL2. Co-expression of CAR did not affect the ability of transgenic CCR2b to mediate migration of ATCs in response to either SK-N-AS supernatant or to rhCCL2 (Figure 4a) and these CCR2b ATCs were able to specifically recognize and kill SK-N-AS in cytotoxicity assays (Figure 4b). Both ATCs transduced with either CAR or CAR-CCR2b eliminated GD2+ GFP-expressing SK-N-AS tumor cells in 7 day co-culture assays compared to non-transduced control ATCs (p<.05) (Figure 4c and d). Hence, SK-N-AS produces CCL2 which can induce CAR-CCR2b ATC migration and is sensitive to killing by CAR expressing ATCs.
Figure 4. CAR-CCR2b ATCs show specificity towards GD2+ neuroblastoma and enhanced CCL2 migration.

A) ATCs modified with CAR-CCR2b shown increased migration towards SK-N-AS supernatant and rhCCL2 compared to NT ATCs. B) ATCs transduced with CAR-CCR2b specifically recognize and kill SK-N-AS tumor cells, whereas non-transduced (NT) control ATCs do not. C and D) Seven day co-culture assays comparing NT ATCs with ATCs transduced with CAR or with CAR-CCR2b at a 10:1 T cell to tumor ratio, demonstrating complete elimination of GFP+ SK-N-AS tumor cells by CAR and CAR-CCR2b transduced ATCs, respectively, but not by NT ATCs.
CAR-CCR2bATCs home to and expand at the site of GD2+CCL2+ neuroblastoma cells in vivo
Binding of GD2 on tumor cells by CAR induces T cell activation which results in the production of IL-2 and proliferation (10). As shown in Figure 3, ATCs transduced with CCR2b rapidly accumulate at the tumor site (1–3 days). To learn whether CCR2b can augment both tumor-specific migration as well as in vivo expansion in response to GD2 recognition, we generated ATCs transduced with EGFPluc and CAR (CAR-EGFPluc) or with EGFPluc, CAR and CCR2b (CAR-CCR2b-EGFPluc) and measured biodistribution and expansion by bioluminescence in our xenograft model. We used flow cytometry to confirm that both ATC populations expressed equivalent levels of EGFPluc and CAR, and that CCR2b was only present in ATCs transduced with all three vectors (Figure 5a). We injected CAR-EGFPluc or CAR-CCR2b-EGFPluc transduced ATCs intravenously into mice with established subcutaneous SK-N-AS tumors. Mice also received i.p. injection of IL-2 every 2–3 days to facilitate T cell expansion, and were imaged on days 1, 7 and 14 (Figures 5b and 5c). At early time points, ATCs expressing CCR2b migrated to the tumor site, whereas ATCs expressing CAR-EGFPluc did not. On subsequent imaging, the signal strength continued to increase but only at the tumor sites of mice receiving CAR-CCR2b-EGFPluc. The intensity reached by 14 days indicated local expansion of the modified T cells (p=.04). Hence, transgenic CCR2b facilitates trafficking of CAR transduced ATCs to CCL2-secreting neuroblastomas thus facilitating their proliferation in response to GD2 antigen stimulation.
Figure 5. CAR-CCR2b ATCs show enhanced homing and expansion at the tumor site.

A) ATCs were transduced with EGFPluc and either CAR (middle panel) or CAR and CCR2b (right panel) and analyzed by FACS for EGFP, CAR (F(ab′)2 Cy5 antibody) and CCR2b (CCR2 PE). B) SCID mice received SK-N-AS tumor cells s.c. in the right flank followed by i.v. injection of CAR-EGFPluc or CAR-CCR2b-EGFPluc gene-modified ATCs 7 days later. C) Mice were imaged for bioluminescence using tumor ROI on days 1, 7, and 14 showing enhanced homing and expansion of CAR-CCR2b-EGFPluc modified ATCs compared to ATCs modified only with CAR-EGFPluc.
