

Abstract
Peroxisome proliferator-activated receptors (PPARs) play a role in oxidative stress and VEGF regulation, which are closely related to age-related macular degeneration (AMD). PPAR γ expression and its downstream molecules were examined in fat-1 mice (transgenic mice that convert n-6 to n-3 fatty acids), Ccl2-/-/Cx3cr1-/- mice (an AMD model), ARPE19 cells (a human retinal pigment epithelial cell line, RPE, a cell type with a critical role in AMD), and human eyes with and without AMD. PPAR α, β, and γ, VEGF and receptors were determined by immunohistochemistry in the mice models, humans, and ARPE19 cells. Transcripts of PPARs, VEGF, MMP-9 and HO-1 were determined by RQ-PCR. PPARs were constitutively expressed in normal neuroretina and RPE of humans and mice. PPAR γ expression was increased in fat-1 and Ccl2-/-/Cx3cr1-/- mice. VEGF was decreased in fat-1 mice but increased in Ccl2-/-/Cx3cr1-/- mice. VEGF receptors were stable. VEGF, MMP9 and HO-1 transcript levels were increased in ARPE19 cells under H2O2 - induced oxidative stress. Human AMD retinas exhibited higher PPAR γ. The findings of increased expression of PPAR γ and its downstream proteins (VEGF, MMP9, and HO-1) in H2O2-treated ARPE19 cells, Ccl2-/-/Cx3cr1-/- mice, and human AMD eyes, but decreased VEGF in fat-1 mice, suggest that PPAR γ may play a role in AMD.
INTRODUCTION
Age-related macular degeneration (AMD) is the leading cause of irreversible blindness in the elderly [1]. As average lifespan increases, the burden of diseases of aging including AMD continues to rise. AMD was estimated to affect over 1.75 million people in 2004 in the US alone, and threatens to grow to 3 million by 2020 [2]. AMD is classified into two clinical types: “dry” and “wet” AMD - the former is characterized by drusen and macular geographic atrophy of RPE and photoreceptor, while the latter manifests as macular neovascular AMD with choroidal neovascularization. As the demand for therapy increases, much effort is being directed toward the elucidation of the mechanisms underlying AMD pathogenesis. Though the pathophysiology has yet to be fully understood, it is postulated that oxidative stress plays a crucial role in disease development [3-5]. Oxidative damage to retinal pigmented epithelial (RPE) cells and photoreceptors has been implicated in the pathogenesis of AMD. The age-associated incr ease in AMD risk and other well-documented risk factors such as, smoking and diet (low omega-3) might be mediated by cumulative damage to the retina from oxidative stress.
The RPE constitute a monolayer of cuboidal cells beneath the neural retina above the Bruch’s membrane and choroidal capillaries. The RPE is involved in phagocytosis of the outer segments of photoreceptors, the vitamin A cycle, and the blood-retinal barrier [6]. Damage to the RPE at the macular region is an early and crucial event in the development of AMD [1]. Loss of normal physiological function in the aging RPE cells and RPE injury from oxidative stress can contribute to drusen formation, the early hallmark of AMD [3, 7].
Peroxisome proliferator-activated receptors (PPARs) are members of the steroid/thyroid nuclear receptor superfamily of ligand-activated transcription factors [8,9]. There are three subtypes of PPARs: α, β, and γ. PPAR γ has been reported to be expressed in ocular tissue, specifically RPE cells [10,11]. All three subtypes share structural and functional similarities. PPAR-γxs activation was recently shown to mitigate inflammation associated with chronic and acute neurological insults [12]. Recently, PPARs have been associated with age-related changes in Alzheimer’s disease and Parkinson’s disease [13,14]. PPARs are also known to interact with molecules that play a critical role in the progression of AMD either directly or via their downstream pathways [8]. Specifically vascular endothelial growth factor (VEGF), matrix metalloproteases (MMPs, which are capable of degrading extracellular matrix proteins), docosahexanoic acid (DHA), heme-oxidase-1 (HO-1, which serves as an antioxidant by sequestering potentially oxidizing metals) and reactive oxygen species (ROS), are all related to PPARs and AMD-associated processes.
