

Abstract
Inhibition of p38MAPK alpha/beta is known to enhance 1,25-dihydroxyvitamin (1,25D)-induced monocytic differentiation, but the detailed mechanism of this effect was not clear. We now show that the enhancement of differentiation becomes apparent with slow kinetics (12-24h). Interestingly, the inhibition of p38MAPK alpha/beta by their selective inhibitor SB202190 (SB) leads to an upregulated expression of p38MAPK isoforms gamma and delta in 1,25D-treated AML cells, in cell lines and in primary culture. Although the expression and activating phosphorylations of p38MAPK alpha are also increased by an exposure of the cells to SB, its kinase activity is blocked by SB, as shown by reduced levels of phosphorylated Hsp27, a downstream target of p38MAPK alpha. A positive role of p38MAPKs in 1,25D-induced differentiation is shown by the inhibition of differentiation by antisense oligonucleotides to all p38MAPK isoforms. Other principal branches of MAPK pathways showed early (6h) activation of MEK/ERK by SB, followed by activation of JNK1/2 pathway and enhanced expression and/or activation of PU.1, ATF-2 differentiation-related transcription factors. Taken together with previous reports, the results indicate that 1,25D-induced differentiation is enhanced by the activation of at least three branches of MAPK pathways (ERK1/2; p38MAPK gamma/delta; JNK1/2). This activation may result from the removal of feedback inhibition of an upstream regulator of those pathways, when p38MAPK alpha and beta are inhibited by SB.
Keywords: Vitamin D, differentiation, p38MAPK, signaling pathways, ATF-2, PU.1
Introduction
Vitamin D-derived compounds, such as the physiologically active hormonal form 1,25-dihydroxyvitamin D3 (1,25D), are being studied as potential agents for differentiation therapy of acute myeloid leukemia (AML). This is based on numerous studies showing that the exposure of human and rodent AML cells to these compounds induces differentiation with monocytic/macrophage phenotype, and results in cell cycle arrest which limits cell proliferation [1-4]. However, this has not as yet been translated to the clinic, because the concentrations used for in vitro and animal experiments far exceed those than can be tolerated by humans, as they cause life-threatening hypercalcemia [5, 6]. Among the approaches considered for improving the status of 1,25D as an anti-leukemia agent are studies designed to elucidate the mechanistic basis for its differentiation-inducing actions, and while there is a considerable body of knowledge on this subject, the total picture is still not clear [7-11].
One focus of such studies has been the role of MAPK pathways in 1,25D induced monocytic differentiation. Previous studies have shown that MEK/ERK pathway is activated during the early stage of 1,25D-induced monocytic differentiation [7, 12], and JNK pathway also contributes to this process, particularly in the later stages of differentiation [13]. However, the role of p38 MAPK has been unclear. Treatment of AML cells with 1,25D can increase the activating phosphorylation of p38MAPK when detected as usually studied using an antibody which recognizes all isoforms of p38MAPK, e.g. [14, 15], suggesting that the p38MAPK pathway may also activate processes that assist the acquisition of differentiated phenotype. Seemingly in contrast, the addition of SB202190, generally regarded as a specific inhibitor of p38MAPK, e.g. [16-19], to 1,25D-treated cells further increased the differentiation process [14, 15, 20, 21]. We now demonstrate that the increased differentiation is due to inhibition of only the alpha/beta isoform of p38MAPK, with an apparently compensatory increase in the expression of gamma and delta isoforms of p38MAPK, along with the previously known activation of ERK and JNK1/2 pathways [14, 15], and suggest a mechanism linking these branches of the MAPKs pathways.
Materials and Methods
Cell culture
HL60 cells, derived from a patient with promyeloblastic leukemia [22], were obtained from the ATCC, and the cells were maintained in Iscove’s Modified Dulbecco’s Medium (IMDM) (ATCC, Manassas, VA), supplemented with 15% heat-inactivated, iron-enriched bovine calf serum (HyClone, Logan, UT). Human myelomonocytic leukemia U937 [23] cells, were maintained in RPMI 1640 medium (Mediatech, Washington, DC), supplemented with 1% glutamine and 10% above mentioned bovine calf serum. All cells were kept in a 37°C incubator supplied with 5% CO2. The cells were passaged and fed with medium 2–3 times per week to maintain the log phase growth. Cell viability was determined using trypan blue (0.25%) exclusion, and cell numbers by counting cells in a Neubauer hemocytometer. Cell viability was also obtained by using gating in an Epics XL-MCL flow cytometry instrument (Beckman Coulter, Fullerton, CA), based on forward and side scatter [24]. All experimental groups were seeded in fresh tissue culture medium at certain proper concentration for indicated time in 25-cm2 flasks, and incubated with 5 μM SB202190, 10 nM 1,25D for the indicated times. Each experiment was repeated at least three times.
Isolation and culture of mononuclear cells from patients’ peripheral blood
Peripheral blood mononuclear cells, obtained following the patient’s informed consent according to the Institutional IRB protocol, were isolated by using Ficoll (Histopaque-1077, Sigma-Aldrich, St Louis, MO) as previously described [25]. The isolated cells were cultured with SB (added at time zero) and 1,25D (added at 30 min dissolved in ethanol) for periods of time ranging from 5-9 days in RPMI plus 10% bovine calf serum. Control groups received equivalent amount of DMSO and ethanol. The duration of incubation depended on culture viability, monitored by Trypan Blue exclusion. Before harvesting, the cultures were observed under an inverted microscope for adherence of the cells to the bottom of the flask. When adherence was noted, the cells were harvested by scraping with a rubber “policeman”.
Chemicals and antibodies
1,25D was a kind gift from Dr. Milan Uskokovic, BioXell, Inc., Nutley, NJ. The p38 MAPK inhibitor SB 202190 (SB) was obtained from Sigma-Aldrich (St. Louis, MO), and the stocks were prepared by dissolving in DMSO. SB was added to the cultures 1 hour before the exposure to 1,25D.
Antibodies against total p38 alpha, PU.1, also known as Spi-1 (T-21), and calregulin (H-170) were purchased from Santa Cruz Inc. Antibodies against p38 beta, p38 gamma, p38 delta, P-p38 (Thr 180/Tyr 182), P-ATF-2 (Thr 71), P-JNK (Thr 183/Tyr 185), P-HSP27 (Ser82), P-MEK (Ser217/221), P-ERK (Thr202/Tyr204), MLK3 and P-MKK3/MKK6 (Ser189/207) were purchased from Cell Signaling.
Antisense oligonucleotides
Antisense oligonuleotides were used to inhibit the product of genes of interest. The scrambled and p38 antisense oligonucleotides were designed and synthesized by Integrated DNA Technologies (IDT, Coralville, IA). The sequences of oligonucleotides used, respectively, were: p38 scrambled, 5’-CCA CGG CCA TTA TGC GT ATC-3’; p38 alpha antisense, 5’-G*A*G* CCC TGT ACC T*A*G *T-3’ (* the three nucleotides at each end were phosphorothioated to stablize the oligonucleotide); p38 beta antisense, 5’-A*G*C*TCCTTCCACTCCT*C*C*A-3’; p38 gamma antisense, 5’-C*C*C* TCT GTC CTT CCT CT*G *C*A-3’; p38 delta antisense, 5’-T*C*C* CAG ATT TCC AGC CT*C *C*A-3’. The final concentration of 5 μM scrambled or p38 antisense oligonucleotides were added together with 1 μM Endo-porter (Gene Tools, Philomath, OR) to the cultures for 24-48 hours, then the whole experiments were set up by supplementing half amount of oligonucleotides and then adding SB and 1,25D.
Determination of markers of differentiation
To analyze the expression of cell surface markers, aliquots of 106 cells were harvested at the indicated times, washed twice with 1× PBS, then incubated for 45 min at room temperature with 0.5 μl MY4-RD-1 and 0.5 μl MO1-FITC antibodies (Beckman Coulter), to detect the expression of surface cell markers CD14 and CD11b, respectively. The cells were then washed three times with ice cold 1× PBS, and resuspended in 0.5 ml 1×PBS. Two-parameter analysis was performed using the same XL-MCL flow cytometer described above Isotypic mouse IgG1 was used to set threshold parameters. Each experiment was repeated at least three times.
In addition, monocytic differentiation was monitored by cytochemical determination of nonspecific esterase (NSE) activity. Smears were made by resuspending 2 × 106 cells in 100 μl 1 × PBS and spreading on slides. The air-dried smears were fixed in formaldehyde and acetone mixture buffer for 30 sec, then washed and stained for 45 min at room temperature with the following solution: 0.067 M sodium phosphate buffer, pH 7.6, 8.9 ml, hexazotized pararosaniline, 0.6 ml, 10 mg alpha-naphthyl acetate, and 0.5 ml ethylene glycol monomethyl ether. The slides were then washed with distilled water for three times and then air-dried. The percentage of positive cells in a total of 500 cells was counted using a hemocytometer. Each experiment was repeated at least three times.
Cell extracts and Western Blotting
These procedures were carried out as previously described except that Calregulin was used as a loading control antibody [26].