CAR and CCR2b ATC have enhanced killing of GD2+CCL2+ neuroblastomas in vivo
To learn whether the increased frequency of CAR-CCR2b ATCs at the tumor site enhances the anti-tumor effects of these T cells, we measured tumor growth in SCID mice using SK-N-AS tumor cells modified to express EGFPluc. We generated two ATC lines, one that was transduced with CAR alone and the second that co-expressed CAR and CCR2b (CAR-CCR2b). Flow cytometric analysis showed that CAR ATCs expressed the GD2 CAR (44%) but not CCR2b (0.4%), whereas dual transduced ATCs expressed both CAR (32%) and CCR2b (43%) or co-expressed CAR and CCR2b (19%) (Figure 6a). Both ATC cultures had equivalent cytotoxicity towards SK-N-AS-EGFPluc (not shown). Following s.c. injection of SK-N-AS, mice received their first injection of CAR or CAR-CCR2b modified ATCs when the tumor was palpable, and subsequent tumor growth was monitored by bioluminescence imaging. We observed significant reduction in tumor growth in mice receiving CAR-CCR2b modified ATCs compared to mice receiving ATCs modified with CAR alone (p=.04 day 18; p=.02 day 22) (Figures 6b and 6c). Hence, improved migration following CCR2b transgene expression is associated with improved elimination of GD2+CCL2+ neuroblastoma tumors by CAR expressing ATCs.
Figure 6. CAR-CCR2b ATCs show enhanced anti-tumor effect.

A) ATCs cells were transduced with either CAR (middle panel) or CAR and CCR2b (right panel) and analyzed by FACS for expression of CAR (by F(ab′)2 FITC antibody; y-axis) and CCR2 (CCR2 PE; x-axis) indicating that both T cell populations express equivalent CAR but only CCR2b gene-modified ATCs express CCR2. B and C) SCID mice received an s.c. injection right flank of SK-N-AS modified to express EGFPluc followed by i.v. injection of either CAR (left panel) or CAR-CCR2b (right panel) and measured for tumor growth by bioluminescence on days 0, 8, 18 and 22. CAR-CCR2b ATCs showed significant (P<.05) inhibition of tumor growth compared to ATCs modified with CAR alone.
DISCUSSION
In this study, we have demonstrated that transgenic expression of CCR2b on ex vivo expanded ATCs dramatically enhances both in vitro and in vivo chemotaxis in response to CCL2 produced by neuroblastoma cell lines and primary tumor cells. When the activated T cells also express a chimeric antigen receptor directed to the tumor associated antigen GD2, co-expression of CCR2b leads to rapid migration to a subcutaneous neuroblastoma xenograft associated with greater anti-tumor activity than CAR expressing ATCs that lack CCR2b
Indium111 labeling and in vivo tracking of ATCs expressing a CAR specific for the α-folate receptor showed that these cells retained the capacity to migrate to ovarian cancer after non-specific activation and expansion (1). Similarly, ATCs transduced with the α and β chains of MART1 and gp100-specific T cells induced disease regression in patients with metastatic melanoma and were present as infiltrates in tumor biopsies (13). In principle, therefore gene modified, activated and expanded T cells can continue to migrate to tumor sites, and the failure of migration observed in patients with tumors such as neuroblastoma suggests instead that their absence at tumor sites may be a consequence of a mismatch between the chemokines the tumors produce and the chemokine receptors expressed by ATC.
CCL2 is a chemokine active for CCR2 expressing T lymphocytes (14, 15), NKT cells (16) and γδ T cells(17, 18), and our results indicate that transgenic expression of the CCR2 receptor is both necessary and sufficient to restore CCL2 responsiveness to T cell subsets in which receptor expression is deficient. Our data demonstrating an absence of CCR2 on ATCs, a lack of CCL2 responsiveness and a failure of migration to tumor sites differ from some earlier findings in which CCR2 was detected on human ATCs (2) at a sufficient level for CCL2-induced T cell migration in tumor bearing immune deficient mice (19). While these differences may be explained on the basis of distinct culture conditions, including the use of anti-CD28 antibody which may facilitate downregulation of CCR2 following TCR activation (20), it is likely that the transgenic CCR2 is expressed at higher levels and on a greater percentage of cells than previously reported and will likely lead to enhanced tumor migrations even in subjects with low constitutive expression of CCR2.
More aggressive MYCN amplified tumors transcriptionally repress production of CCL2 (16, 21) suggesting that CCR2b modification may be of limited value in high-risk disease. However, of these high-risk patients(stage 3,4), approximately 70% lack MYCN amplification (22). Further, while MYCN status is used for risk stratification, Matthay et al showed that in those patients receiving high dose chemotherapy followed by autologous hematopoietic stem cell rescue (AHSCT) as consolidation therapy (the current standard of care), MYCN amplification proved to be a negative prognostic factor in overall survival (OS) but not event-free survival (EFS) (23). This indicates that a substantial number of high-risk patients with relapsed/refractory disease may ultimately benefit from CCL2-directed T cell therapies.
