

Abstract
Molecularly defined P2Y receptor subtypes are known to regulate the functions of neurons through an inhibition of KV7 K+ and CaV2 Ca2+ channels and via an activation or inhibition of Kir3 channels. Here, we searched for additional neuronal ion channels as targets for P2Y receptors. Rat P2Y1 receptors were expressed in PC12 cells via an inducible expression system, and the effects of nucleotides on membrane currents and intracellular Ca2+ were investigated. At a membrane potential of −30 mV, ADP induced transient outward currents in a concentration-dependent manner with half-maximal effects at 4 μm. These currents had reversal potentials close to the K+ equilibrium potential and changed direction when extracellular Na+ was largely replaced by K+, but remained unaltered when extracellular Cl− was changed. Currents were abolished by P2Y1 antagonists and by blockade of phospholipase C. ADP also caused rises in intracellular Ca2+, and ADP-evoked currents were abolished when inositol trisphosphate-sensitive Ca2+ stores were depleted. Blockers of KCa2, but not those of KCa1.1 or KCa3.1, channels largely reduced ADP-evoked currents. In hippocampal neurons, ADP also triggered outward currents at −30 mV which were attenuated by P2Y1 antagonists, depletion of Ca2+ stores, or a blocker of KCa2 channels. These results demonstrate that activation of neuronal P2Y1 receptors may gate Ca2+-dependent K+ (KCa2) channels via phospholipase C-dependent increases in intracellular Ca2+ and thereby define an additional class of neuronal ion channels as novel effectors for P2Y receptors. This mechanism may form the basis for the control of synaptic plasticity via P2Y1 receptors.
Introduction
Flow of information within or between different neurons depends on electrical activity provided by ligand- and voltage-gated ion channels. Accordingly, changes in the responsiveness of a neuron are most frequently brought about by alterations in the opening and closure of ion channels, and such effects are in most instances mediated by heptahelical G protein-coupled receptors. This principle also holds true for G protein-coupled nucleotide (P2Y) receptors expressed in neurons (Hussl & Boehm, 2006). In fact, nucleotides have been reported to control ion channels in a large number of different neuronal tissues (Lechner & Boehm, 2004). For example, nucleotides have been suggested to inhibit currents through voltage-gated Ca2+ channels via native P2Y1 (Gerevich et al. 2004), P2Y2 (Abe et al. 2003), P2Y12 (Vartian & Boehm, 2001) and P2Y13 (Wirkner et al. 2004) receptors, and currents through KV7 channels via native P2Y1 (Filippov et al. 2006), P2Y2 (Filippov et al. 1994), P2Y4 (Meng et al. 2003) and P2Y6 (Boehm, 1998) receptors. In addition, several nucleotides have been reported to either activate or inhibit K+ currents in a variety of neurons, but the receptor and channel subtypes involved remained unidentified (Lechner & Boehm, 2004).
Most neurons express more than one subtype of P2Y receptor, but the precise pharmacological identification of the receptor subtype(s) involved in specific responses as observed in primary neurons is often difficult (Hussl & Boehm, 2006). Therefore, the regulation of neuronal ion channels via P2Y receptors has frequently been investigated using recombinant receptors expressed either in neurons or in neuron-like cell lines (Boehm, 2003). The P2Y receptor subtype being expressed in most, if not all, neuronal tissues, is the P2Y1 receptor (Hussl & Boehm, 2006).
In sympathetic neurons, recombinant P2Y1 receptors have been found to mediate (i) an inhibition of members of the family of endogenously expressed CaV2 channels (Filippov et al. 2000), (ii) an inhibition of endogenous KV7 channels (Brown et al. 2000), (iii) and an activation and inhibition of heterologously expressed Kir3.1/3.2 channels (Filippov et al. 2004). In PC12 cells, which are ontogenetically related to sympathetic neurons and widely used as a model for the investigation of neuronal ion channels (Greene & Tischler, 1976), activation of recombinant P2Y1 receptors also leads to the closure of endogenous KV7 channels (Moskvina et al. 2003). One difference between sympathetic neurons and PC12 cells is the fact that the primary neurons (Moskvina et al. 2003), but not the PC12 cells (Arslan et al. 2000; Unterberger et al. 2002), do express endogenous P2Y1 receptors. Therefore, the present study was initiated to further investigate the repertoire of the coupling of P2Y1 receptors to neuronal ion channels on a neuronal background that lacks this receptor subtype, i.e. in PC12 cells (Unterberger et al. 2002). The results reveal that recombinant P2Y1 receptors mediate an activation of KCa2 channels in this cell line. These data are confirmed in primary hippocampal neurons, where endogenous P2Y1 receptors had been found before to couple to KV7 channels (Filippov et al. 2006). As KCa2 channels are major determinants of spike timing precision in neurons (Stocker, 2004), these data describe a novel and important effector system for neuronal P2Y receptors and thereby broaden the spectrum of neuronal ion channels that are controlled by P2Y receptors.
Methods
Cell culture, molecular cloning and generation of stable cell lines
Primary cultures of rat hippocampal neurons were prepared as described previously (Boehm & Betz, 1997) with minor modifications. Hippocampi were dissected from neonatal Sprague–Dawley rats which had been killed by decapitation in full accordance with all rules of the Austrian animal protection law and the Austrian animal experiment by-laws. These rules are also in accordance with the general rules described by Drummond in The Journal of Physiology (Drummond, 2009). The tissue was cut into small pieces, incubated in papain (30 min at 36°C; Worthington; 1 mg ml−1 in L-15 Leibovitz Medium, supplemented with 1 mm kynurenic acid), and dissociated by trituration in Dulbecco's modified Eagle's medium (Invitrogen, Austria) containing 10% fetal calf serum and 5 μg ml−1 insulin, 5 μg ml−1 transferrin, 5 ng ml−1 sodium selenite (Boehringer, Mannheim, Germany), 10 nm progesterone, 2 mm MgCl2, 25,000 IU l−1 penicillin, and 25 mg l−1 streptomycin (Sigma, Vienna, Austria). Approximately 300,000 cells were seeded into 35 mm culture dishes (Nunc; Roskilde, Denmark) coated with poly-d-lysine (Sigma; 1 mg ml−1). After 3–5 days, 1 μm cytosine arabinoside was added to the culture medium to reduce the proliferation of non-neural cells.
Rat phaeochromocytoma PC12 cells (Greene & Tischler, 1976) were grown on collagen-coated (Trevigen, Gaithersburg, MD, USA) culture dishes (Nunc, Roskilde, Denmark) in Optimem I growth medium (Invitrogen) containing 2 mm l-glutamine (HyClone, Aalst, Belgium), 10% heat inactivated horse serum and 5% tetracycline free fetal calf serum (both Invitrogen). Cells were split twice a week.
As P2Y1 receptors expressed in PC12 cells are tonically activated by endogenously released nucleotides (Moskvina et al. 2003), an inducible expression system was used in this study. Accordingly, the rat P2Y1 receptor DNA sequence (Moskvina et al. 2003) was subcloned into a pcDNA4/TO vector (Invitrogen). The construct was inserted between the BamHI and the EcoRI site of the target vector. Upstream of the start codon, a Kozak sequence was added using the forward primer CGGATCCACCATGACCGAGGTGCCTTG and the reverse primer CGAATTCTCACAAACTTGTGTCTCCGTTC to ensure proper expression in mammalian systems. The sequence of the resulting construct was verified by DNA sequencing.
For the generation of a tetracycline inducible stable cell line, the TREX system (Invitrogen) was used. Briefly, cells were transfected using ExGen500 (Fermentas, St Leon-Rot, Germany) with the pcDNA6/TR vector containing a blasticidin resistance and the pcDNA4/TO vector containing a Zeocin resistance and the rat P2Y1 receptor sequence as described above. Clones containing both vectors were selected by the addition of blasticidin (10 μg ml−1) and Zeocin (200 μg ml−1) to the culture medium. The medium was exchanged every 48 h until distinct islands of surviving cells were visible.