Immunoprecipitation
Immunoprecipitation was performed by using Pierce Direct IP Kit (Pierce Biotechnology, Rockford, IL) according to the manufacturer’s recommended procedure. Briefly, 10 μg of anti-p38 alpha or anti-p38 gamma or anti-p38 delta antibody was coupled to 20 μl of AminoLink Plus Coupling Resin in a spin column by adding 3 μl of the sodium cyanoborohydride solution for every 200 μl reaction maintained in 1 × coupling buffer, and incubated on a rotator at room temperature for 90-120 minutes, then a 300 μl aliquot of the whole cell extract which contains 500 μg total protein was incubated with the antibody-coupling resin with gentle end-over-end mixing or shaking at 4°C overnight. Next, the immunoprecipitated proteins were eluted from the column and run on 10% SDS–PAGE gel, transferred to a membrane, and probed with specified antibodies. The protein bands were visualized with a chemiluminescence assay system (Amersham Pharmacia Biotech, Inc).
p38 Kinase enzyme assay
The kinase assay was performed by using the p38 MAP Kinase Assay Kit (Nonradioactive) (Cell Signaling Technology) according to the manufacture’s protocol. Briefly, 45 μl eluted IP proteins as described above prepared from 500 μg whole cell lysate of HL60 or U937 cells treated as indicated for individual experiments, 1 μl of ATF-2 fusion proteins and 1 μl 100 μM ATP was suspended in 5 μl 5 × kinase buffer (25 mM Tris-HCl, pH 7.5, 5 mM beta-glycerophosphate, 2 mM DTT, 0.1 mM Na3VO4, 10 mM MgCl2), and incubated for 30 min at 30°C. For “in vitro” kinase assay groups, the final concentration of 5 μM SB202190 was added to 50 μl reaction volumn. The reaction was terminated by 25 μl 3 × SDS sampling buffer, boiled for 5 min, vortexed, and then microcentrifuged for 2 min. Samples (30 μl) were loaded on 10% SDS-PAGE gel and analyzed by western blotting using phospho-ATF antibody.
RNA extraction and Real-time PCR (qRT-PCR)
RNA was extracted from cells using TRIzol® Reagent (Invitrogen, Carlsbad, CA), which is a mono-phasic solution of phenol and guanidine isothiocyanate, an improvement to the single-step RNA isolation method developed by Chomczynski and Sacchi [27]. The procedures were done as previously described. Briefly, 5-10 × 106 cells were homogenized in 1 ml Trizol and incubated for 5 min at RT. After addition of chloroform followed by centrifugation at 14,000 rpm, the aqueous phase was transferred to a fresh tube. The RNA was precipitated by mixing with 500 μl isopropyl alcohol, and incubated 10 min at RT. RNA pellet was obtained by centrifugation, the supernatant discarded, and the pellet washed with 75% ethanol in DEPC water, then briefly dried for 10 min. The RNA pellet was then dissolved in 50-100 μl DEPC water. The forward and reverse primers sequences used, respectively, were: p38 alpha, 5’-TGC CAA GCC ATG AGG CAA GAA A-3’ and 5’-TGG TGG CAC AAA GCT GAT GAC T-3’, p38 beta, 5’-AGC CAT ATC TGG CAA GAA GCT GGA-3’ and 5’-ACA AGG AAA GAG GAC TGA CCC ACA-3’, p38 gamma, 5’-TTG AAT TGG ATG CGC TAC ACG CAG-3’ and 5’-AGG GCT TGC ATT GGT CAG GAT AGA-3’, p38 delta, 5’-ACA GCC TTT CAA GCA GAG GAC AGA-3’ and 5’-GAA ACC AAC ACA GCA TCA CTG CCA-3’, CD11b, 5’-GCA AGT GTC TGT GTG CAA GTG TGT-3’ and 5’-TCA GTG GAG AGA AGC TGC TGT GTT-3’, CD14, 5’-AAC TCC CTC AAT CTG TCG TTC GCT-3’ and 5’-GGG CAA AGG GTT GAA TTG GTC GAA-3’, ARP0, 5’-AGA TGC AGC AGA TCC GCA T-3’ and 5’-GTG GTG ATA CCT AAA GCC TG-3’. For reverse transcription, samples were incubated in an Eppendorf PCR system at 42°C for 15 min, then 99°C for 5 min, and 5°C for 5 min. For real-time PCR, relative quantification of target cDNA was performed using a Roche LightCycler instrument with Faststart DNA Masterplus Syber Green I kit (Hoffmann-La Roche) and gene-specific primers. The parameters for PCR were as follows: 45 cycles of 1) 95°C for 15 s, 2) 58°C for 10 s, and 3) 72°C for 15 s followed by an incremental increase (0.1°C/s) of temperature from 65 °C to 95°C to analyze the melting curve of the products. A mixture of forward and reverse primers at a final concentration of 1.5 μM was used to detect target genes and ARP0 (acidic ribosomal protein p0, internal control). The Ct value, the cycle number at which signal fluorescence surpassed fluorescence background noise, was recorded, and relative fold values were calculated based on the equation 2-ΔΔCt, where ΔΔCt=ΔCt treated-ΔCt calibrator (untreated samples) and ΔCt= Ct target gene-Ct ARP0.
Statistical analysis
Significance of the differences between mean values of two compared groups was assessed by a two tailed student’s test. P < 0.05 was considered statistically significant. The computations were performed with an IBM-compatible personal computer and used Microsoft EXCEL.
Results and Discussion
SB increases p38MAPKγ and δ levels in AML cells induced to differentiate by 1,25D
It is known that SB202190 and SB203580 p38 inhibitors enhance 1,25D-induced monocytic differentiation of human AML cells, and that SB202190 is more potent in this action [15, 28], but the rapidity of the enhancement was not characterized. We now show that in both HL60 and U937 cells the surface marker of myeloid cells CD11b, and the monocytic marker CD14, are not evident at 6h after exposure of the cells to 1,25D or to 1,25D/SB, but CD14 significantly increased at 12h of such treatments, while the increase in CD11b was evident by 24h (Fig 1). While SB alone did not significantly induce differentiation of HL60 and U937 cells, SB enhanced the intensity of the differentiation marker CD11b induced by 1,25D in both cell types. In contrast, no significant enhancement was noted for surface expression of CD14 in HL60 cells, and there was only a variable decrease of CD14 expression in U937 cells, the reason for which needs to be further investigated (Fig 1). However, the enhancement of differentiation by SB was confirmed in both HL60 and U937 cells by the increased presence of the cytoplasmic esterase characteristic of monocytes, known as NSE (Supplementary Fig S1).
Fig.1. SB ehances effect of 1,25D with kinetics corresponding to the kinetics of 1,25D induction of differentiation, and mainly increases the cell surface differentiation marker CD11b.

HL60 and U937 cells were treated with SB (5 μM) and 1,25D (10 nM) for the indicated times, and cell surface differentiation markers CD11b and CD14 were determined by flow cytometry. Note that the expression of differentiation markers was increased in these two cell lines treated with 1,25D, and SB potentiated the 1,25D-induced expression of CD11b by 12 hours treatment in HL60, but by 24 hours in U937 cells. Means ± SD are shown; n=3. * signifies p<0.05 for CD11b upregulation comparing 1,25D with SB-1,25D treated groups.
To initiate an analysis of the basis for the synergy between SB and 1,25D on differentiation-related events, we examined the effects of these compounds on the expression of three (alpha, gamma and delta) of the four known isoforms of p38MAPK, as the beta form is expressed at low levels in HL60 cells, and shares properties with isoform alpha, the most studied p38MAPK [29]. As shown in Fig 2, little change in mRNA levels of the isoforms examined was noted at early (6h) times of exposure, while at the late stage of differentiation (72h) we observed a dramatic elevation (16-fold) in the level of p38MAPK delta isoform in HL60 cells exposed to 1,25D, which was enhanced by the addition of SB, resulting in a 43-fold increase. Less marked, but also highly significant increases by 1,25D and enhancement by SB were observed in U937 cells, here evident in both gamma and delta isoforms. No significant increases in isoform alpha were noted in these experiments (Fig 2).
Fig.2. SB increases 1,25D-induced p38MAPKγ and δ mRNA levels.


(A) Quantiative RT-PCR shows that SB dramatically enhanced 1,25D-induced expression of p38MAPKδ mRNA in HL60 cells after 72 hours treatment, indicating an effect of SB at the transcriptional level. Means ± SD are shown; n=3. *** signifies p<0.0001 for the dramatic upregulation of p38MAPKδ, comparing 1,25D with SB-1,25D treated groups. (B) Similarly, SB significantly enhanced 1,25D-induced mRNA levels of both p38MAPKγ and δ mRNAs in U937 cells after 72 hours treatment. Means ± SD are shown; n=3. * signifies p<0.05 for p38γ and p38δ upregulation comparing 1,25D with SB-1,25D treated groups.
Determination of the protein levels of p38MAPK isoforms by Western blot analysis during several stages of differentiation showed that, consistent with little change in mRNA levels (Fig 2), there were no sustained variations in protein levels of p38MAPK isoform alpha in either cell line, and SB had only a modest effect on the abundance of this isoform (Fig 3, and S2,A and B). In contrast, the other isoforms examined showed gradual increases that to greater or lesser extent paralleled the differentiation process. These were gamma and delta in HL60 cells, and beta, gamma and delta in U937 cells. Perhaps of functional significance, SB further increased the protein levels of gamma and delta isoforms (Fig 3, and S2, A and B). In addition, SB increased the levels of phosphorylated p38MAPK, measured as all isoforms collectively, as an antibody for specific isoforms is not available.
Fig.3. Expression of p38MAPK isoforms increases variably in human AML cell lines after exposure to 1,25D for 6 and 72 hours, and protein levels of at least two isoforms other than isoform p38MAPKα are enhanced by SB.


(A) Western blots showing that in HL60 cells SB upregulated the 1,25D-induced expression of p38MAPKγ and p38MAPKδ after 72 hours, and that of phosphorylated p38 (all isoforms). p38MAPKα changed only slightly following the exposure to 1,25D or SB-1,25D. Expression of p38MAPKβ at protein level was not detectable in the HL60 cell strain used for these experiments. (B) Western blots showing that in U937 cells SB similarly upregulated 1,25D-induced expression of p38MAPKγ and p38MAPKδ after 72 hours exposure, although p38MAPKδ expression was relatively low in this cell line. The expression of p38MAPKβ was upregulated by SB only to a minor extent, while the expression of p38MAPKα changed little as a result of 1,25D or SB exposure. The O.D. values are shown below each panel as the ratio of the signal of each band to the corresponding signal for calregulin, the loading control.