CCL2 has also been found to be highly secreted by the tumor microenvironment in a variety of malignancies including melanoma, sarcoma, glioma, ovarian, prostate and breast cancer (24–29). CCL2 production is associated with increased infiltrating tumor-associated macrophage (TAM) that promote tumor growth via cytokines such as IL-6 (30), enhance angiogenesis (31–33), and contribute to immune suppression through production of IL-10 (34) and recruitment of Th2 and regulatory T cells (35, 36). Moreover, CCL2 is produced by TAMs themselves, suggesting a self-amplifying loop(37). The abundance of CCL2 in solid tumors, produced both by tumor cells and infiltrating immune cells, implies that this chemokine pathway might be used as a generic mechanism to induce tumor-specific migration of cytotoxic T cells for cancer treatment, irrespective of whether CCL2 is produced by the tumor cells themselves.
Under physiological or stress conditions, CCL2 is secreted by non-malignant cells including endothelium, fibroblasts, smooth muscle, and astrocytic and microglial cells(38). CCR2b-modified T cells may therefore migrate to these tissues resulting in decreased antitumor effects or unintended toxicity. The amounts and concentration of CCL2 produced at tumor sites, however, make it probable that the bulk of effector cells will go to the tumor site. The use of autologous virus-specific T cells transduced with the GD2-CAR may further reduce the potential for off-target toxicity against non-tumor tissues (4).
In summary, using neuroblastoma as a model, we show that transgenic expression of a chemokine receptor complementary to tumor-associated chemokines will enhance the ability of ATCs to traffic to tumors and improves their anti-tumor efficacy. In principle, this approach should be applicable to any tumor directed T cell lacking a receptor that matches the chemokines the tumor produces.
Acknowledgments
This research was funded by The Hope Street Kids Foundation Research Award (AEF) and the NIH/NCI 2 P01 CA094237-06 (MKB and AEF). JAC was supported by T32 HL092332 and K12 CA090433.
Footnotes
CONFLICT OF INTEREST
The authors declare no conflict of interest.
AUTHORSHIP
AEF and JAC designed and performed research, analyzed data and wrote the paper. AL, AB, and MP performed experiments and analyzed data. MKB and CMR contributed to research analysis, discussion and critical review of the manuscript.
References
- 1.Kershaw MH, Westwood JA, Parker LL, et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin Cancer Res. 2006;12:6106–6115. doi: 10.1158/1078-0432.CCR-06-1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Park JR, Digiusto DL, Slovak M, et al. Adoptive transfer of chimeric antigen receptor re-directed cytolytic T lymphocyte clones in patients with neuroblastoma. Mol Ther. 2007;15:825–833. doi: 10.1038/sj.mt.6300104. [DOI] [PubMed] [Google Scholar]
- 3.Lamers CH, Langeveld SC, Groot-van Ruijven CM, et al. Gene-modified T cells for adoptive immunotherapy of renal cell cancer maintain transgene-specific immune functions in vivo. Cancer Immunol Immunother. 2007;56:1875–1883. doi: 10.1007/s00262-007-0330-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pule MA, Savoldo B, Myers GD, et al. Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nat Med. 2008;14:1264–1270. doi: 10.1038/nm.1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Till BG, Jensen MC, Wang J, et al. Adoptive immunotherapy for indolent non-Hodgkin lymphoma and mantle cell lymphoma using genetically modified autologous CD20-specific T cells. Blood. 2008;112:2261–2271. doi: 10.1182/blood-2007-12-128843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kershaw MH, Wang G, Westwood JA, et al. Redirecting migration of T cells to chemokine secreted from tumors by genetic modification with CXCR2. Hum Gene Ther. 2002;13:1971–1980. doi: 10.1089/10430340260355374. [DOI] [PubMed] [Google Scholar]