Electrophysiology
For electrophysiological recordings, PC12 cells were seeded at low density onto 3.5 cm dishes coated with rat tail collagen (Vartian & Boehm, 2001). For differentiation into a neuron-like phenotype, nerve growth factor (NGF; 50 ng ml−1) was added to the culture medium and cells were grown for another 7–10 days; the medium was exchanged after 4 days. Primary cultures of hippocampal neurons were used 14–21 days after plating.
In order to determine Ca2+-dependent K+ currents, single PC12 cells or hippocampal neurons were voltage clamped using the perforated patch technique as described before (Moskvina et al. 2003). Briefly, patch pipettes were pulled (Flaming-Brown puller, Sutter Instrument Co., Novato, CA, USA) from borosilicate glass capillaries (Science Products, Frankfurt/Main, Germany) and front-filled with a solution consisting of (mm): K2SO4 (75), KCl (55), MgCl2 (8), and Hepes (10), adjusted to pH 7.3 with KOH. Then, the electrodes were back-filled with the same solution containing 240 μg ml−1 amphotericin B (in 0.8% DMSO) which yielded tip resistencies of 1–3 MΩ. PC12 cells were incubated in and continuously superfused with external solution containing (in mm) NaCl (140), KCl (6), CaCl2 (2), MgCl2 (2), Hepes (10), glucose (20), adjusted to pH 7.4 with NaOH. The Kv7 channel inhibitor Xe991 (3 μm) was added in order to prevent the contribution of these channels, which are known to be inhibited by P2Y1 receptors (Moskvina et al. 2003; Filippov et al. 2006).
Currents were recorded at room temperature (20–24°C) using an Axopatch 200B amplifier and the pCLAMP 10.2 software (Molecular Devices, Sunnyvale, CA, USA). Unless stated otherwise, cells were voltage clamped to a holding potential of −30 mV. Current traces were filtered using the built in 8-pole Bessel low pass filter of the amplifier (fc = 1–2 kHz) and subsequently digitized at 5–10 kHz using a Digidata 1320A (Molecular Devices). The liquid junction potential was calculated to be +8 mV and was not compensated for. Drugs were applied using a DAD-12 drug application device (Adams & List, Westbury, NY, USA), which permits complete solution exchange around the cells within 100 ms (Boehm, 1999).
Current amplitudes in response to the application of nucleotides were quantified using the Clampfit 10.2 software (Molecular Devices). For statistical evaluation, passive holding currents were subtracted and mean current amplitudes in a time window of 100 ms around the peak were measured to reduce the impact of noise. For quantification of the amount of inhibition by different K+ channel blockers, 100 μm ADP was applied to each cell for periods of 10 s three times to evoke three consecutive currents (I1, I2, I3), the three application periods being separated by 10 min washout periods. For I1 and I2, ADP was applied alone (control), whereas before and during I3 specific blockers were present. The percentage of inhibition was then calculated as 100 – 100 × ((I3/I2)/(I2/I1)) to account for a potential rundown of ADP induced currents.
Voltage-activated Ca2+ currents were recorded in the whole-cell configuration as described before (Vartian & Boehm, 2001). In these experiments, the pipette solution consisted of (mm): CsCl (130), tetraethylammonium chloride (20), CaCl2 (0.24), glucose (10), Hepes (10), EGTA (5), Mg-ATP (2), and Li-GTP (2), adjusted to pH 7.3 with CsOH, to yield tip resistances of 2–3 MΩ. The external bathing solution contained (mm): NaCl (120), tetraethylammonium chloride (20), KCl (3), MgCl2 (2), CaCl2 (5), glucose (20), Hepes (10), adjusted to pH 7.3 with NaOH. This combination of solutions results in small liquid junction potentials of about +2 mV which, however, were neglected.
Whole-cell Ca2+ currents were elicited by 30 ms depolarizations from a holding potential of −80 mV to 0 mV at a frequency of 4 min−1. Leakage currents were corrected for by an on-line leak subtraction protocol which applies four hyperpolarizing pulses prior to the depolarization to 0 mV in order to determine the extent of leakage. These currents were quantified by measuring peak current amplitudes. To account for time-dependent changes, drug effects were evaluated by evoking currents in the presence of test drugs (B) and by comparing them to control currents recorded before (A) and after (washout, C) the application of the drugs, according to the equation: 100 − (200B/(A + C)) =% inhibition of Ca2+ currents (Vartian & Boehm, 2001). In order to generate concentration–response curves for the inhibition of Ca2+ currents by ADP in different cells, all values of inhibition observed in the presence of different ADP concentrations were normalized to the value obtained with 100 μm of the nucleotide in the very same cell (normalized ICa inhibition).
Determination of intracellular Ca2+
Measurements of intracellular Ca2+ concentrations were performed as described before (Boehm et al. 1997). Briefly, primary rat hippocampal or PC12 cell cultures on glass coverslips coated with rat tail collagen were incubated in culture medium containing 2% bovine serum albumin (instead of serum) and 5 mm fura-2 AM for 30 min at 36°C in 5% CO2. Thereafter, the coverslips were transferred to a coverslip chamber (Adams & List), which was placed on an inverted microscope (Nikon Eclipse TE200). The cultures were washed with and incubated in the same buffer as used for the recording of K+ currents (see above). Drugs were applied again via a DAD-12 device. Changes in intracellular Ca2+ were determined in single cells by the two-wavelength method (Grynkiewicz et al. 1985) with excitation at 340 and 380 nm, and emission at 500 nm, where increases in the ratio of the fluorescence signals obtained with excitation at 340 and 380 nm (F340/F380), respectively, reflect rises in the Ca2+ concentration.
Statistics
All values are expressed as arithmetic means ± standard error of the mean; n values represent numbers of individual cells in electrophysiological measurements and Ca2+ imaging experiments. Unless indicated otherwise, differences between groups were evaluated using the Kruskal–Wallis non-parametric ANOVA followed by a Dunn's post hoc test; P values < 0.05 were considered as indicating statistical significance.
Materials
Bulk chemicals, amphotericin B, dimethyl sulfoxide (DMSO), adenosine diphosphate (ADP), adenosine-3′-phosphate-5′-phosphate (A3P5P), 2′-deoxy-N6-methyl adenosine 3′,5′-diphosphate diammonium salt (MRS 2179), thapsigargin, U73122 (1-[6-[((17β)-3-methoxyestra-1,3,5[10]-trien-17-yl)amino]hexyl]-1H-pyrrole-2,5-dione), U73343 (1-[6-[((17β)-3-methoxyestra-1,3,5[10]-trien-17-yl)amino]hexyl]-2, 5-pyrrolidinedione), and bicuculline methiodide (BMI) were obtained from Sigma (Vienna, Austria); apamin, Xe991 (10,10-bis(4-pyridinylmethyl)-9(10H)-anthracenone dihydrochloride), and 1-[(2-chlorophenyl)diphenylmethyl]-1H-pyrazole (TRAM 34) were purchased from Tocris (Bristol, UK); iberiotoxin (IBTX) and tetrodotoxin (TTX) were bought from Latoxan (Rosans, France). Fura-2 AM was obtained from Molecular Probes (via Invitrogen; Lofer, Austria). Primers were purchased from VBC Biotech (Vienna, Austria); MRS2216 (2-chloro-N6-methyldeoxyadensoine 3′,5′-biphosphate) was a kind gift from Dr K. A. Jacobson (Bethesda, MD, USA); ExGen500 in vitro transfection reagent, pfu polymerase, BamHI and EcoRI restriction enzymes were purchased from Fermentas (St-Leon Rot, Germany); blasticidin and Zeocin were obtained from InvivoGen (Toulouse, France); tetracycline was bought from Boehringer Mannheim (Mannheim, Germany).