* Expression of p38 beta was not detectable in HL60 cells.
During this study we had an opportunity to evaluate the responses of leukemic blasts obtained from the blood of 3 patients with AML. These cells responded by monocytic differentiation to both SB and 1,25D, and the combination of SB and 1,25D produced an additive effect illustrated for one patient in Fig 4. Importantly, these responses correlated with an increased expression of p38MAPK isoforms beta and gamma, while isoform delta was only upregulated by SB, and isoform alpha showed no significant increase. In particular, p38 gamma level was markedly increased by the combination of SB/1,25D. Since there was only a slight increase in differentiation markers following this treatment, the implication is that while p38 isoforms contribute to the induction of differentiation, they are not sufficient to determine the phenotype.
Fig.4. SB can potentiate 1,25D-induced differentiation and upregulate expression of p38MAPK isoforms in ex vivo patient specimens.
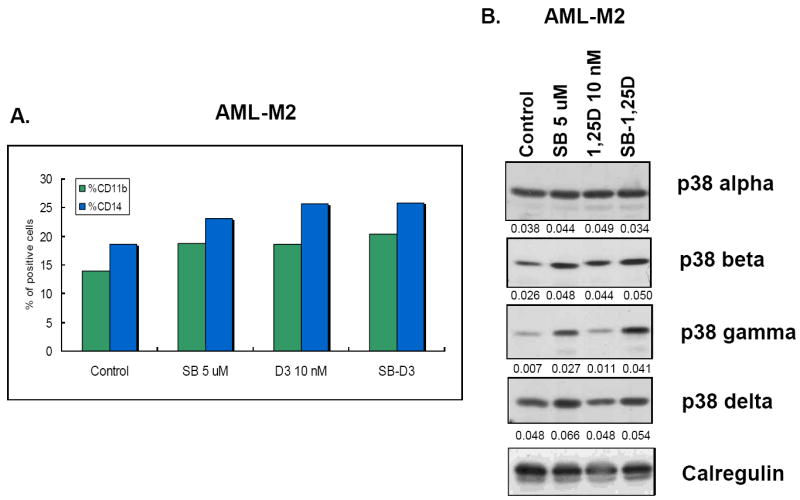
(A) Differentiation markers CD11b and CD14 were increased by an exposure to SB alone, 1,25D, and their combination for 5 days. (B) Western blots showing that SB alone increased the expression of p38MAPKβ, p38MAPKγ and p38MAPKδ, and potentiated the 1,25D-induced expression of these isoforms. The expression of p38MAPKα essentially unchanged. The O.D. values are shown below each panel as the ratio of the signal of each band to the signal of the corresponding loading control, calregulin.
The experiment with patient blasts adds significance to the studies with cell lines, and strengthens the conclusion that the potentiation of 1,25D-induced differentiation of human AML cells by SB is associated with an upregulated expression of two or more p38MAPK isoforms.
SB inhibits p38MAPKα but not p38MAPKγ or p38MAPKδ kinase activity in human leukemia cell lines
The observation noted above that SB increases the total level of phosphorylated p38MAPK isoforms in HL60 cells, with similar but less obvious increases in U937 cells (Fig 3), raises the question whether this indicates increased kinase activity of any of the four isoforms. Since this takes place in the intracellular presence of SB, a p38MAPK inhibitor, this is not a simple question to answer. However, as pointed out before, although SB is frequently referred to as a specific inhibitor of p38MAPK, this applies only to p38MAPK alpha and beta, with isoforms gamma and delta not being inhibited at all by SB at concentration up to 50 μM [28]. Further, it is also known that binding of SB to p38MAPK does not prevent its phosphorylation by upstream kinases such as MKK3/6, though this phosphorylation does not activate the enzyme activity of p38MAPK, when the p38MAPK kinase activity is assessed in vivo by the levels of phosphorylation of downstream targets of p38MAPK, such as Hsp27 [28]. This was described in HeLa cells, and is apparently cell-type specific, as in LPS-stimulated THP-1 and arsenate-activated 293T cells SB inhibits p38MAPK activation as well as phosphorylation [30, 31]. Therefore, to determine if in AML cells SB actually inhibits p38MAPKα/β activity we checked the activating phosphorylation levels of p38MAPKs (all isoforms) and the phosphorylation levels of Hsp27, a documented downstream target of p38MAPKα/β [32]. The results illustrated in Fig 5A demonstrate that “in vivo” (intracellular) kinase activity of p38MAPKα/β is indeed inhibited in AML cells, even though the activating phosphorylations are present, and actually their levels are increased in HL60 cells by the presence of SB.
Fig.5. SB inhibits p38MAPKα but not p38MAPKγ or p38MAPKδ kinase activity in human AML cell lines.
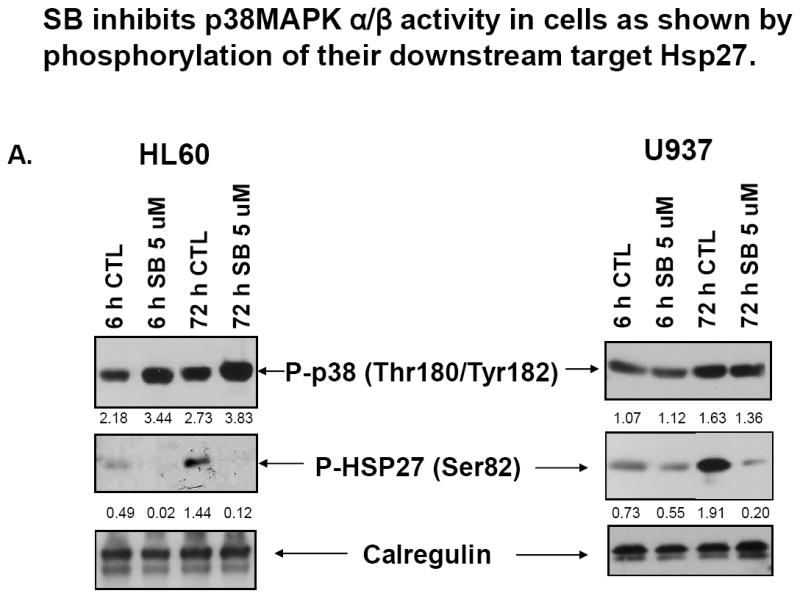
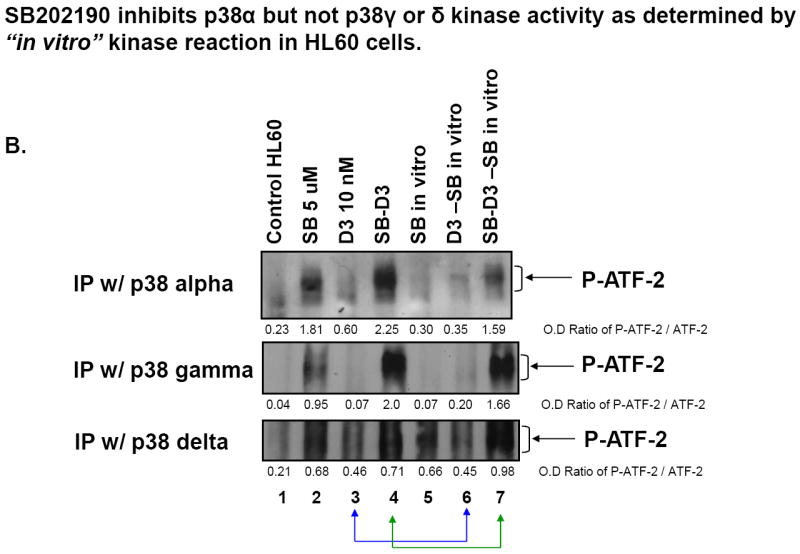
(A) Western blots showing that SB induces higher levels of activating phosphorylation (Thr180/Tyr182) of p38MAPKs in HL60 cells, but inhibits p38MAPK α/β activity in both HL60 and U937 cells, as shown by reduced phosphorylation of their downstream target, Hsp27. The O.D. values shown below each panel represent the ratio of the signal of each band to the signal of the corresponding loading control, calregulin. (B) Western blots of kinase assays showing that SB202190 inhibits p38MAPKα but not p38MAPKγ or p38MAPKδ kinase activity, determined by “in vitro” kinase reaction in HL60 cells. Lane 5-7 cells were treated, harvested and the protein extracted in exactly the same way as the cells shown in lanes 2-4. The “SB in vitro” indicates that during the kinase reaction, the same concentration of SB was added again to the 50 μl reaction buffer. ATF-2 was used as the substrate for each p38MAPK isoform, and phosphorylation of ATF-2 was detected by western blot analysis for activating phosphorylations (indicated by bracket on the right side of each panel). The O.D. values are shown as the ratio of the signal of phospho-ATF-2 of each band to the signal of the corresponding loading control, ATF-2.
To address the question whether p38MAPK isoforms gamma and delta in AML cells resemble the previously studied cell types, and are not inhibited by 5 μM SB, we performed the standard in vitro assay in which the individual p38MAPK isoforms are immunoprecipitated, and the phosphorylation of ATF-2, a known immediate target of p38MAPKs [33, 34], is measured in the presence and absence of SB added directly to the assay mixture, since the active site of the p38MAPK is washed free of SB in the preparation of the p38MAPK containing IP. The results shown in Fig 5B and S3 illustrate that, using IPs from HL60 cells, phosphorylation of the p38MAPK substrate ATF-2 by the isoform alpha is inhibited, as expected, by the addition of SB in vitro (compare lane 3 with lane 6, and lane 4 with lane 7 in Fig 5B). However, in the same assay, isoforms gamma and delta are not inhibited by SB. Similar results were seen when U937 cell extracts were tested (data not shown), indicating that in AML cells, as in HeLa cells, isoforms gamma and delta retain kinase activity and have potential function in the presence of moderate concentrations of SB.