VEGF is recognized as an essential regulator of normal and abnormal vessel growth [15]. VEGF is expressed in response to hypoxia, oncogenes, and cytokines [16], binding to and stimulating the autophosphorylation of two distinct receptor tyrosine kinases, VEGFR1 or Flt-1 (fms-related tyrosine kinase 1) and VEGFR2 or KDR/Flk-1 (kinase insert domain containing receptor/fetal liver kinase 1) [17]. This process activates formation of choroidal neovascularization or development of wet AMD [18,19].
The aim of our study was to investigate the different levels of expression of PPAR molecules among different models related to AMD. In addition, to evaluating PPAR expression in the human retina, we also examined two mouse strains: the fat-1 transgenic mouse and the Ccl2-/-/Cx3cr1-/- mouse. The fat-1 transgenic mouse expresses a C. elegans fat-1 gene encoding an n-3 fatty acid desaturase that converts n-6 to n-3 fatty acids. This is usually absent in mammals and creates a situation in which endogenously produced n-3 fatty acids are in high supply [20]. The Ccl2-/-/Cx3cr1-/- mice are a murine model of AMD [21].
METHODS
Animals
All animal experiments were performed under the protocols approved by the NEI Institutional Animal Care and Use Committee. Mice were treated according to the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research.
Fat-1 mice were generated as reported previously [20]. Briefly, a gene from Caenorhabditis elegans was introduced into F0 C57BL/6 mice to support endogenous conversion of n-6 to n-3 fatty acids. Generation of Ccl2-/-/Cx3cr1-/- mice has been previously described [21, 22]. Briefly, the Ccl2-/- and Cx3cr1-/- mice were bred to obtain homozygous Ccl2-/-/Cx3cr1-/- mice. Wild-type (WT) mice were C57BL/6. All mice were provided food and water ad libitum and kept on a 12 h light-dark cycle.
Cells
Human RPE cells (ARPE-19) were obtained from the American Type Culture Collection (Manassas, VA). This cell line is not transformed and has structural and functional properties characteristic of RPE cells in vivo. The RPE cells were cultured in DMEM/F12 medium (1:1) (Sigma, St Louis, MO, USA) containing 10% fetal bovine serum (Invitrogen Corp, Grand Island, NY), 100 U/mL penicillin G potassium, and 0.1 mg/mL streptomycin sulfate (Sigma, St Louis, MO, USA). The cells were cultured at 37°C in humidified 5% CO2 conditions and split when approximately 90% confluent. The cells propagated rapidly and were used for experiments at passages 1 to 3.
Chemicals
To induce oxidative stress, 1mM, 0.5mM and 0.1mM hydrogen peroxide (H2O2) conditioned medium was made freshly by diluting 30% H2O2 (Fisher Scientific, Fair lawn, NJ, USA) in growth medium just before use.
Quantitative Determination of Cell Viability by MTT Assay
The tetrazolium dye-reduction assay (MTT; 3-[4,5-dimethylthiazol- 2-yl]-2,5-diphenyl tetrazolium bromide) was used to determine cell survival rate with different concentrations of hydrogen peroxide (H2O2), at different time points (in the MTT assay, living cells are defined by functional mitochondrial dehydrogenase) [23]. The modified MTT test was performed for the assessment of cell viability [24]. H2O2, a non-radical of the reactive oxygen species (ROS), is a common oxidant for evaluation of RPE cells, which are under constant ROS flux in vivo [25]. After exposure to the control or one of the test solutions (1mM H2O2, 0.5mM H2O2, 0.1mM H2O2) for the indicated time periods, 96-well cultures were incubated with 50 μg/mL MTT at a dilution of 1:10 based on the volume of culture medium for 4 hours at 37°C. At the end of incubation, the MTT solution was removed, and the cells were dissolved in 200 l DMSO. The proportion of viable cells (those with mitochondria capable of cleaving the MTT molecule to produce the dark purple substance, formazan) was determined by measuring the optical density (OD) of each sample at 570nm with an ELISA plate reader (GE Healthcare, Uppsala, Sweden). For repeat studies, 12 wells were exposed to each solution. The mean optical densities for each group of cultures were compared.