Results
ADP induces transient outward currents in PC12 cells stably expressing P2Y1 receptors
During experiments focusing on the inhibition of KV7 channels via recombinant P2Y1 receptors expressed in PC12 cells (Moskvina et al. 2003), the occurrence of transient outward currents had been observed at membrane potentials of −30 mV (not shown). Similar effects occur when PC12 cells are exposed to bradykinin (Villarroel et al. 1989). Therefore, NGF-differentiated PC12 cells were clamped at that holding potential, and increasing concentrations of ADP were applied (Fig. 1). In cells stably expressing rat P2Y1 receptors under the control of a tetracycline-sensitive promoter and exposed to 1 mg ml−1 tetracycline for 24 h, ADP consistently triggered outward currents in a concentration-dependent manner (Fig. 1); such currents were found in several different clones of P2Y1 expressing cells, but never in PC12 cells that had not been transfected with plasmids coding for P2Y1 receptors or in non-differentiated cells (not shown). The peak amplitudes of the currents evoked by ADP in P2Y1 expressing cells increased in a concentration-dependent manner (Fig. 1A and B). The amplitudes were half-maximal at an ADP concentration of 4.0 ± 0.6 μm and reached a maximum of 237.5 ± 10.2 pA (Fig. 1B). Between the start of the ADP application and the resulting peak amplitude, there were short delays in the range of 5–10 s, and these delays decreased in a concentration-dependent manner (Fig. 1C).
Figure 1. Induction of outward currents by ADP in PC12 cells stably expressing rat P2Y1 receptors.
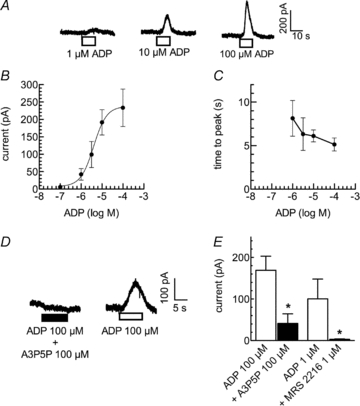
Cells were voltage clamped to −30 mV, and ADP was present for 10 s as indicated by the bars in A and D with washout periods of 10 min between the applications. A, original current traces. B, concentration–response curve for the peak amplitudes of outward currents induced by ADP (n= 16). The continuous line represents the least square fit of the data to a Hill equation with the following resulting parameters: log EC50−5.40 ± 0.06, nH 1.40 ± 0.24. C, concentration–response curve for the delay of peak outward currents induced by ADP (n= 16). D, ADP was first applied together with the specific P2Y1 antagonists A3P5P and then alone; original current traces are shown. E, ADP was first applied together with the specific P2Y1 antagonists A3P5P or MRS 2216 and then alone; peak amplitudes in either the presence or absence of A3P5P or MRS 2216 are depicted. *Significant difference at P < 0.05 (n= 5).
The ADP-evoked currents are mediated by P2Y1 receptors
PC12 cells express all known P2Y receptors but P2Y1 (Unterberger et al. 2002). Moreover, the inhibition of noradrenaline release from and the reduction of voltage-gated Ca2+ currents in PC12 cells are both mediated by P2Y12 receptors (Lechner et al. 2004). Thus, the effects of ADP as described above might involve endogenous receptors, but not the recombinant P2Y1 receptor. Therefore, ADP (100 μm) was applied in the presence and absence of the specific P2Y1 antagonists A3P5P (Boyer et al. 1996) and MRS 2216 (Nandanan et al. 1999). Currents in the presence of the antagonists were determined first, and the currents in their absence thereafter following a 10 min washout period; thus, the actions of these antagonists were reversible. Current amplitudes in the presence of A3P5P (100 μm) were >80% smaller than those in its absence (Fig. 1D and E), and MRS 2216 (1 μm) entirely abolished the currents triggered by 1 μm ADP (Fig. 1E). Together, these data confirm that ADP did evoke the K+ currents by an activation of the recombinant P2Y1 receptors.
Outward currents elicited by ADP are carried by K+ ions
To elucidate the ionic nature of the currents evoked by ADP, current to voltage relations were investigated. First, 100 μm ADP was applied to cells tonically clamped to −60, −90 or −120 mV. In these experiments, the reversal potential for ADP-induced currents was found to be close to −90 mV (Fig. 2A). Under quasi physiological conditions, only K+ and Cl− have their reversal potentials in the negative voltage range. Therefore, we either raised the external K+ concentration (by replacing NaCl with KCl) from 6 mm to 100 mm or reduced the external Cl− concentration (by replacing NaCl with sodium gluconate) from 154 to 84 mm. As shown in Fig. 2B, only the change in the K+, but not that in the Cl−, concentration caused a conspicuous change in ADP-evoked currents and even reversed its direction. This indicated that the ADP-evoked currents were carried by K+ ions.
Figure 2. Reversal potential and current–voltage relation of ADP induced currents.
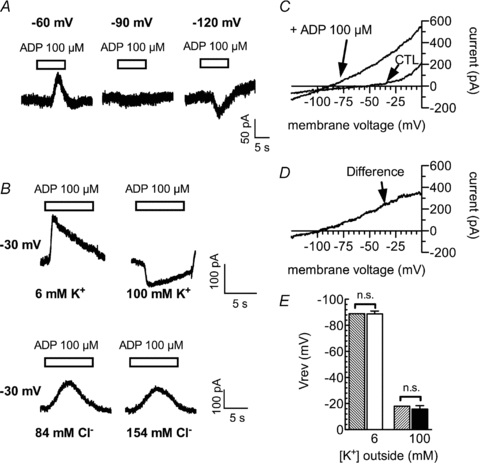
A, sample currents in a cell clamped to −60 mV, −90 mV or −120 mV with 6 mm extracellular K+. ADP was applied as indicated by the bars. B, sample currents from cells clamped to −30 mV. Upper panel, currents were recorded from one cell in either 6 mm (left) or 100 mm (right) extracellular K+. Lower panel, currents were recorded from another cell in either 84 mm (left) or 154 mm (right) extracellular Cl−. C, the cell was subjected to rapid (1 mV ms−1) ramp depolarizations from −120 mV to 0 mV in the absence (CTL) or presence of ADP. D, the difference between these two currents. E, the reversal potentials (Vrev) of ADP induced currents in either 6 mm (open bar; n= 6) or 100 mm (filled bar; n= 4) extracellular K+ as well as the calculated K+ equilibrium potentials for these two concentrations (hatched bars). n.s. indicates no significant differences between the experimental results and the calculated values (P > 0.05; Wilcoxon's signed-rank test).
To be able to determine the reversal potential of the ADP-induced currents more precisely, fast ramp depolarizations from −120 mV to 0 mV with a rate of 1 mV ms−1 were applied, while currents were induced by 100 μm ADP. To calculate the voltage dependence of the net currents evoked by ADP, the ramps were also applied in the absence of ADP (Fig. 2C). The subtraction of the currents in the absence of ADP from those in the presence of ADP (difference current in Fig. 2D) revealed linear current–voltage relations and a reversal potential for the ADP-evoked currents of −88.8 ± 2.2 mV (n= 6) under control conditions (i.e. 6 mm K+ outside of and 205 mm K+ inside the PC12 cells). This was not different from the calculated K+ equilibrium of −89 mV. When the external K+ concentration was then raised again from 6 mm to 100 mm, the reversal potential of the ADP induced current shifted to −15.8 ± 2.7 mV (n= 4). Again, this was not different from the calculated equilibrium potential for K+ which is −18 mV under these conditions (Fig. 2E). This confirms that the ADP-evoked currents are K+ currents.
The ADP-evoked currents rely on an activation of phospholipase C
Ca2+-dependent K+ currents in neurons are most commonly activated by transmembrane Ca2+ entry via either voltage-activated Ca2+ channels or ligand-gated ion channels (Pedarzani & Stocker, 2008). In PC12 cells, Ca2+ entry via the ionophores of P2X receptors has been reported to gate Ca2+-dependent K+ channels (Fujii et al. 1999), and high ADP concentrations might activate P2X receptors. To prove that the actions of ADP are mediated via a G protein-dependent signalling cascade and not by ionotropic receptors, PC12 cells were treated for 3 min with 3 μm of the phospholipase C inhibitor U73122. For comparison with a negative control, the cells were first exposed for the same period of time to the inactive analogue U73343. This latter treatment did not significantly influence the amplitudes of ADP-evoked currents. The subsequent application of U73122, however, reduced the current amplitudes by >90% (Fig. 3A and B). Thus, the ADP-evoked currents require an activation of phospholipase C.