Isoforms p38MAPKγ and p38MAPKδ play a positive role in 1,25D- and SB-1,25D-induced cell differentiation
The increases in the expression of isoforms p38MAPKγ and p38MAPKδ during 1,25D-induced differentiation demonstrated above, and the retention of the kinase activity of these isoforms when SB was added to enhance the effect of 1,25D, raised the possibility that the isoforms not inhibited by SB have a functional role in 1,25D-induced differentiation, and its enhancement by SB. To test this, we employed the approach of incubating the cells with antisense oligonucleotides (AS), since AML cells, particularly HL60 cells, lose viability when subjected to transfections necessary for knock-down of gene expression by siRNA and similar technique. Transfections appear to be especially difficult when stress-related genes are knocked-down, and SB has been shown to cause cell death [17]. Thus, we initiated these experiments using AS, to the individual isoforms, and found that each reduced 1,25D-induced differentiation to some extent, irrespective if SB was added or not (Figs S4 and S5). In HL60 cells no significant effect of p38MAPKα AS on CD11b levels was noted, but the expression of CD14 was moderately but significantly reduced (14-20%) by the AS oligos to p38MAPKα in cells exposed to 1,25D +/-SB (Fig S4A). In U937 cells AS oligos to isoforms α and β both reduced CD11b and CD14; AS to isoform α by 44-69%, and AS to isoform β by 78-79% for 1,25D +/-SB in each case (Fig S4B). We also performed a pilot experiment using AS to isoforms γ and δ and found marked reductions in the expression of both CD11b and CD14 (Fig S5).
However, since isoforms p38MAPKα and p38MAPKβ are inhibited by SB and therefore cannot have a functional role in the SB-induced enhancement of differentiation, for further studies we focused on isoforms p38MAPKγ and p38MAPKδ, used in combination to resemble the presumably physiological situation. Fig 6 shows that the expression of the differentiation markers was reduced by the AS oligos to γ and δ with high significance (p< 0.01) in both 1,25D and SB-1,25D treated groups. At this time it is not possible to assign the degree of relative importance to these isoforms to the differentiation process, since their expression appears to be cell-type specific, especially at protein level (Fig 3), but since the knock-down of either isoform decreases differentiation, their functions appear redundant. Importantly, however, there was no difference in the level of differentiation markers due to SB enhancement when the isoforms p38MAPKγ and p38MAPKδ were knocked-down, indicating that the enhancement is mediated by these isoforms. While this was evident in both cell lines, the effect was numerically greater in U937 cells, most likely to due the greater uptake of the AS oligos by the cells. We have therefore used these cells to further explore the underlying mechanisms of AS-oligo action.
Fig.6. Isoforms p38MAPKγ and p38MAPKδ play a positive role in 1,25D and SB-1,25D-induced cell differentiation.
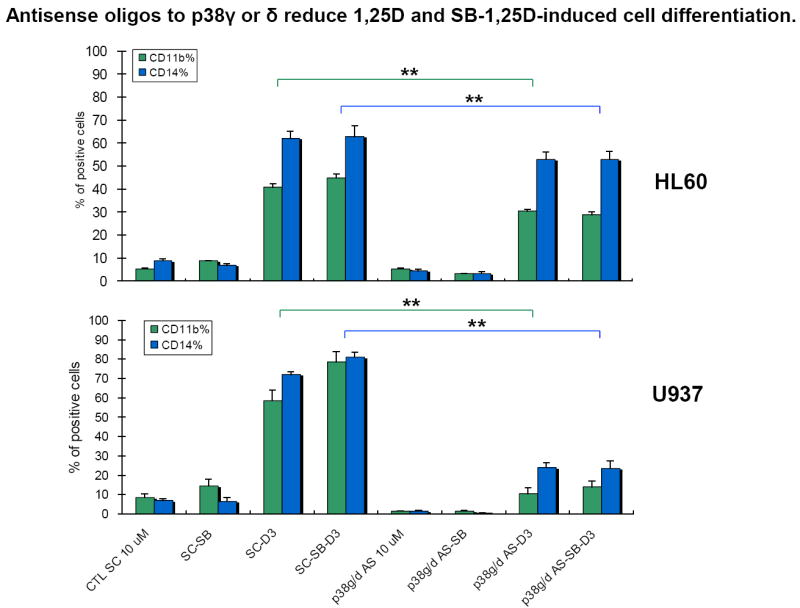
Combined p38MAPKγ and p38MAPKδ antisense oligos (indicated as p38g/d in the figure, final concentration 10 μM, were added 48 hours before exposure to SB (5 μM) and 1,25D (10 nM). Cells were treated with SB and 1,25D for 72 hours. Note that the expression of both differentiation markers was significantly reduced by the antisense oligos in 1,25D and SB-1,25D treated groups in both cell lines. SC: scrambled oligos, p38g/d AS: p38MAPKγ/δ antisense oligos. The reductions for HL60 cells were, CD11b and CD14, respectively: SC-D3 vs p38MAPKγ/δ AS-D3, 25% and 15%; SC-SB-D3 vs p38MAPKγ/δ AS-SB-D3 36% and 16%. For U937 cells SC-D3 vs p38MAPKγ/δ AS-D3, 82% and 66%; SC-SB-D3 vs p38MAPKγ/δ AS-SB-D3 82% and 71%. Means ± SD are shown; n=3. ** signifies p<0.01 for reduction of CD11b and CD14 expression comparing 1,25D with SB-1,25D-treated groups.
AS oligos to p38MAPKγ and p38MAPKδ reduce phosphorylation of differentiation-related MAP kinases
We have confirmed that AS oligos can reduce the levels of their cognate proteins, and this is most apparent in 1,25D-treated cells, with reductions at least by 60% for combined knock-down of p38MAPKγ and p38MAPKδ, the isoforms not inhibited by SB (Fig S6). We have therefore tested if the MAPKs representative of the three principal MAPK pathways, are affected by these reductions. As shown in Fig 7, robust reductions in the phosphorylated, active forms of representatives of all these pathways (P-MKK3/6, P-MEK, and P-JNK) are indeed evident, of similar magnitude as the knock-down of p38MAPKγ and p38MAPKδ isoforms. This links the p38MAPKγ and p38MAPKδ isoforms to the previously known participation of MEK/ERK and JNK/c-jun pathways in 1,25D-induced differentiation of AML cells [7, 13, 35], and provides an explanation for finding activated p38MAPKs in SB-treated cells (Fig 3, and Fig S2A), as P-MKK3/6 directly activates p38MAPKs [36-39].
Fig.7. In 1,25D-treated cells p38MAPKγ and p38MAPKδ antisense oligos reduce phosphorylation of differentiation-related MAP kinases.
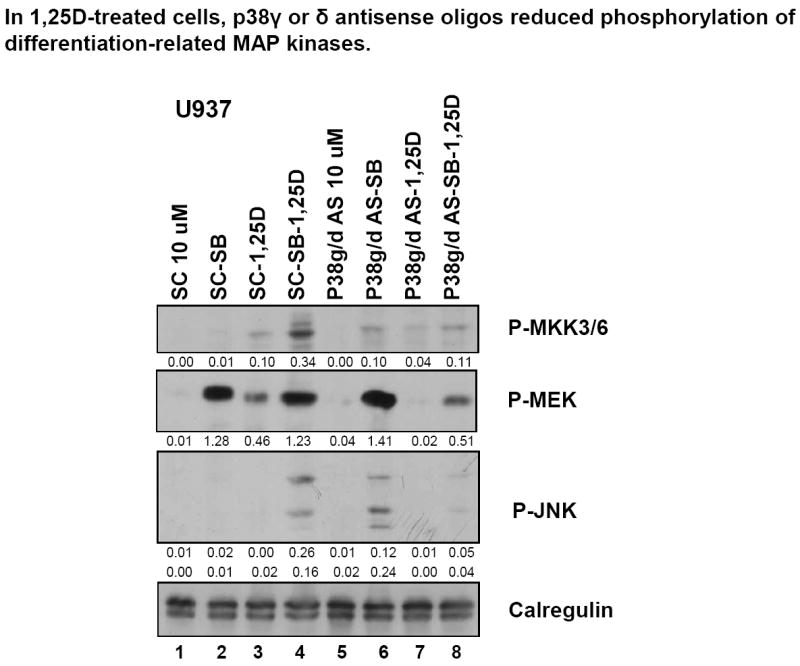
Western blots showing that p38MAPKγ and p38MAPKδ antisense oligos reduced expression of phospho-MKK3, an upstream regulator of p38MAPK, as well as that of phospho-MEK and phospho-JNK in 1,25D-treated cells.
Exposure of AML cells to SB enhances 1,25D-induced activation of MAPK pathways downstream and upstream of p38MAPK, as well as of differentiation-related transcription factors PU.1 and ATF-2
To further link the current finding that the isoforms not inhibited by SB, p38MAPKγ and p38MAPKδ, play a positive role in 1,25D-induced differentiation and the enhancement by SB of this process, we determined the effect of SB alone, and in combination with 1,25D, on the MAPKs representative of the MEK/ERK and JNK MAPK pathways (Fig 8). It can be seen that SB alone activates MEK/ERK starting at an early stage of differentiation (6h) in both HL60 and U937 cells, but it does not increase the effect of 1,25D on this pathway (Fig 8 and data not shown). In contrast, SB alone modestly activates the JNK pathway, principally at a late stage of differentiation (72h), but an enhancement of the effect of 1,25D can be seen at 6h already. These findings show that SB influences the early, presumably causative, events in 1,25D- induced differentiation, in which the ERK pathway is primarily involved [7].
Fig.8. SB activates MEK/ERK at the early stage, while JNK and the myeloid differentiation-related transcription factor expression PU.1 are activated principally at a late stage of differentiation.