ARPE19 cells of the same passage incubated in DMEM-F12 medium without hydrogen peroxide treatment served as controls.
Immunohistochemistry
The eyes of 4 month old age-matched WT and Ccl2-/- Cx3cr1-/- mice and 14 month old fat-1 mice were snap-frozen. All 5 strains of mice eyes were examined using the avidin-biotin-complex immunoperoxidase (ABC) system. Frozen sections 4 μm thick were fixed in acetone, and immunolabeled with goat anti-mouse PPAR α (Santa Cruz Biotechnology, Inc), goat anti-mouse PPAR β (Santa Cruz Biotechnology, Inc), rabbit anti-mouse PPAR γ (Santa Cruz Biotechnology, Inc), rabbit anti-mouse VEGFR1 (Santa Cruz Biotechnology, Inc), rabbit anti-mouse VEGFR2 (Santa Cruz Biotechnology, Inc) or goat anti-mouse VEGF antibody (R and D Systems, Inc). Biotin labeled goat-anti-rabbit IgG (VECTOR laboratory, Burlingame, CA) or horse anti-goat IgG (VECTOR laboratory, Burlingame, CA) was used as the secondary antibody. The sections were treated using the ABC immunoperoxidase system with 3.3′-diaminobenzidine as the substrate and counterstained with methyl green [21, 22, 26].
ARPE19 cells were plated on Lab-Tek Chamber Slides with eight culture chambers (Intermed Nunc, Naperville, Illinois) treated with H2O2 and stained using the methods and antibodies (PPAR γ and VEGF) at 90% confluency.
Immunohistochemistry analyzing 5 human ocular paraffin-embedded sections from two wet AMD, two dry AMD, and one normal eye was conducted in conformance with the policies and principles stated in the Federal Policy for the Protection of Human Subjects (US Office of Science and Technology Policy) and in the Declaration of Helsinki. Formalin-fixed human ocular paraffin embedded sections were deparaffinized [27, 28]. Detection of PPAR γ on human sections was carried out as described above and repeated at least once in each eye.
For immunofluorescence, sections the frozen sections were fixed briefly in 4% formaldehyde and in 0.1 M glycine, and blocked briefly in 10% serum. The primary anti-VEGF antibody was applied followed by incubation with Alexa 555 rabbit anti-goat IgG (Molecular Probes, Eugene, OR). DNA-binding dye DAPI was used to fluorescently label nuclei (blue color). Slides were examined using a confocal microscope (Leica TCS SP-2, Leica Microsystems, Exton, PA). At least three sections from each eye and two different eyes for each mouse strain were viewed.
Detection of PPAR α, PPAR β, PPAR γ, VEGF, MMP-9, and HO-1 transcripts by quantitative real-time PCR (RQ-PCR)
Ten micrograms of RNA taken from the eyes of 4-month old WT, Ccl2-/- Cx3cr1-/- mice, and 14-month old Fat-1 transgenic mice, were used for cDNA synthesis using RNeasy Mini Kit (Qiagen, Valencia, CA). One million ARPE19 cells were used for RNA isolation using RNeasy Mini Kit (Qiagen, Valencia, CA). RQ-PCR was performed using an Applied Biosystems 7500 Real-Time PCR System and validated and inventoried Taqman gene expression kits (Applied Biosystems, Foster City, CA). The gene expression kits PPAR α (Mm00627559_m1 and Hs00231882_m1), PPAR β (Mm00803186_g1 and Hs00602622_m1), PPAR γ (Mm01184323_m1 and Hs01115513_m1), VEGF (Hs0090 054_m1), MMP-9 (Hs00234579_m1), HO-1 (Hs00157965_m1), Ribosomal protein 29 (Mm02342448_gH), and Ribosomal protein 13 (Hs01945436_u1) were used according to the manufacturer’s instructions. The comparative Ct method was used to establish relative quantification of the fold changes in gene expression according to User Bulletin #2: ABI Prism 7700 Sequence Detection System, PE Applied Biosystems, 1997. Fold changes were normalized first by the level of RSP13 and RSP29, accordingly. The average fold change due to the gene manipulation was again normalized to the transcript level of WT mouse and presented graphically. Each sample was analyzed at least three times.