Figure 3. Roles of phospholipase C and intracellular Ca2+ stores in ADP-induced currents.
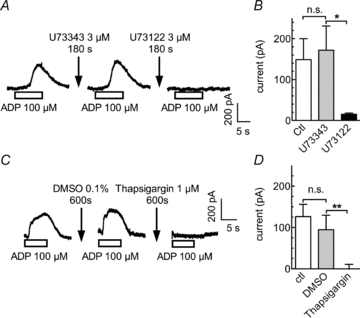
A and B, after an initial application of ADP, cells were treated for 420 s with control solution followed by a 180 s application of U73343 (3 μm) and a second ADP application. This was followed by a 420 s washout, a 180 s treatment with U73122 (3 μm), and a third ADP application. A, sample currents recorded from a single PC12 cell. B, peak current amplitudes in the absence or presence of either U73343 or U73122 (n= 4); *significant difference at P < 0.05. C and D, cells were continuously superfused with control solution (ctl), DMSO (0.1%) or 1 μm thapsigargin, each for 10 min. Outward currents were elicited by 10 s applications of ADP at the end of these treatments. Original current traces (A) and peak amplitudes of the currents (B) evoked by ADP under control conditions (ctl) and in the presence of either 0.1% DMSO or 1 μm thapsigargin (**P < 0.01; n.s. no significant difference; n= 5).
Depletion of intracellular Ca2+ stores prevents the activation of outward currents by ADP
One of the consequences of phospholipase C activation is the liberation of Ca2+ from intracellular stores via inositol trisphosphate (Berridge, 2009). Therefore, experiments were designed to directly demonstrate the involvement of such a mechanism. In PC12 cells, the inositol trisphosphate-sensitive intracellular Ca2+ stores can be depleted of Ca2+ by the Ca2+-ATPase inhibitor thapsigargin (Fasolato et al. 1991). Accordingly, in PC12 cells displaying unequivocal current responses to 100 μm ADP, whether in the absence or presence of solvent (0.1% DMSO), the nucleotide failed to cause any change in holding current after exposure of the cells to 1 μm thapsigargin (Fig. 3C and D). Thus, inositol trisphosphate-sensitive intracellular Ca2+ stores are involved in the activation of currents via P2Y1 receptors.
ADP raises intracellular Ca2+ concentrations in PC12 cells expressing P2Y1 receptors
The above results indicate that the activation of the recombinant P2Y1 receptors may lead to increases in cytosolic Ca2+ concentrations. To directly demonstrate such an effect, Ca2+ responses to increasing concentrations of ADP were tested in untransfected PC12 cells as well as in PC12 cells expressing P2Y1 receptors, both loaded with the Ca2+ indicator fura-2. As reported before (Arslan et al. 2000), native PC12 cells did not show any changes in intracellular Ca2+ when exposed to ADP. Nevertheless, these cells clearly displayed Ca2+ rises when challenged by depolarizing K+ concentrations (Fig. 4A). However, in P2Y1 receptor expressing cells, ADP raised intracellular Ca2+ in a concentration-dependent manner (Fig. 4B and C) with half-maximal effects at 18.2 ± 3.4 μm. Hence, activation of the recombinant P2Y1 receptors leads to increases in intracellular Ca2+ concentrations.
Figure 4. ADP-induced rises in intracellular Ca2+ and activation of KCa2 channels.
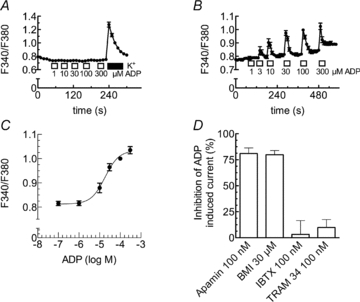
A–C, either untransfected (A) or PC12 cells stably expressing rP2Y1 receptors (B and C) were loaded with fura-2 AM and continuously superfused; ADP was applied as indicated by the bars. Changes in intracellular Ca2+ were evaluated by the changes in the ratio of fluorescence at 340 nm and 380 nm (F340/F380). A, changes in the ratio of fluorescence in untransfected PC12 cells due to the application of either the indicated ADP concentrations or 100 mm K+. B, changes in the ratio of fluorescence in PC12 cells stably expressing rP2Y1 receptors due to the application of the indicated ADP concentrations (n= 42). C, concentration–response curve for the maximal increase of intracellular Ca2+ caused by the indicated concentrations of ADP. Data points were fitted to a Hill equation using a non-linear least square algorithm; the fitted parameters are: log EC50=−4.74 ± 0.09, nH= 1.29 ± 0.38 (n= 42). D, PC12 cells stably expressing rP2Y1 receptors were voltage clamped to −30 mV, and currents evoked by ADP (100 μm) three times (I1, I2, I3) at 10 min intervals either under control conditions or in the presence of apamin (100 nm, n= 8), biccuculine methiodide (BMI, 30 μm, n= 8), iberiotoxin (IBTX, 100 nm, n= 10), or TRAM 34 (100 nm, n= 6). After the first 25 min, cells were exposed for 5 min to one of these channel blockers and ADP was applied again in the presence of these blockers. To assess the block of ADP induced currents by these drugs, the percentage of inhibition was calculated as 100 – 100 × ((I3/I2)/(I2/(I1)).
ADP-evoked currents are carried by KCa2 channels
The results shown above strongly suggest that the ADP-evoked currents are mediated by Ca2+-dependent K+ channels. There are several ion channel proteins that mediate Ca2+-dependent K+ currents in neurons (Sah & Faber, 2002) which can be discriminated by specific blockers: the scorpion toxin iberiotoxin (IBTX) is highly specific for KCa1.1 channels, the bee venom toxin apamin and biccuculin methiodide (BMI) for KCa2 channels, and TRAM 34 for KCa3 channels (Wei et al. 2005). To elucidate which KCa channels may underlie the currents evoked by ADP, 100 μm of the nucleotide was applied three times for periods of 10 s to each cell (see Methods) in either the absence or the presence of the aforementioned blockers. Apamin (0.1 μm) and BMI (30 μm) inhibited the ADP-induced currents by 81.0 ± 5.4% and 79.9 ± 4.0%, respectively. In contrast, neither 0.1 μm IBTX nor 0.1 μm TRAM 34 caused unequivocal reductions in current amplitudes (3.2 ± 13.3% and 9.8 ± 7.7% inhibition; Fig. 4D). These results indicate that the ADP-evoked currents were mediated by members of the KCa2 channel family.
P2Y1 receptors do not contribute to the inhibition of voltage activated Ca2+ currents by ADP
In PC12 cells, endogenous P2Y12 receptors mediate the inhibition of voltage-gated Ca2+ channels by ADP (Kubista et al. 2003). In sympathetic neurons, both recombinant P2Y1 (Brown et al. 2000) and P2Y12 (Simon et al. 2002) receptors mediate an inhibition of voltage-gated Ca2+ channels. Hence, it appeared obvious to investigate whether P2Y1 receptors heterologously expressed in PC12 cells might contribute to the inhibition of voltage-activated Ca2+ currents by ADP. As described in detail before (Vartian & Boehm, 2001), ADP did not only reduce the amplitudes of Ca2+ currents evoked by 30 ms depolarizations from −80 to 0 mV, but also slowed the activation kinetics (Fig. 5). This indicates a direct inhibition of the channels via G protein βγ subunits (Vartian & Boehm, 2001). This effect appeared to be identical whether the PC12 cells under investigation expressed P2Y1 receptors (Fig. 5B) or did not (Fig. 5A). This conclusion is further confirmed by two additional findings: (i) concentration–response curves for the inhibition of Ca2+ current amplitudes were superimposable whether obtained in P2Y1 receptor expressing or non-transfected PC12 cells (Fig. 5C); (ii) 100 μm ADP reduced Ca2+ current amplitudes to the very same extent in P2Y1 expressing and non-expressing cells (Fig. 5D). Taken together, these results show that the heterologous expression of P2Y1 receptors in PC12 cells does not alter the inhibition of voltage-gated Ca2+ channels via endogenous P2Y12 receptors (Kubista et al. 2003) and suggest that the recombinant P2Y1 receptors are not involved in that effect.