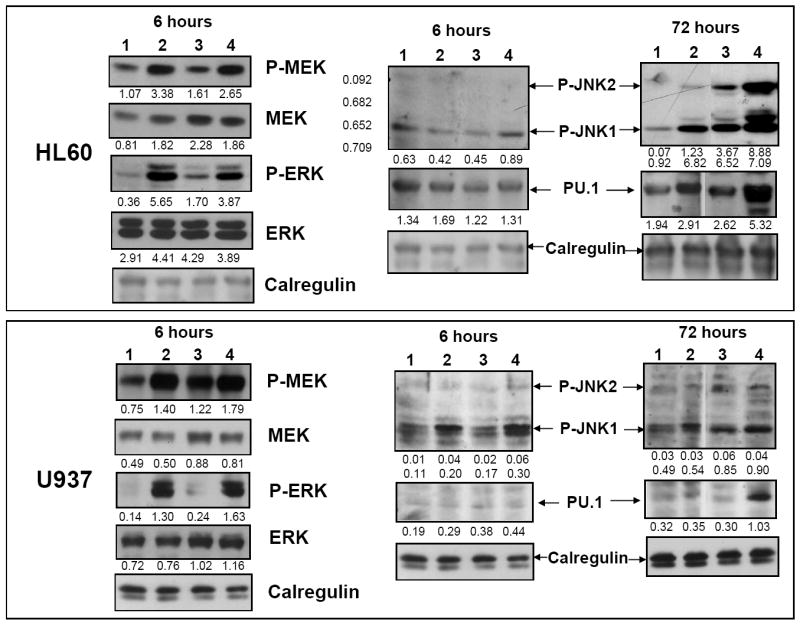
Enhancement by SB of 1,25D-activation of these signaling molecules also takes place. Western blots showing that SB alone increased phospho-MEK/ERK after 6 h treatment in both cell lines. In HL60 cells phospho-JNK and the expression of differentiation-related transcription factor PU.1 did not increase at that time; however, after 72 hours at the later stage of cell differentiation, SB enhanced 1,25D-activated phosphorylation of JNK and 1,25D-induced expression of PU.1. In U937 cells an increase in P-JNK1/2 was already noted at 6h. The lanes represent cells treated for 6h or 72h, as indicated, with: 1. Vehicle control. 2. SB (5 μM). 3. 1,25D (10 nM). 4. SB-1,25D. The O.D. values shown below each panel represent the ratios of the signal of each band to the signal of the corresponding loading control, calregulin.
Since MAPK signaling impinges on transcription factors which mediate the expression of myeloid/monocytic differentiation-related genes [40, 41], in these experiments we monitored the expression of two such transcription factors. One is PU.1, involved in upregulation of the myeloid differentiation marker CD11b [42], the expression of which is enhanced by SB (Fig 1), and another one is the activated ATF-2, a principal component of the AP-1 transcription factor essential for monocytic differentiation [35]. Fig 8 shows that SB and 1,25D induce a synergistic increase in the expression of PU.1 at the late stage of differentiation, consistent with the effect of this combination on the appearance of the surface CD11b (Fig 1). Similarly, treatment of both HL60 and U937 cells with SB and 1,25D for 72h results in an increased level of activated ATF-2 (Fig 9), which is also known to have a positive effect on differentiation [35].
Fig.9. Inhibition of p38MAPKα by SB results in increased expression of its upstream regulator MLK3 and the activation of MKK3/6.
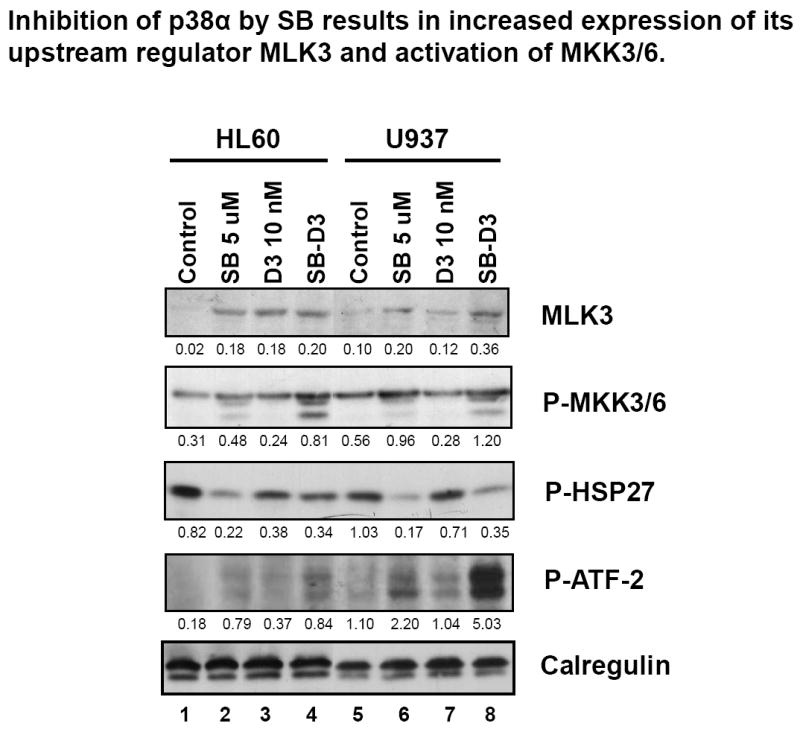
Western blots showing that SB (5 μM, 48h) increased the expression of MLK, an upstream regulator of p38MAPK, and increased phosphorylation of MKK3/6, but reduced phosphorylation of Hsp27, a downstream target of p38MAPKα. However, SB increased 1,25D-induced phospho-ATF-2, a component of transcription factor AP-1, which contributes to 1,25D-induced cell differentiation. The O.D. values are shown below each panel as the ratio of the signal of each band to the signal of the corresponding loading control, calregulin.
We propose that MAPK pathways become over-activated when SB inhibits the kinase activity of p38MAPK alpha/beta due to a lack of negative feedback inhibition of an up-stream regulator, normally provided by a down-stream target of p38MAPK alpha/beta (Fig 10). Such down-stream target is probably Hsp27, and the MAPK that fits the role of the “master” regulator is a kinase upstream of Mixed Lineage Kinase 3 (MLK3). MLK3 has been shown to regulate JNK and p38 MAPK pathways [43, 44], and possibly also the MEK/ERK pathway [45, 46]. The evidence supporting this hypothesis is shown in Fig 9. SB alone increased the expression of MLK3 in both HL60 and U937 cells, and enhanced the effect of 1,25D on MLK3 expression. These effect were paralleled by a similar increase in the level of a downstream target of MLK3, P- MKK3/6, while phosphorylation of Hsp27, a down-stream target of p38MAPK alpha/beta showed inhibition when the cells were exposed to SB, as discussed before. Interestingly, the level of phosphorylated ATF-2 increased in cells treated with SB/1,25D combination (Fig 9), showing that the inhibition of p38MAPK alpha/beta activity by SB can be more than compensated by the increased activity of its isoforms and/or JNK1/2.
Fig.10. A hypothetical scheme for the mechanisms of enhancement of 1,25D-induced cell differentiation by a p38α/β kinase inhibitor SB202190.
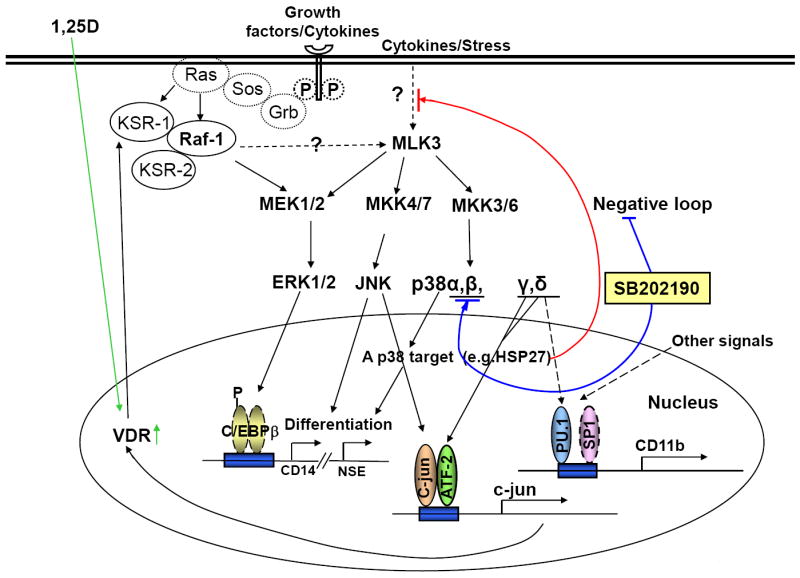
It is proposed that in proliferating cells a target of p38MAPKα, e.g. Hsp27, has a negative feedback on a kinase, perhaps upstream of MLK3, that regulates p38MAPK and JNK pathways, and to a lesser extent the MEK/ERK pathway. Removal of this restraint (the negative feedback loop) by the selective p38α/β kinase inhibitor SB activates all three MAPK pathways, which are involved in 1,25D-induced cell differentiation. In addition, SB increases the expression of the other two p38MAPK isoforms, γ and δ, which activate one of the principal components of AP-1, ATF-2, and perhaps also PU.1. Upregulation of c-jun and ATF-2 results in the expression of VDR, which in turn enhances the expression of other genes required for differentiation such as and C/EBPbeta. Dotted lines denote pathways which are speculative.
Fig 10 illustrates the concepts generated by the present data, integrated with some of the existing knowledge of the molecular basis of the signaling circuitry that can lead to the normalization of the malignant phenotype of AML cells. A more complete picture has been presented in a recent review, in which the complexity of the signaling networks and temporal changes are discussed [10].