- 7.Bromley SK, Mempel TR, Luster AD. Orchestrating the orchestrators: chemokines in control of T cell traffic. Nat Immunol. 2008;9:970–980. doi: 10.1038/ni.f.213. [DOI] [PubMed] [Google Scholar]
- 8.Springer TA. Traffic signals for lymphocyte recirculation and leukocyte emigration: the multistep paradigm. Cell. 1994;76:301–314. doi: 10.1016/0092-8674(94)90337-9. [DOI] [PubMed] [Google Scholar]
- 9.Di SA, De AB, Rooney CM, et al. T lymphocytes coexpressing CCR4 and a chimeric antigen receptor targeting CD30 have improved homing and antitumor activity in a Hodgkin tumor model. Blood. 2009;113:6392–6402. doi: 10.1182/blood-2009-03-209650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pule MA, Straathof KC, Dotti G, et al. A chimeric T cell antigen receptor that augments cytokine release and supports clonal expansion of primary human T cells. Mol Ther. 2005;12:933–941. doi: 10.1016/j.ymthe.2005.04.016. [DOI] [PubMed] [Google Scholar]
- 11.Foster AE, Leen AM, Lee T, et al. Autologous designer antigen-presenting cells by gene modification of T lymphocyte blasts with IL-7 and IL-12. J Immunother. 2007;30:506–516. doi: 10.1097/CJI.0b013e318046f3b1. [DOI] [PubMed] [Google Scholar]
- 12.Vera J, Savoldo B, Vigouroux S, et al. T lymphocytes redirected against the kappa light chain of human immunoglobulin efficiently kill mature B lymphocyte-derived malignant cells. Blood. 2006;108:3890–3897. doi: 10.1182/blood-2006-04-017061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Johnson LA, Morgan RA, Dudley ME, et al. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood. 2009;114:535–546. doi: 10.1182/blood-2009-03-211714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Roth SJ, Carr MW, Springer TA. C-C chemokines, but not the C-X-C chemokines interleukin-8 and interferon-gamma inducible protein-10, stimulate transendothelial chemotaxis of T lymphocytes. Eur J Immunol. 1995;25:3482–3488. doi: 10.1002/eji.1830251241. [DOI] [PubMed] [Google Scholar]
- 15.Zhang T, Somasundaram R, Berencsi K, et al. Migration of cytotoxic T lymphocytes toward melanoma cells in three-dimensional organotypic culture is dependent on CCL2 and CCR4. Eur J Immunol. 2006;36:457–467. doi: 10.1002/eji.200526208. [DOI] [PubMed] [Google Scholar]
- 16.Metelitsa LS, Wu HW, Wang H, et al. Natural killer T cells infiltrate neuroblastomas expressing the chemokine CCL2. J Exp Med. 2004;199:1213–1221. doi: 10.1084/jem.20031462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Penido C, Vieira-de-Abreu A, Bozza MT, et al. Role of monocyte chemotactic protein-1/CC chemokine ligand 2 on gamma delta T lymphocyte trafficking during inflammation induced by lipopolysaccharide or Mycobacterium bovis bacille Calmette-Guerin. J Immunol. 2003;171:6788–6794. doi: 10.4049/jimmunol.171.12.6788. [DOI] [PubMed] [Google Scholar]
- 18.Penido C, Costa MF, Souza MC, et al. Involvement of CC chemokines in gammadelta T lymphocyte trafficking during allergic inflammation: the role of CCL2/CCR2 pathway. Int Immunol. 2008;20:129–139. doi: 10.1093/intimm/dxm128. [DOI] [PubMed] [Google Scholar]
- 19.Brown CE, Vishwanath RP, Aguilar B, et al. Tumor-derived chemokine MCP-1/CCL2 is sufficient for mediating tumor tropism of adoptively transferred T cells. J Immunol. 2007;179:3332–3341. doi: 10.4049/jimmunol.179.5.3332. [DOI] [PubMed] [Google Scholar]
- 20.Loetscher P, Seitz M, Baggiolini M, et al. Interleukin-2 regulates CC chemokine receptor expression and chemotactic responsiveness in T lymphocytes. J Exp Med. 1996;184:569–577. doi: 10.1084/jem.184.2.569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Song L, Ara T, Wu HW, et al. Oncogene MYCN regulates localization of NKT cells to the site of disease in neuroblastoma. J Clin Invest. 2007;117:2702–2712. doi: 10.1172/JCI30751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brodeur GM. Neuroblastoma: biological insights into a clinical enigma. Nat Rev Cancer. 2003;3:203–216. doi: 10.1038/nrc1014. [DOI] [PubMed] [Google Scholar]