Figure 5. Roles of P2Y1 receptors in the inhibition of Ca2+ currents by ADP.
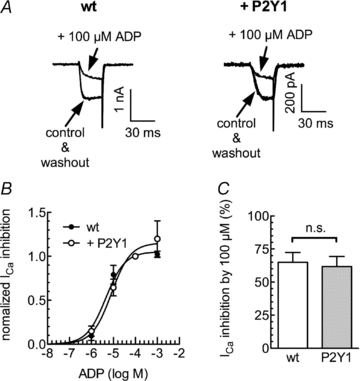
Either untransfected PC12 cells or PC12 cells stably expressing P2Y1 receptors were voltage clamped to −80 mV and depolarized to 0 mV for 30 ms once every 15 s in the presence of TEA (20 mm) and tetrodotoxin (500 nm). A, original current traces in wild-type (wt; left) or P2Y1 expressing (right) cells obtained before (control), during and after (washout) the application of ADP (100 μm). B, concentration–response curves for the inhibition of peak Ca2+ currents (ICa) by the indicated concentrations of ADP. Data points were fitted to a Hill equation yielding the following values for half maximal inhibition: log EC50: 5.34 ± 0.06 for wt cells of −5.09 ± 0.05 for P2Y1 expressing PC12 cells (R2= 0.99 each). C, inhibition of peak Ca2+ currents (ICa) by 100 μm ADP in wild type (wt) or P2Y1 expressing PC12 cells. n.s. indicates no significant difference, P > 0.1 (Mann–Whitney U test; n= 4–6).
ADP induces outward currents via KCa channels in hippocampal neurons
To confirm the above results in a true neuronal environment and with endogenously expressed P2Y receptors, we employed primary cultures of rat hippocampal neurons. There, ADP has also been found to reduce currents through KV7 channels via endogenous P2Y1 receptors (Filippov et al. 2006). At a holding potential of −30 mV, ADP (10–100 μm) induced outward currents (Fig. 6A) that were attenuated by 30 μm BMI by about 50% (Fig. 6B); the unspecific K+ channel blocker tetraethylammonium (1 mm) further reduced the currents that remained in the presence of BMI. Moreover, ADP-induced currents were largely reduced by 30 μm of the specific P2Y1 antagonist MRS 2179 (von Kugelgen, 2006) (Fig. 6C) and by the depletion of intracellular Ca2+ stores by thapsigargin (Fig. 6D). To directly demonstrate that ADP can induce rises in intracellular Ca2+, hippocampal cultures were also loaded with fura-2 and exposed to the nucleotide, which clearly resulted in a positive signal. However, the ADP-induced increase in the fluorescence ratio was much smaller than that in PC12 cells and also much smaller than the rise triggered by 100 mm K+ (Fig. 6E).
Figure 6. Effects of ADP in hippocampal neurons in primary cell culture.
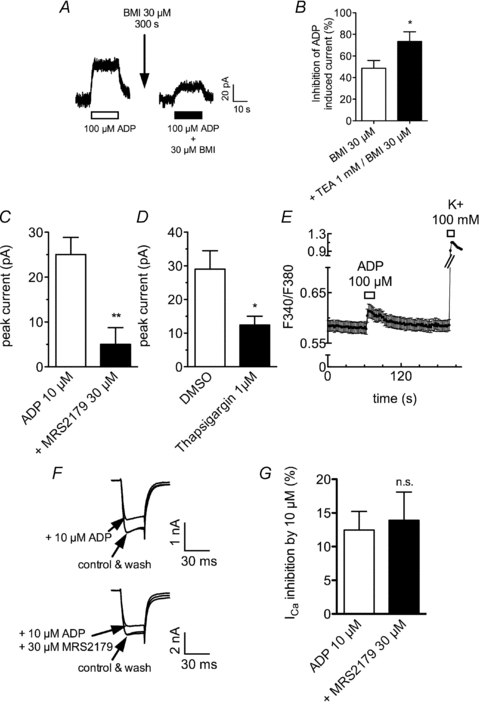
Hippocampal neurons in primary cell culture were voltage clamped to either −30 mV using the perforated patch clamp technique (A–D) or to −80 mV using the whole cell patch clamp technique (F and G); alternatively, the neurons were loaded with fura-2 AM (E). A, sample recording of currents evoked by ADP at −30 mV; ADP was applied once every 600 s either alone (left panel) or after a 300 s pretreatment with BMI (30 μm; right panel). B, ADP was applied alone, together with BMI, or together with BMI and TEA; the inhibition of the amplitudes of ADP induced outward current is shown (*P < 0.05 vs. BMI alone, n= 8–9, Mann–Whitney U test). C, ADP (10 μm) was applied either alone or together with the P2Y1 antagonist MRS2179 (30 μm); the amplitudes of ADP induced currents are shown (**P < 0.01, n= 6, Mann–Whitney U test). D, ADP was applied either before or after the depletion of intracellular Ca2+ stores with 1 μm thapsigargin for 30 min; the amplitudes of ADP induced outward currents are shown (*P < 0.05, n= 7–9, Mann–Whitney U test). E, fura-2-loaded hippocampal neurons were sequentially exposed to ADP and K+ for the periods of time indicated by the bars; the changes in the ratio of fluorescence at 340 nm and 380 nm (F340/F380) are shown (n= 40). F, original traces of Ca2+ currents evoked by 30 ms depolarizations from −80 mV to 0 mV once every 15 s in the presence of TEA (20 mm) and tetrodotoxin (500 nm); ADP was applied alone (upper panel) or together with the P2Y1 antagonist MRS 2179 (30 μm). F, inhibition of Ca2+ current amplitudes by ADP in the absence or presence of MRS 2179; n.s. indicates no significant difference, P > 0.05 (n= 6, Mann–Whitney U test).
For further comparison with the results obtained with the recombinant P2Y1 receptor in PC12 cells, voltage-activated Ca2+ currents were also determined in hippocampal neurons. ADP (10 μm) reduced Ca2+ current amplitudes by more than 10%, but this effect was not altered by MRS 2179 (Fig. 6E and F), thus indicating that it was not mediated by P2Y1 receptors.
Discussion
Ca2+-dependent K+ channels are important elements in the control of neuronal functions as they are involved in action potential repolarization and afterhyperpolarization and thereby in the fine tuning of the varying firing patterns in different types of neurons (Sah & Faber, 2002). In particular, the coordinated gating of KCa2 channels is highly important for spike timing precision (Stocker, 2004) and this gating has been found to be controlled by G protein-coupled receptors, such as bradykinin (Villarroel et al. 1989), metabotropic glutamate (Shirasaki et al. 1994; Fiorillo & Williams, 1998), muscarinic acetylcholine (Wakamori et al. 1993; Fiorillo & Williams, 2000), and adenosine A1 (Clark et al. 2009) receptors. The present results add P2Y1 receptors to the list of G protein-coupled receptors that mediate an activation of KCa2 channels and thereby broaden the signalling repertoire of neuronal P2Y receptors (Hussl & Boehm, 2006).
Activation of P2Y1 receptors heterologously expressed in PC12 cells leads to an accumulation of inositol phosphates and to an inhibition of KV7 channels (Moskvina et al. 2003). The present results demonstrate that ADP acts via these recombinant P2Y1 receptors to trigger currents that are carried by K+ ions. Considering that the activation of the P2Y1 receptors in PC12 cells generates inositol phosphates (Moskvina et al. 2003), it appeared obvious to test for the role of phospholipase C. Indeed, blockade of phospholipase C prevented the activation of currents by ADP. Moreover, ADP was found to raise intracellular Ca2+ in a concentration-dependent manner via the recombinant P2Y1 receptors, and this is a response typically mediated by inositol trisphosphate. In line with this conclusion, the depletion of the inositol trisphosphate-sensitive Ca2+ stores of PC12 cells by thapsigargin (Fasolato et al. 1991) abolished ADP-evoked currents. Thus, the signalling cascade mediating the activation of K+ currents by ADP involved phospholipase C, inositol trisphosphate, and the release of Ca2+ from appropriate intracellular stores. In this context, it should be noted that ADP concentrations required to trigger outward currents were about fourfold lower than those required to elevate the intracellular Ca2+ concentration (compare Fig. 1B and 4C). This apparent discrepancy is most likely due to the fact that our measurements of intracellular Ca2+ provide averages for the entire cytosol, whereas the signalling between intracellular Ca2+ stores and transmembrane ion channels relies on Ca2+ concentrations within microdomains underneath the membrane (Delmas & Brown, 2002). Alternatively, this discrepancy can be explained by the high cooperativity of Ca2+ ions in the gating of KCa channels (Xia et al. 1998).