In conclusion, we show for the first time that isoforms of p38MAPK alpha can positively contribute to differentiation of AML cells induced by 1,25D, and that their expression is markedly increased when the activity of p38MAPK alpha/beta is inhibited. The data provided are consistent with the hypothesis that phosphorylation of a target of p38MAPK alpha/beta normally results in the inhibition of an upstream regulator of multiple branches of MAPK pathways. Thus, lack of the inhibition of the candidate regulator upstream of MLK3 results in upregulation of MEK/ ERK (early) and JNK (principally late) pathways, which participate in 1,25D–induced differentiation (Fig 10). The role of ERK appears to be in the control of p27 expression which leads to G1 cell cycle block associated with differentiation [47-50], and of JNK to promote expression of VDR which initiates 1,25D signals. Since the effects of SB on 1,25D-induced differentiation resemble in many respects those of silibinin [25], a plant antioxidant with anti-tumor properties [51], our elucidation of the mode of action of SB in AML cells should also facilitate studies of the anti-leukemia actions of plant antioxidants, by comparing perturbations of signaling pathways induced by these compounds with those elicited by a pharmacological agent with an established mechanism of action. Thus, an improved understanding of the molecular basis for the interaction between the hormonal form of vitamin D and this pharmacological agent is likely to be valuable for the design of therapy of human diseases.
Supplementary Material
Fig.S1 SB enhances 1,25D-induced expression of the cytoplasmic differentiation marker NSE. HL60 and U937 cells were treated with SB (5 μM) and 1,25D (10 nM) for 48 hours, and the cells positive for NSE were enumerated using a light microscope. The percentage differences of positive cells between treated groups and untreated controls are shown here. Note that the expression of NSE was increased in both cell lines treated with 1,25D, and SB potentiated the effect of 1,25D. Means ± SD are shown; n=3.
Fig. S2 Kinetics of the expression of p38MAPK isoforms in human AML cell lines after exposure to 1,25D for various times. The protein levels of at least two isoforms other than isoform p38MAPKα are enhanced by SB. (A) Western blots showing that in HL60 cells SB upregulated the 1,25D-induced expression of p38MAPKγ and p38MAPKδ, best seen after 48 hours, and that of phosphorylated p38 (all isoforms). Isoform p38MAPKα changed only slightly following the exposure to 1,25D or SB-1,25D. Expression of p38MAPKβ at protein level was not detectable in the HL60 cell strain used for these experiments. (B) Western blots showing that in U937 cells SB upregulated 1,25D-induced expression of p38MAPKγ and p38MAPKδ similar to HL60 cells, although p38MAPKδ expression was relatively low in this cell line. The expression of p38MAPKβ was upregulated by SB only to a minor extent, while the expression of p38MAPKα changed little as a result of 1,25D or SB exposure. The O.D. values are shown below each panel as the ratio of the signal of each band to the corresponding signal for calregulin, the loading control.
Fig.S3 Western blots of kinase assays including the reaction substrate unphosphorylated ATF-2, which serves here as the loading control. When present in the reaction mixture SB202190 inhibited p38MAPKα but not p38MAPKγ or p38MAPKδ kinase activity, determined by “in vitro” kinase reaction in HL60 cells. Lane 5-7 cells were treated, harvested and the protein extracted in exactly the same way as the cells shown in lanes 2-4. The “SB in vitro” indicates that during the kinase reaction, the same concentration of SB was added to the 50 μl reaction buffer. Phosphorylation of ATF-2 was detected by Western blot analysis for activating phosphorylations. The O.D. values are shown as the ratio of the signal of phospho-ATF-2 of each band to the signal of the corresponding loading control, ATF-2.
Fig.S4 Isoforms p38MAPKα and p38MAPKβ contribute to 1,25D-induced cell differentiation. In these experiments AS oligos (final concentration 10 μM) were added 48 hours before the exposure of cells to SB (5 μM) and/or 1,25D (10 nM) and incubated for 72 hours. SC: scrambled oligos, p38a AS: p38MAPKα antisense oligos, p38b AS: p38MAPKβ antisense oligos. (A) HL60 cells. Only AS to p38MAPKα was studied in these cells, as they express very low levels of p38MAPKβ. The expression of CD14 was moderately but significantly reduced by the AS to p38MAPKα in both 1,25D (by 20%) and SB-1,25D (by 14%) treated groups. No significant effect of p38MAPKα AS on CD11b levels was noted in these cells. Means ± SD are shown; n=3. * signifies p<0.05 for reduction of CD14 expression comparing 1,25D with SB-1,25D-treated groups. (B) p38MAPKα or p38MAPKβ AS oligos were added separately to U937 cell cultures. Note that the expression of both cell differentiation markers CD11b and CD14 was markedly and highly significantly reduced by AS oligos in 1,25D and SB-1,25D treated groups. (The reductions were, CD11b and CD14, respectively: SC-D3 vs p38MAPKα AS-D3, 69% and 44%; SC-SB-D3 vs p38MAPKα AS-SB-D3 59% and 44%; SC-D3 vs p38MAPKβ AS-D3 79% and 55%; SC-SB-D3 vs p38MAPKβ AS-SB-D3 78% and 55%).The means ± SD are shown; n=3. ** signifies p<0.01 for reduction of both CD11b and CD14 expression comparing 1,25D with SB-1,25D-treated groups.
Fig.S5 Isoforms p38MAPKγ and p38MAPKδ play a positive role in 1,25D- and SB-1,25D-induced cell differentiation. SC: scrambled oligos, p38g AS: p38MAPKγ antisense oligos, p38d AS: p38MAPKδ antisense oligos. In both HL60 and U937 cells, p38MAPKγ or p38MAPKδ antisense oligos, final concentrations 10 μM) were added 48 hours before exposure to SB and 1,25D. Cells were then treated with SB and 1,25D for 72 hours. Note that the expression of both differentiation markers was reduced by the antisense oligos in 1,25D and SB-1,25D treated groups in both cell lines. The reductions for HL60 cells were, CD11b and CD14, respectively: SC-D3 vs p38MAPKγ AS-D3, 74% and 50%; SC-SB-D3 vs p38MAPKγ AS-SB-D3 70% and 49%; SC-D3 vs p38MAPKδ AS-D3 57% and 40%; SC-SB-D3 vs p38MAPKδ AS-SB-D3 30% and 15%. For U937 cells, SC-D3 vs p38MAPKγ AS-D3, 62% and 55%; SC-SB-D3 vs p38MAPKγ AS-SB-D3 51% and 43%; SC-D3 vs p38MAPKδ AS-D3 51% and 42%; SC-SB-D3 vs p38MAPKδ AS-SB-D3 40% and 40%. Results of one individual experiment for each cell line are presented in this Figure, as combined γ/δ isoforms were studied subsequently, and are shown as text Figure 6.
Fig.S6 Western blots showing that in 1,25D-treated (10 nM, 72h) cells, p38MAPKγ and p38MAPKδ antisense oligos reduced expression of target genes but not of non-target genes such as p38MAPKα.
Acknowledgments
We thank Dr. Milan Uskokovic, BioXell, Nutley, NJ, for the gift of 1,25-dihydroxyvitamin D3, and Dr. Michael Danilenko and Ms. Xiangwen Chen-Deutsch for helpful discussions, and Mr. S.M.Saad Hussain for technical help. This work was supported by grants RO1-CA-44722-21 and RO1-CA-117942-04 from the NIH/National Cancer Institute.