- 23.Matthay KK, Reynolds CP, Seeger RC, et al. Long-term results for children with high-risk neuroblastoma treated on a randomized trial of myeloablative therapy followed by 13-cis-retinoic acid: a children’s oncology group study. J Clin Oncol. 2009;27:1007–1013. doi: 10.1200/JCO.2007.13.8925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Graves DT, Barnhill R, Galanopoulos T, et al. Expression of monocyte chemotactic protein-1 in human melanoma in vivo. Am J Pathol. 1992;140:9–14. [PMC free article] [PubMed] [Google Scholar]
- 25.Kuratsu J, Yoshizato K, Yoshimura T, et al. Quantitative study of monocyte chemoattractant protein-1 (MCP-1) in cerebrospinal fluid and cyst fluid from patients with malignant glioma. J Natl Cancer Inst. 1993;85:1836–1839. doi: 10.1093/jnci/85.22.1836. [DOI] [PubMed] [Google Scholar]
- 26.Negus RP, Stamp GW, Relf MG, et al. The detection and localization of monocyte chemoattractant protein-1 (MCP-1) in human ovarian cancer. J Clin Invest. 1995;95:2391–2396. doi: 10.1172/JCI117933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Valkovic T, Lucin K, Krstulja M, et al. Expression of monocyte chemotactic protein-1 in human invasive ductal breast cancer. Pathol Res Pract. 1998;194:335–340. doi: 10.1016/S0344-0338(98)80057-5. [DOI] [PubMed] [Google Scholar]
- 28.Sciacca FL, Sturzl M, Bussolino F, et al. Expression of adhesion molecules, platelet-activating factor, and chemokines by Kaposi’s sarcoma cells. J Immunol. 1994;153:4816–4825. [PubMed] [Google Scholar]
- 29.Mizutani K, Sud S, McGregor NA, et al. The chemokine CCL2 increases prostate tumor growth and bone metastasis through macrophage and osteoclast recruitment. Neoplasia. 2009;11:1235–1242. doi: 10.1593/neo.09988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Song L, Asgharzadeh S, Salo J, et al. Valpha24-invariant NKT cells mediate antitumor activity via killing of tumor-associated macrophages. J Clin Invest. 2009;119:1524–1536. doi: 10.1172/JCI37869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nishie A, Ono M, Shono T, et al. Macrophage infiltration and heme oxygenase-1 expression correlate with angiogenesis in human gliomas. Clin Cancer Res. 1999;5:1107–1113. [PubMed] [Google Scholar]
- 32.Leek RD, Lewis CE, Whitehouse R, et al. Association of macrophage infiltration with angiogenesis and prognosis in invasive breast carcinoma. Cancer Res. 1996;56:4625–4629. [PubMed] [Google Scholar]
- 33.Solinas G, Germano G, Mantovani A, et al. Tumor-associated macrophages (TAM) as major players of the cancer-related inflammation. J Leukoc Biol. 2009;86:1065–1073. doi: 10.1189/jlb.0609385. [DOI] [PubMed] [Google Scholar]
- 34.Sica A, Schioppa T, Mantovani A, et al. Tumour-associated macrophages are a distinct M2 polarised population promoting tumour progression: potential targets of anti-cancer therapy. Eur J Cancer. 2006;42:717–727. doi: 10.1016/j.ejca.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 35.Balkwill F. Cancer and the chemokine network. Nat Rev Cancer. 2004;4:540–550. doi: 10.1038/nrc1388. [DOI] [PubMed] [Google Scholar]
- 36.Mantovani A, Allavena P, Sozzani S, et al. Chemokines in the recruitment and shaping of the leukocyte infiltrate of tumors. Semin Cancer Biol. 2004;14:155–160. doi: 10.1016/j.semcancer.2003.10.001. [DOI] [PubMed] [Google Scholar]
- 37.Colombo MP, Mantovani A. Targeting myelomonocytic cells to revert inflammation-dependent cancer promotion. Cancer Res. 2005;65:9113–9116. doi: 10.1158/0008-5472.CAN-05-2714. [DOI] [PubMed] [Google Scholar]
- 38.Deshmane SL, Kremlev S, Amini S, et al. Monocyte chemoattractant protein-1 (MCP-1): an overview. J Interferon Cytokine Res. 2009;29:313–326. doi: 10.1089/jir.2008.0027. [DOI] [PMC free article] [PubMed] [Google Scholar]