PC12 cells express endogenous P2Y2, P2Y4, P2Y6 and P2Y12 receptors (Unterberger et al. 2002). To verify that the current responses to ADP were indeed mediated by the recombinant P2Y1 receptors, ADP was applied in the presence of either A3P5P (Boyer et al. 1996) or MRS 2216 (Nandanan et al. 1999), both of which are antagonists with a selectivity for P2Y1. As expected, the two P2Y1 antagonists largely attenuated or even abolished the current responses towards ADP. Together, the present results indicate that activation of P2Y1 receptors leads to the generation of inositol trisphosphate via phospholipase C, to the release of Ca2+ from inositol trisphosphate-sensitive stores and to the subsequent activation of a K+ current. Hence, it was straightforward to assume that these currents were mediated by Ca2+-dependent K+ channels. This was confirmed by the finding that the ADP-induced currents were largely reduced by blockers of KCa2 channels. As these currents were not altered by blockers of KCa1.1 or KCa3.1 channels, one may conclude that these currents were mediated by KCa2 channels. This conclusion is further supported by the fact that the ADP-evoked currents displayed a linear current–voltage relation: KCa2 channels are gated by intracellular Ca2+ independently of the membrane voltage (Stocker, 2004), whereas KCa1.1 channels are gated by intracellular Ca2+ and voltage, with increasing Ca2+ concentrations shifting the current–voltage curve to the left (Cox et al. 1997). Moreover, the channels gated by ADP were open at voltages more negative than −80 mV, but even with high intracellular Ca2+ concentrations KCa1.1 channels rather remain closed at such negative membrane potentials (Cox et al. 1997) even though this depends on the co-expressed β subunit (Brenner et al. 2000). Thus, these biophysical characteristics of the ADP-evoked currents confirm a major contribution of KCa2 channels.
In line with this conclusion, PC12 cells have been reported to express high levels of apamin-sensitive Ca2+-dependent K+ channels (Schmid-Antomarchi et al. 1986), but no KCa1.1 channels (Terstappen, 1999). Furthermore, bradykinin activates a Ca2+-dependent K+ current in PC12 cells that is blocked by tubocurarin, a preferential blocker of channels of the KCa2 family (Villarroel et al. 1989). In contrast to these results, ATP was found to activate a K+ current in PC12 cells through an activation of ionotropic P2X receptors and transmembrane Ca2+ entry, and this current was blocked by 100 nm charybdotoxin or iberiotoxin, but not by 200 nm apamin (Fujii et al. 1999). Thus, KCa1.1 channels may be present in PC12 cells to varying degrees depending on the clonal line under investigation.
The fact that activation of P2X receptors has been reported to gate Ca2+-dependent K+ channels leads to the question as to whether similar effects have also been observed for P2Y receptors. Indeed, in pigment epithelial cells of the rat retina ATP, ADP, UTP and 2-methylthio-ATP were shown to raise intracellular Ca2+ and to induce K+ currents that were blocked by iberiotoxin (Ryan et al. 1999). However, the receptor subtypes involved remained unidentified. Similarly, in Vero cells ATP, ADP and UTP were reported to raise intracellular Ca2+ and to induce currents through large conductance K+ channels, but the receptor subtype as well as the channels involved remained elusive (Hafting & Sand, 2000). Later on, on the basis of the antagonism by suramin and PPADS, the P2Y receptor subtype involved was suggested to be P2Y1, but more selective ligands had not been tested (Hafting et al. 2006). In addition, recombinant human P2Y4 receptors were found to mediate an enhancement of currents through human KCa1.1 as well as KCa3.1 channels. The P2Y2 counterparts, in contrast, mediated an inhibition of KCa1.1, but an activation of KCa3.1 channels (Hede et al. 2005). Thus, P2Y receptors have been demonstrated to control the gating of KCa1.1 and KCa3.1 channels, which are mostly expressed in non-neural tissues. The present results, in contrast, provide the first evidence that P2Y receptors, in particular P2Y1 which is expressed in various types of neurons, mediate an activation of neuronal KCa2 channels.
Before, recombinant P2Y1 receptors had been found to mediate an inhibition of KV7 channels (Brown et al. 2000), activation and inhibition of Kir3.1/3.2 channels (Filippov et al. 2004) and an inhibition of CaV2 channels (Filippov et al. 2000) as observed in rat sympathetic neurons. This multitude of signalling mechanisms described for one receptor type raises concerns about potentially aberrant signalling due to receptor overexpression. The following of the above results strongly suggest that the activation of Ca2+-dependent K+ channels via P2Y1 is not due to artificially high levels of receptor expression: (i) in PC12 cells, the recombinant P2Y1 receptors mediated both the activation of KCa channels and an inhibition of KV7 channels (Moskvina et al. 2003), but left voltage-activated Ca2+ currents unaltered; (ii) in primary hippocampal neurons, endogenous P2Y1 receptors also mediated the activation of KCa channels and an inhibition of KV7 channels (Filippov et al. 2006), but failed to affect voltage-gated Ca2+ channels; (iii) the inhibition of KV7 channels (Moskvina et al. 2003) and the induction of Ca2+-dependent K+ currents (present results), both via recombinant P2Y1 receptors expressed in PC12 cells, occurred at similar ADP concentrations with half-maximal effects at 2 and 4 μm, respectively. For comparison, the inhibition of KV7 channels via native P2Y1 receptors in hippocampal neurons was half-maximal at 0.08 μm (Filippov et al. 2006), and the agonist used in that case (ADPβS) is equipotent to ADP at P2Y1 receptors (von Kugelgen, 2006). Thus, nucleotides are about 50-fold less potent in regulating ion channels in PC12 cells via recombinant P2Y1 receptors than via endogenous P2Y1 receptors in hippocampal neurons. This together with the congruence of results obtained with native and recombinant P2Y1 receptors renders the likelihood of overexpression artefacts minimal. Nevertheless, it remains to be clarified why recombinant P2Y1 receptors mediate an inhibition of voltage-gated Ca2+ channels in rat sympathetic neurons (Filippov et al. 2000), but not in PC12 cells (see above).
Taken together, this report provides the first evidence that P2Y receptors, in particular P2Y1, may be linked to KCa2 channels in a neuronal environment. Thus, the group of neuronal P2Y receptors are as versatile G protein-coupled neurotransmitter receptors as, for instance, members of the family of muscarinic acetylcholine receptors, which also control KCa2 channels (Fiorillo & Williams, 2000) in addition to KV7, CaV2 (Shapiro et al. 1999) and Kir3 channels (Kofuji et al. 1995). Moreover, the activation of KCa2 channels may form the basis for the influence of P2Y1 receptor activation on synaptic plasticity, such as the inhibition of long-term depression (Guzman et al. 2005; Hopf et al. 2010).
Acknowledgments
This study was supported by grants P17611, P19710 and W1205 from the Austrian Science Funds (FWF). G.K.C. is member of the doctoral school ‘Cell Communication in Health and Disease, CCHD’ at the Medical University of Vienna. The perfect technical assistance of Gabi Gaupmann and Martina Molin is gratefully acknowledged.