Abbreviations
- 1,25D
1α,25-dihydroxyvitamin D
- AML
Acute Myeloid Leukemia
- AS
Antisense oligonucleotide
- IP
Immunoprecipitate
- MAPK
Mitogen-activated Protein Kinase
- NSE
nonspecific esterase
- OD
Optical Density
- SB
SB202190
- SC
Scrambled oligonucleotide
- TF
Transcription Factor
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Abe E, Miyaura C, Sakagami H, Takeda M, Konno K, Yamazaki T, Yoshiki S, Suda T. Differentiation of mouse myeloid leukemia cells induced by 1 alpha,25-dihydroxyvitamin D3. Proc Natl Acad Sci U S A. 1981;78:4990–4994. doi: 10.1073/pnas.78.8.4990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mangelsdorf DJ, Koeffler HP, Donaldson CA, Pike JW, Haussler MR. 1,25-Dihydroxyvitamin D3-induced differentiation in a human promyelocytic leukemia cell line (HL-60): receptor-mediated maturation to macrophage-like cells. J Cell Biol. 1984;98:391–398. doi: 10.1083/jcb.98.2.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Studzinski GP, Bhandal AK, Brelvi ZS. A system for monocytic differentiation of leukemic cells HL 60 by a short exposure to 1,25-dihydroxycholecalciferol. Proc Soc Exp Biol Med. 1985;179:288–295. doi: 10.3181/00379727-179-42098. [DOI] [PubMed] [Google Scholar]
- 4.Luong QT, Koeffler HP. Vitamin D compounds in leukemia. J Steroid Biochem Mol Biol. 2005;97:195–202. doi: 10.1016/j.jsbmb.2005.06.017. [DOI] [PubMed] [Google Scholar]
- 5.Pakkala S, de Vos S, Elstner E, Rude RK, Uskokovic M, Binderup L, Koeffler HP. Vitamin D3 analogs: effect on leukemic clonal growth and differentiation, and on serum calcium levels. Leuk Res. 1995;19:65–72. doi: 10.1016/0145-2126(94)00065-i. [DOI] [PubMed] [Google Scholar]
- 6.Koeffler HP, Hirji K, Itri L. 1,25-Dihydroxyvitamin D3: in vivo and in vitro effects on human preleukemic and leukemic cells. Cancer Treat Rep. 1985;69:1399–1407. [PubMed] [Google Scholar]
- 7.Wang X, Studzinski GP. Activation of extracellular signal-regulated kinases (ERKs) defines the first phase of 1,25-dihydroxyvitamin D3-induced differentiation of HL60 cells. J Cell Biochem. 2001;80:471–482. doi: 10.1002/1097-4644(20010315)80:4<471::aid-jcb1001>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 8.Studzinski GP, Wang X, Ji Y, Wang Q, Zhang Y, Kutner A, Harrison JS. The rationale for deltanoids in therapy for myeloid leukemia: role of KSR-MAPK-C/EBP pathway. J Steroid Biochem Mol Biol. 2005;97:47–55. doi: 10.1016/j.jsbmb.2005.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tse AK, Wan CK, Shen XL, Zhu GY, Cheung HY, Yang M, Fong WF. 1,25-dihydroxyvitamin D3 induces biphasic NF-kappaB responses during HL-60 leukemia cells differentiation through protein induction and PI3K/Akt-dependent phosphorylation/degradation of IkappaB. Exp Cell Res. 2007;313:1722–1734. doi: 10.1016/j.yexcr.2007.02.022. [DOI] [PubMed] [Google Scholar]
- 10.Gocek E, Studzinski GP. Vitamin D and differentiation in cancer. Crit Rev Clin Lab Sci. 2009;46:190–209. doi: 10.1080/10408360902982128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hughes PJ, Marcinkowska E, Gocek E, Studzinski GP, Brown G. Vitamin D3-driven signals for myeloid cell differentiation--implications for differentiation therapy. Leuk Res. 2010;34:553–565. doi: 10.1016/j.leukres.2009.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marcinkowska E. Evidence that activation of MEK1,2/erk1,2 signal transduction pathway is necessary for calcitriol-induced differentiation of HL-60 cells. Anticancer Res. 2001;21:499–504. [PubMed] [Google Scholar]
- 13.Wang Q, Wang X, Studzinski GP. Jun N-terminal kinase pathway enhances signaling of monocytic differentiation of human leukemia cells induced by 1,25-dihydroxyvitamin D3. J Cell Biochem. 2003;89:1087–1101. doi: 10.1002/jcb.10595. [DOI] [PubMed] [Google Scholar]
- 14.Wang X, Studzinski GP. Inhibition of p38MAP kinase potentiates the JNK/SAPK pathway and AP-1 activity in monocytic but not in macrophage or granulocytic differentiation of HL60 cells. J Cell Biochem. 2001;82:68–77. doi: 10.1002/jcb.1141. [DOI] [PubMed] [Google Scholar]
- 15.Wang X, Rao J, Studzinski GP. Inhibition of p38 MAP kinase activity up-regulates multiple MAP kinase pathways and potentiates 1,25-dihydroxyvitamin D(3)-induced differentiation of human leukemia HL60 cells. Exp Cell Res. 2000;258:425–437. doi: 10.1006/excr.2000.4939. [DOI] [PubMed] [Google Scholar]
- 16.Manthey CL, Wang SW, Kinney SD, Yao Z. SB202190, a selective inhibitor of p38 mitogen-activated protein kinase, is a powerful regulator of LPS-induced mRNAs in monocytes. J Leukoc Biol. 1998;64:409–417. doi: 10.1002/jlb.64.3.409. [DOI] [PubMed] [Google Scholar]
- 17.Karahashi H, Nagata K, Ishii K, Amano F. A selective inhibitor of p38 MAP kinase, SB202190, induced apoptotic cell death of a lipopolysaccharide-treated macrophage-like cell line, J774.1. Biochim Biophys Acta. 2000;1502:207–223. doi: 10.1016/s0925-4439(00)00045-4. [DOI] [PubMed] [Google Scholar]
- 18.Parmar S, Katsoulidis E, Verma A, Li Y, Sassano A, Lal L, Majchrzak B, Ravandi F, Tallman MS, Fish EN, Platanias LC. Role of the p38 mitogen-activated protein kinase pathway in the generation of the effects of imatinib mesylate (STI571) in BCR-ABL-expressing cells. J Biol Chem. 2004;279:25345–25352. doi: 10.1074/jbc.M400590200. [DOI] [PubMed] [Google Scholar]
- 19.Blank VC, Pena C, Roguin LP. STAT1, STAT3 and p38MAPK are involved in the apoptotic effect induced by a chimeric cyclic interferon-alpha2b peptide. Exp Cell Res. 2010;316:603–614. doi: 10.1016/j.yexcr.2009.11.016. [DOI] [PubMed] [Google Scholar]
- 20.Ji Y, Studzinski GP. Retinoblastoma protein and CCAAT/enhancer-binding protein beta are required for 1,25-dihydroxyvitamin D3-induced monocytic differentiation of HL60 cells. Cancer Res. 2004;64:370–377. doi: 10.1158/0008-5472.can-03-3029. [DOI] [PubMed] [Google Scholar]
- 21.Wang Q, Harrison JS, Uskokovic M, Kutner A, Studzinski GP. Translational study of vitamin D differentiation therapy of myeloid leukemia: effects of the combination with a p38 MAPK inhibitor and an antioxidant. Leukemia. 2005;19:1812–1817. doi: 10.1038/sj.leu.2403916. [DOI] [PubMed] [Google Scholar]
- 22.Gallagher R, Collins S, Trujillo J, McCredie K, Ahearn M, Tsai S, Metzgar R, Aulakh G, Ting R, Ruscetti F, Gallo R. Characterization of the continuous, differentiating myeloid cell line (HL-60) from a patient with acute promyelocytic leukemia. Blood. 1979;54:713–733. [PubMed] [Google Scholar]
- 23.Sundstrom C, Nilsson K. Establishment and characterization of a human histiocytic lymphoma cell line (U-937) Int J Cancer. 1976;17:565–577. doi: 10.1002/ijc.2910170504. [DOI] [PubMed] [Google Scholar]
- 24.Darzynkiewicz Z, Bruno S, Del Bino G, Gorczyca W, Hotz MA, Lassota P, Traganos F. Features of apoptotic cells measured by flow cytometry. Cytometry. 1992;13:795–808. doi: 10.1002/cyto.990130802. [DOI] [PubMed] [Google Scholar]
- 25.Zhang J, Harrison JS, Uskokovic M, Danilenko M, Studzinski GP. Silibinin can induce differentiation as well as enhance vitamin D(3)-induced differentiation of human AML cells ex vivo and regulates the levels of differentiation-related transcription factors. Hematol Oncol. 2009 doi: 10.1002/hon.929. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang J, Posner GH, Danilenko M, Studzinski GP. Differentiation-inducing potency of the seco-steroid JK-1624F2-2 can be increased by combination with an antioxidant and a p38MAPK inhibitor which upregulates the JNK pathway. J Steroid Biochem Mol Biol. 2007;105:140–149. doi: 10.1016/j.jsbmb.2007.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 28.Lee JC, Kassis S, Kumar S, Badger A, Adams JL. p38 mitogen-activated protein kinase inhibitors--mechanisms and therapeutic potentials. Pharmacol Ther. 1999;82:389–397. doi: 10.1016/s0163-7258(99)00008-x. [DOI] [PubMed] [Google Scholar]
- 29.Jiang Y, Chen C, Li Z, Guo W, Gegner JA, Lin S, Han J. Characterization of the structure and function of a new mitogen-activated protein kinase (p38beta) J Biol Chem. 1996;271:17920–17926. doi: 10.1074/jbc.271.30.17920. [DOI] [PubMed] [Google Scholar]
- 30.Frantz B, Klatt T, Pang M, Parsons J, Rolando A, Williams H, Tocci MJ, O’Keefe SJ, O’Neill EA. The activation state of p38 mitogen-activated protein kinase determines the efficiency of ATP competition for pyridinylimidazole inhibitor binding. Biochemistry. 1998;37:13846–13853. doi: 10.1021/bi980832y. [DOI] [PubMed] [Google Scholar]
- 31.Ben-Levy R, Hooper S, Wilson R, Paterson HF, Marshall CJ. Nuclear export of the stress-activated protein kinase p38 mediated by its substrate MAPKAP kinase-2. Curr Biol. 1998;8:1049–1057. doi: 10.1016/s0960-9822(98)70442-7. [DOI] [PubMed] [Google Scholar]
- 32.Clifton AD, Young PR, Cohen P. A comparison of the substrate specificity of MAPKAP kinase-2 and MAPKAP kinase-3 and their activation by cytokines and cellular stress. FEBS Lett. 1996;392:209–214. doi: 10.1016/0014-5793(96)00816-2. [DOI] [PubMed] [Google Scholar]
- 33.Zayzafoon M, Botolin S, McCabe LR. P38 and activating transcription factor-2 involvement in osteoblast osmotic response to elevated extracellular glucose. J Biol Chem. 2002;277:37212–37218. doi: 10.1074/jbc.M200129200. [DOI] [PubMed] [Google Scholar]
- 34.Tindberg N, Porsmyr-Palmertz M, Simi A. Contribution of MAP kinase pathways to the activation of ATF-2 in human neuroblastoma cells. Neurochem Res. 2000;25:527–531. doi: 10.1023/a:1007520311457. [DOI] [PubMed] [Google Scholar]