Glossary
Abbreviations
- GPCR
G-protein coupled receptor
Author contributions
All experiments were performed at the Centre for Physiology and Pharmacology, Medical University of Vienna. The contributions of the authors were as follows: conception and design of the experiments: K.S., H.K. and S.B.; collection, analysis and interpretation of data: K.S., G.K.C., P.G., S.B.; drafting the article or revising it critically for important intellectual content: all authors.
References
- Abe M, Endoh T, Suzuki T. Extracellular ATP-induced calcium channel inhibition mediated by P1/P2Y purinoceptors in hamster submandibular ganglion neurons. Br J Pharmacol. 2003;138:1535–1543. doi: 10.1038/sj.bjp.0705174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arslan G, Filipeanu CM, Irenius E, Kull B, Clementi E, Allgaier C, Erlinge D, Fredholm BB. P2Y receptors contribute to ATP-induced increases in intracellular calcium in differentiated but not undifferentiated PC12 cells. Neuropharmacology. 2000;39:482–496. doi: 10.1016/s0028-3908(99)00141-0. [DOI] [PubMed] [Google Scholar]
- Berridge MJ. Inositol trisphosphate and calcium signalling mechanisms. Biochim Biophys Acta. 2009;1793:933–940. doi: 10.1016/j.bbamcr.2008.10.005. [DOI] [PubMed] [Google Scholar]
- Boehm S, Betz H. Somatostatin inhibits excitatory transmission at rat hippocampal synapses via presynaptic receptors. J Neurosci. 1997;17:4066–4075. doi: 10.1523/JNEUROSCI.17-11-04066.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehm S, Harvey RJ, von Holst A, Rohrer H, Betz H. Glycine receptors in cultured chick sympathetic neurons are excitatory and trigger neurotransmitter release. J Physiol. 1997;504:683–694. doi: 10.1111/j.1469-7793.1997.683bd.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehm S. Selective inhibition of M-type potassium channels in rat sympathetic neurons by uridine nucleotide preferring receptors. Br J Pharmacol. 1998;124:1261–1269. doi: 10.1038/sj.bjp.0701956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehm S. ATP stimulates sympathetic transmitter release via presynaptic P2X purinoceptors. J Neurosci. 1999;19:737–746. doi: 10.1523/JNEUROSCI.19-02-00737.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehm S. P2Ys go neuronal: modulation of Ca2+ and K+ channels by recombinant receptors. Br J Pharmacol. 2003;138:1–3. doi: 10.1038/sj.bjp.0705044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer JL, Romero-Avila T, Schachter JB, Harden TK. Identification of competitive antagonists of the P2Y1 receptor. Mol Pharmacol. 1996;50:1323–1329. [PubMed] [Google Scholar]
- Brenner R, Jegla TJ, Wickenden A, Liu Y, Aldrich RW. Cloning and functional characterization of novel large conductance calcium-activated potassium channel beta subunits, hKCNMB3 and hKCNMB4. J Biol Chem. 2000;275:6453–6461. doi: 10.1074/jbc.275.9.6453. [DOI] [PubMed] [Google Scholar]
- Brown DA, Filippov AK, Barnard EA. Inhibition of potassium and calcium currents in neurones by molecularly-defined P2Y receptors. J Auton Nerv Syst. 2000;81:31–36. doi: 10.1016/s0165-1838(00)00150-8. [DOI] [PubMed] [Google Scholar]
- Clark BD, Kurth-Nelson ZL, Newman EA. Adenosine-evoked hyperpolarization of retinal ganglion cells is mediated by G-protein-coupled inwardly rectifying K+ and small conductance Ca2+-activated K+ channel activation. J Neurosci. 2009;29:11237–11245. doi: 10.1523/JNEUROSCI.2836-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox DH, Cui J, Aldrich RW. Allosteric gating of a large conductance Ca-activated K+ channel. J Gen Physiol. 1997;110:257–281. doi: 10.1085/jgp.110.3.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmas P, Brown DA. Junctional signaling microdomains: bridging the gap between the neuronal cell surface and Ca2+ stores. Neuron. 2002;36:787–790. doi: 10.1016/s0896-6273(02)01097-8. [DOI] [PubMed] [Google Scholar]
- Drummond GB. Reporting ethical matters in The Journal of Physiology: standards and advice. J Physiol. 2009;587:713–719. doi: 10.1113/jphysiol.2008.167387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fasolato C, Zottini M, Clementi E, Zacchetti D, Meldolesi J, Pozzan T. Intracellular Ca2+ pools in PC12 cells. Three intracellular pools are distinguished by their turnover and mechanisms of Ca2+ accumulation, storage, and release. J Biol Chem. 1991;266:20159–20167. [PubMed] [Google Scholar]
- Filippov AK, Brown DA, Barnard EA. The P2Y1 receptor closes the N-type Ca2+ channel in neurones, with both adenosine triphosphates and diphosphates as potent agonists. Br J Pharmacol. 2000;129:1063–1066. doi: 10.1038/sj.bjp.0703185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippov AK, Choi RC, Simon J, Barnard EA, Brown DA. Activation of P2Y1 nucleotide receptors induces inhibition of the M-type K+ current in rat hippocampal pyramidal neurons. J Neurosci. 2006;26:9340–9348. doi: 10.1523/JNEUROSCI.2635-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippov AK, Fernandez-Fernandez JM, Marsh SJ, Simon J, Barnard EA, Brown DA. Activation and inhibition of neuronal G protein-gated inwardly rectifying K+ channels by P2Y nucleotide receptors. Mol Pharmacol. 2004;66:468–477. doi: 10.1124/mol.66.3.. [DOI] [PubMed] [Google Scholar]
- Filippov AK, Selyanko AA, Robbins J, Brown DA. Activation of nucleotide receptors inhibits M-type K current [IK(M)] in neuroblastoma x glioma hybrid cells. Pflugers Arch. 1994;429:223–230. doi: 10.1007/BF00374316. [DOI] [PubMed] [Google Scholar]
- Fiorillo CD, Williams JT. Glutamate mediates an inhibitory postsynaptic potential in dopamine neurons. Nature. 1998;394:78–82. doi: 10.1038/27919. [DOI] [PubMed] [Google Scholar]
- Fiorillo CD, Williams JT. Cholinergic inhibition of ventral midbrain dopamine neurons. J Neurosci. 2000;20:7855–7860. doi: 10.1523/JNEUROSCI.20-20-07855.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii F, Kimura J, Tase C. Ca2+-activated K+ current induced by external ATP in PC12 cells. Clin Exp Pharmacol Physiol. 1999;26:39–47. doi: 10.1046/j.1440-1681.1999.02980.x. [DOI] [PubMed] [Google Scholar]
- Gerevich Z, Borvendeg SJ, Schroder W, Franke H, Wirkner K, Norenberg W, Furst S, Gillen C, Illes P. Inhibition of N-type voltage-activated calcium channels in rat dorsal root ganglion neurons by P2Y receptors is a possible mechanism of ADP-induced analgesia. J Neurosci. 2004;24:797–807. doi: 10.1523/JNEUROSCI.4019-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene LA, Tischler AS. Establishment of a noradrenergic clonal line of rat adrenal pheochromocytoma cells which respond to nerve growth factor. Proc Natl Acad Sci U S A. 1976;73:2424–2428. doi: 10.1073/pnas.73.7.2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- Hafting T, Sand O. Purinergic activation of BK channels in clonal kidney cells (Vero cells) Acta Physiol Scand. 2000;170:99–109. doi: 10.1046/j.1365-201x.2000.00766.x. [DOI] [PubMed] [Google Scholar]
- Guzman SJ, Gerevich Z, Hengstler JG, Illes P, Kleemann W. P2Y1 receptors inhibit both strength and plasticity of glutamatergic synaptic neurotransmission in the rat prefrontal cortex. Synapse. 2005;57:235–238. doi: 10.1002/syn.20177. [DOI] [PubMed] [Google Scholar]