- 35.Wang X, Studzinski GP. The requirement for and changing composition of the activating protein-1 transcription factor during differentiation of human leukemia HL60 cells induced by 1,25-dihydroxyvitamin D3. Cancer Res. 2006;66:4402–4409. doi: 10.1158/0008-5472.CAN-05-3109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Han J, Lee JD, Jiang Y, Li Z, Feng L, Ulevitch RJ. Characterization of the structure and function of a novel MAP kinase kinase (MKK6) J Biol Chem. 1996;271:2886–2891. doi: 10.1074/jbc.271.6.2886. [DOI] [PubMed] [Google Scholar]
- 37.Wang XS, Diener K, Manthey CL, Wang S, Rosenzweig B, Bray J, Delaney J, Cole CN, Chan-Hui PY, Mantlo N, Lichenstein HS, Zukowski M, Yao Z. Molecular cloning and characterization of a novel p38 mitogen-activated protein kinase. J Biol Chem. 1997;272:23668–23674. doi: 10.1074/jbc.272.38.23668. [DOI] [PubMed] [Google Scholar]
- 38.Jiang Y, Gram H, Zhao M, New L, Gu J, Feng L, Di Padova F, Ulevitch RJ, Han J. Characterization of the structure and function of the fourth member of p38 group mitogen-activated protein kinases, p38delta. J Biol Chem. 1997;272:30122–30128. doi: 10.1074/jbc.272.48.30122. [DOI] [PubMed] [Google Scholar]
- 39.Ono K, Han J. The p38 signal transduction pathway: activation and function. Cell Signal. 2000;12:1–13. doi: 10.1016/s0898-6568(99)00071-6. [DOI] [PubMed] [Google Scholar]
- 40.Treisman R. Regulation of transcription by MAP kinase cascades. Curr Opin Cell Biol. 1996;8:205–215. doi: 10.1016/s0955-0674(96)80067-6. [DOI] [PubMed] [Google Scholar]
- 41.Thompson T, Danilenko M, Vassilev L, Studzinski GP. Tumor suppressor p53 status does not determine the differentiation-associated G(1) cell cycle arrest induced in leukemia cells by 1,25-dihydroxyvitamin D(3) and antioxidants. Cancer Biol Ther. 2010;10:344–350. doi: 10.4161/cbt.10.4.12366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pahl HL, Scheibe RJ, Zhang DE, Chen HM, Galson DL, Maki RA, Tenen DG. The proto-oncogene PU.1 regulates expression of the myeloid-specific CD11b promoter. J Biol Chem. 1993;268:5014–5020. [PubMed] [Google Scholar]
- 43.Tibbles LA, Ing YL, Kiefer F, Chan J, Iscove N, Woodgett JR, Lassam NJ. MLK-3 activates the SAPK/JNK and p38/RK pathways via SEK1 and MKK3/6. EMBO J. 1996;15:7026–7035. [PMC free article] [PubMed] [Google Scholar]
- 44.Brancho D, Ventura JJ, Jaeschke A, Doran B, Flavell RA, Davis RJ. Role of MLK3 in the regulation of mitogen-activated protein kinase signaling cascades. Mol Cell Biol. 2005;25:3670–3681. doi: 10.1128/MCB.25.9.3670-3681.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hartkamp J, Troppmair J, Rapp UR. The JNK/SAPK activator mixed lineage kinase 3 (MLK3) transforms NIH 3T3 cells in a MEK-dependent fashion. Cancer Res. 1999;59:2195–2202. [PubMed] [Google Scholar]
- 46.Shen YH, Godlewski J, Zhu J, Sathyanarayana P, Leaner V, Birrer MJ, Rana A, Tzivion G. Cross-talk between JNK/SAPK and ERK/MAPK pathways: sustained activation of JNK blocks ERK activation by mitogenic factors. J Biol Chem. 2003;278:26715–26721. doi: 10.1074/jbc.M303264200. [DOI] [PubMed] [Google Scholar]
- 47.Zi X, Agarwal R. Modulation of mitogen-activated protein kinase activation and cell cycle regulators by the potent skin cancer preventive agent silymarin. Biochem Biophys Res Commun. 1999;263:528–536. doi: 10.1006/bbrc.1999.1398. [DOI] [PubMed] [Google Scholar]
- 48.Das D, Pintucci G, Stern A. MAPK-dependent expression of p21(WAF) and p27(kip1) in PMA-induced differentiation of HL60 cells. FEBS Lett. 2000;472:50–52. doi: 10.1016/s0014-5793(00)01416-2. [DOI] [PubMed] [Google Scholar]
- 49.Bernardi RJ, Johnson CS, Modzelewski RA, Trump DL. Antiproliferative effects of 1alpha,25-dihydroxyvitamin D(3) and vitamin D analogs on tumor-derived endothelial cells. Endocrinology. 2002;143:2508–2514. doi: 10.1210/endo.143.7.8887. [DOI] [PubMed] [Google Scholar]
- 50.Chen F, Kim E, Wang CC, Harrison LE. Ciglitazone-induced p27 gene transcriptional activity is mediated through Sp1 and is negatively regulated by the MAPK signaling pathway. Cell Signal. 2005;17:1572–1577. doi: 10.1016/j.cellsig.2005.03.012. [DOI] [PubMed] [Google Scholar]
- 51.Cheung CW, Gibbons N, Johnson DW, Nicol DL. Silibinin--a promising new treatment for cancer. Anticancer Agents Med Chem. 2010;10:186–195. doi: 10.2174/1871520611009030186. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig.S1 SB enhances 1,25D-induced expression of the cytoplasmic differentiation marker NSE. HL60 and U937 cells were treated with SB (5 μM) and 1,25D (10 nM) for 48 hours, and the cells positive for NSE were enumerated using a light microscope. The percentage differences of positive cells between treated groups and untreated controls are shown here. Note that the expression of NSE was increased in both cell lines treated with 1,25D, and SB potentiated the effect of 1,25D. Means ± SD are shown; n=3.
Fig. S2 Kinetics of the expression of p38MAPK isoforms in human AML cell lines after exposure to 1,25D for various times. The protein levels of at least two isoforms other than isoform p38MAPKα are enhanced by SB. (A) Western blots showing that in HL60 cells SB upregulated the 1,25D-induced expression of p38MAPKγ and p38MAPKδ, best seen after 48 hours, and that of phosphorylated p38 (all isoforms). Isoform p38MAPKα changed only slightly following the exposure to 1,25D or SB-1,25D. Expression of p38MAPKβ at protein level was not detectable in the HL60 cell strain used for these experiments. (B) Western blots showing that in U937 cells SB upregulated 1,25D-induced expression of p38MAPKγ and p38MAPKδ similar to HL60 cells, although p38MAPKδ expression was relatively low in this cell line. The expression of p38MAPKβ was upregulated by SB only to a minor extent, while the expression of p38MAPKα changed little as a result of 1,25D or SB exposure. The O.D. values are shown below each panel as the ratio of the signal of each band to the corresponding signal for calregulin, the loading control.
Fig.S3 Western blots of kinase assays including the reaction substrate unphosphorylated ATF-2, which serves here as the loading control. When present in the reaction mixture SB202190 inhibited p38MAPKα but not p38MAPKγ or p38MAPKδ kinase activity, determined by “in vitro” kinase reaction in HL60 cells. Lane 5-7 cells were treated, harvested and the protein extracted in exactly the same way as the cells shown in lanes 2-4. The “SB in vitro” indicates that during the kinase reaction, the same concentration of SB was added to the 50 μl reaction buffer. Phosphorylation of ATF-2 was detected by Western blot analysis for activating phosphorylations. The O.D. values are shown as the ratio of the signal of phospho-ATF-2 of each band to the signal of the corresponding loading control, ATF-2.
Fig.S4 Isoforms p38MAPKα and p38MAPKβ contribute to 1,25D-induced cell differentiation. In these experiments AS oligos (final concentration 10 μM) were added 48 hours before the exposure of cells to SB (5 μM) and/or 1,25D (10 nM) and incubated for 72 hours. SC: scrambled oligos, p38a AS: p38MAPKα antisense oligos, p38b AS: p38MAPKβ antisense oligos. (A) HL60 cells. Only AS to p38MAPKα was studied in these cells, as they express very low levels of p38MAPKβ. The expression of CD14 was moderately but significantly reduced by the AS to p38MAPKα in both 1,25D (by 20%) and SB-1,25D (by 14%) treated groups. No significant effect of p38MAPKα AS on CD11b levels was noted in these cells. Means ± SD are shown; n=3. * signifies p<0.05 for reduction of CD14 expression comparing 1,25D with SB-1,25D-treated groups. (B) p38MAPKα or p38MAPKβ AS oligos were added separately to U937 cell cultures. Note that the expression of both cell differentiation markers CD11b and CD14 was markedly and highly significantly reduced by AS oligos in 1,25D and SB-1,25D treated groups. (The reductions were, CD11b and CD14, respectively: SC-D3 vs p38MAPKα AS-D3, 69% and 44%; SC-SB-D3 vs p38MAPKα AS-SB-D3 59% and 44%; SC-D3 vs p38MAPKβ AS-D3 79% and 55%; SC-SB-D3 vs p38MAPKβ AS-SB-D3 78% and 55%).The means ± SD are shown; n=3. ** signifies p<0.01 for reduction of both CD11b and CD14 expression comparing 1,25D with SB-1,25D-treated groups.
Fig.S5 Isoforms p38MAPKγ and p38MAPKδ play a positive role in 1,25D- and SB-1,25D-induced cell differentiation. SC: scrambled oligos, p38g AS: p38MAPKγ antisense oligos, p38d AS: p38MAPKδ antisense oligos. In both HL60 and U937 cells, p38MAPKγ or p38MAPKδ antisense oligos, final concentrations 10 μM) were added 48 hours before exposure to SB and 1,25D. Cells were then treated with SB and 1,25D for 72 hours. Note that the expression of both differentiation markers was reduced by the antisense oligos in 1,25D and SB-1,25D treated groups in both cell lines. The reductions for HL60 cells were, CD11b and CD14, respectively: SC-D3 vs p38MAPKγ AS-D3, 74% and 50%; SC-SB-D3 vs p38MAPKγ AS-SB-D3 70% and 49%; SC-D3 vs p38MAPKδ AS-D3 57% and 40%; SC-SB-D3 vs p38MAPKδ AS-SB-D3 30% and 15%. For U937 cells, SC-D3 vs p38MAPKγ AS-D3, 62% and 55%; SC-SB-D3 vs p38MAPKγ AS-SB-D3 51% and 43%; SC-D3 vs p38MAPKδ AS-D3 51% and 42%; SC-SB-D3 vs p38MAPKδ AS-SB-D3 40% and 40%. Results of one individual experiment for each cell line are presented in this Figure, as combined γ/δ isoforms were studied subsequently, and are shown as text Figure 6.
Fig.S6 Western blots showing that in 1,25D-treated (10 nM, 72h) cells, p38MAPKγ and p38MAPKδ antisense oligos reduced expression of target genes but not of non-target genes such as p38MAPKα.