- Hafting T, Haug TM, Ellefsen S, Sand O. Hypotonic stress activates BK channels in clonal kidney cells via purinergic receptors, presumably of the P2Y subtype. Acta Physiol (Oxf) 2006;188:21–31. doi: 10.1111/j.1748-1716.2006.01601.x. [DOI] [PubMed] [Google Scholar]
- Hede SE, Amstrup J, Klaerke DA, Novak I. P2Y2 and P2Y4 receptors regulate pancreatic Ca2+-activated K+ channels differently. Pflugers Arch. 2005;450:429–436. doi: 10.1007/s00424-005-1433-3. [DOI] [PubMed] [Google Scholar]
- Hopf FW, Seif T, Mohamedi ML, Chen BT, Bonci A. The small-conductance calcium-activated potassium channel is a key modulator of firing and long-term depression in the dorsal striatum. Eur J Neurosci. 2010;31:1946–1959. doi: 10.1111/j.1460-9568.2010.07231.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussl S, Boehm S. Functions of neuronal P2Y receptors. Pflugers Arch. 2006;452:538–551. doi: 10.1007/s00424-006-0063-8. [DOI] [PubMed] [Google Scholar]
- Kofuji P, Davidson N, Lester HA. Evidence that neuronal G-protein-gated inwardly rectifying K+ channels are activated by Gβγ subunits and function as heteromultimers. Proc Natl Acad Sci U S A. 1995;92:6542–6546. doi: 10.1073/pnas.92.14.6542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubista H, Lechner SG, Wolf AM, Boehm S. Attenuation of the P2Y receptor-mediated control of neuronal Ca2+ channels in PC12 cells by antithrombotic drugs. Br J Pharmacol. 2003;138:343–350. doi: 10.1038/sj.bjp.0705037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lechner SG, Boehm S. Regulation of neuronal ion channels via P2Y receptors. Purinergic Signal. 2004;1:31–41. doi: 10.1007/s11302-004-4746-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lechner SG, Dorostkar MM, Mayer M, Edelbauer H, Pankevych H, Boehm S. Autoinhibition of transmitter release from PC12 cells and sympathetic neurons through a P2Y receptor-mediated inhibition of voltage-gated Ca2+ channels. Eur J Neurosci. 2004;20:2917–2928. doi: 10.1111/j.1460-9568.2004.03760.x. [DOI] [PubMed] [Google Scholar]
- Meng H, Sakakibara M, Nakazawa H, Tokimasa T. Pyridoxalphosphate-6-azophenyl-2′,4′-disulfonic acid can antagonize the purinoceptor-mediated inhibition of M-current in bullfrog sympathetic neurons. Neurosci Lett. 2003;337:93–96. doi: 10.1016/s0304-3940(02)01314-9. [DOI] [PubMed] [Google Scholar]
- Moskvina E, Unterberger U, Boehm S. Activity-dependent autocrine-paracrine activation of neuronal P2Y receptors. J Neurosci. 2003;23:7479–7488. doi: 10.1523/JNEUROSCI.23-20-07479.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nandanan E, Camaioni E, Jang SY, Kim YC, Cristalli G, Herdewijn P, Secrist JA, 3rd, Tiwari KN, Mohanram A, Harden TK, Boyer JL, Jacobson KA. Structure-activity relationships of bisphosphate nucleotide derivatives as P2Y1 receptor antagonists and partial agonists. J Med Chem. 1999;42:1625–1638. doi: 10.1021/jm980657j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedarzani P, Stocker M. Molecular and cellular basis of small- and intermediate-conductance, calcium-activated potassium channel function in the brain. Cell Mol Life Sci. 2008;65:3196–3217. doi: 10.1007/s00018-008-8216-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan JS, Baldridge WH, Kelly ME. Purinergic regulation of cation conductances and intracellular Ca2+ in cultured rat retinal pigment epithelial cells. J Physiol. 1999;520:745–759. doi: 10.1111/j.1469-7793.1999.00745.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sah P, Faber ES. Channels underlying neuronal calcium-activated potassium currents. Prog Neurobiol. 2002;66:345–353. doi: 10.1016/s0301-0082(02)00004-7. [DOI] [PubMed] [Google Scholar]
- Schmid-Antomarchi H, Hugues M, Lazdunski M. Properties of the apamin-sensitive Ca2+-activated K+ channel in PC12 pheochromocytoma cells which hyper-produce the apamin receptor. J Biol Chem. 1986;261:8633–8637. [PubMed] [Google Scholar]
- Shapiro MS, Loose MD, Hamilton SE, Nathanson NM, Gomeza J, Wess J, Hille B. Assignment of muscarinic receptor subtypes mediating G-protein modulation of Ca2+ channels by using knockout mice. Proc Natl Acad Sci U S A. 1999;96:10899–10904. doi: 10.1073/pnas.96.19.10899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirasaki T, Harata N, Akaike N. Metabotropic glutamate response in acutely dissociated hippocampal CA1 pyramidal neurones of the rat. J Physiol. 1994;475:439–453. doi: 10.1113/jphysiol.1994.sp020084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon J, Filippov AK, Goransson S, Wong YH, Frelin C, Michel AD, Brown DA, Barnard EA. Characterization and channel coupling of the P2Y12 nucleotide receptor of brain capillary endothelial cells. J Biol Chem. 2002;277:31390–31400. doi: 10.1074/jbc.M110714200. [DOI] [PubMed] [Google Scholar]
- Stocker M. Ca2+-activated K+ channels: molecular determinants and function of the SK family. Nat Rev Neurosci. 2004;5:758–770. doi: 10.1038/nrn1516. [DOI] [PubMed] [Google Scholar]
- Terstappen GC. Functional analysis of native and recombinant ion channels using a high-capacity nonradioactive rubidium efflux assay. Anal Biochem. 1999;272:149–155. doi: 10.1006/abio.1999.4179. [DOI] [PubMed] [Google Scholar]
- Unterberger U, Moskvina E, Scholze T, Freissmuth M, Boehm S. Inhibition of adenylyl cyclase by neuronal P2Y receptors. Br J Pharmacol. 2002;135:673–684. doi: 10.1038/sj.bjp.0704514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vartian N, Boehm S. P2Y receptor-mediated inhibition of voltage-activated Ca2+ currents in PC12 cells. Eur J Neurosci. 2001;13:899–908. doi: 10.1046/j.1460-9568.2001.01461.x. [DOI] [PubMed] [Google Scholar]
- Villarroel A, Marrion NV, Lopez H, Adams PR. Bradykinin inhibits a potassium M-like current in rat pheochromocytoma PC12 cells. FEBS Lett. 1989;255:42–46. doi: 10.1016/0014-5793(89)81057-9. [DOI] [PubMed] [Google Scholar]
- von Kugelgen I. Pharmacological profiles of cloned mammalian P2Y-receptor subtypes. Pharmacol Ther. 2006;110:415–432. doi: 10.1016/j.pharmthera.2005.08.014. [DOI] [PubMed] [Google Scholar]
- Wakamori M, Hidaka H, Akaike N. Hyperpolarizing muscarinic responses of freshly dissociated rat hippocampal CA1 neurones. J Physiol. 1993;463:585–604. doi: 10.1113/jphysiol.1993.sp019612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei AD, Gutman GA, Aldrich R, Chandy KG, Grissmer S, Wulff H. International Union of Pharmacology. LII. Nomenclature and molecular relationships of calcium-activated potassium channels. Pharmacol Rev. 2005;57:463–472. doi: 10.1124/pr.57.4.9. [DOI] [PubMed] [Google Scholar]
- Wirkner K, Schweigel J, Gerevich Z, Franke H, Allgaier C, Barsoumian EL, Draheim H, Illes P. Adenine nucleotides inhibit recombinant N-type calcium channels via G protein-coupled mechanisms in HEK 293 cells; involvement of the P2Y13 receptor-type. Br J Pharmacol. 2004;141:141–151. doi: 10.1038/sj.bjp.0705588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia XM, Fakler B, Rivard A, Wayman G, Johnson-Pais T, Keen JE, Ishii T, Hirschberg B, Bond CT, Lutsenko S, Maylie J, Adelman JP. Mechanism of calcium gating in small-conductance calcium-activated potassium channels. Nature. 1998;395:503–507. doi: 10.1038/26758. [DOI] [PubMed] [Google Scholar]