For detection of murine VEGF mRNA RQ-PCR was performed using a Stratagene Mx3000™ Real-Time PCR System and Brilliant SYBR Green QPCR Master Mix (Stratagene, CA). Primers for VEGF were synthesized by SuperArray and supplied as the RT2 Real-Time™ Gene Expression Assay Kit. Reactions were performed in a final volume of 50 μl with 2 μl of single-stranded cDNA. The RQ-PCR cycling conditions were: 95 °C for 10 min followed by 45 cycles of 30 s at 95 °C, 60 s at 55 °C and 60 s at 72 °C and finally fluorescence measurement. For the internal control, β-actin was amplified using primers 5′-CCCAGCACAATGAAGATCAA-3′ and 5′-ACATCTGCTGGAAGGTGGAC-3′. For the internal control, all PCR conditions were the same as for VEGF except that the annealing temperature was 58 °C. Following PCR, a thermal melt profile was performed for amplicon identification. To determine the Ct, the threshold level of fluorescence was set manually in the early phase of PCR amplification. Each sample was analyzed at least three times. ABI SDS 1.3.1 software and the 10−ΔΔCt/k analysis method were used to determine relative amounts of product using β-actin as an endogenous control. The average fold change was presented graphically.
RESULTS
In vivo Mouse Eyes
1. PPAR γ Expression
PPAR α, β and γ protein are diffusely reactive in the neuroretina and RPE of normal adult mice (Fig. 1). PPAR α and β are expressed equally in the Ccl2-/-/Cx3cr1-/- and WT mice, respectively. Therefore all future experiments focused on PPAR γ. PPAR γ protein and transcript expression is mildly increased in the fat-1 and Ccl2-/-/Cx3cr1-/- mice as compared to the WT.
Fig. (1).

PPAR γ protein expression. PPAR γ (black color) is highest in the Ccl2-/-/Cx3cr1-/- (DKO) group, but still, slightly higher in the Fat-1 than the WT. (ABC staining, original magnification, 100x; R: Retina. C: Choroid).
PPAR γ mRNA levels were higher in the two genetically engineered mouse models than WT. Fat-1 mice had a 1.89 fold increase in PPAR γ transcript expression, whereas the Ccl2-/-/Cx3cr1-/- group had a 2.90 fold higher expression as compared to the WT (Fig. 2). PPAR expression did not change significantly in the mice fed with omega-3 enriched diets compared to those on regular diet (Fig. 2). This is further supported by the fact that the Ccl2-/-/Cx3cr1-/- mice fed with high omega-3 diet had significant reduction of their retinal lesions as compared to those on low omega-3 diet [29]. PPAR α and β transcript levels were also unchanged between Ccl2-/-/Cx3cr1-/- and WT mice.
Fig. (2).

PPAR γ transcript expression. Fat-1 mice expressed PPAR γ mRNA1.89 fold higher than WT mice. The DKO mice exhibited the highest expression of PPAR γ transcript at 2.90 fold higher than WT. PPAR γ transcript levels were not changed significantly in the DKO mice fed with high or low n-3 LCPUFA as compared to those on regular diet.
2. Downstream Effects of PPAR γ - VEGF expression
To evaluate downstream markers of PPAR γ activity we examined VEGF expression and found that VEGF protein expression was slightly lower in the fat-1 mice than in the WT. However, VEGF protein expression was higher in the Ccl2-/-/Cx3cr1-/- group than in the WT mice (Fig. 3A). VEGF receptors 1 and 2 expression was also detected. Levels were similar as compared with that in the WT mice.
Fig. (3).

VEGF expression. (A) VEGF is slightly lower in Fat-1 mice than in WT mice. VEGF protein expression is higher in the DKO mice (red: VEGF, blue: DAPI; original magnification, left and middle panels, 250x, right panel, 400x). (B) VEGF transcript expression supports protein expression. VEGF mRNA is downregulated in the Fat-1 mouse in comparison to the WT mouse (0.78 fold). However, VEGF mRNA is 2.55 fold higher in the DKO than in the WT mice.
VEGF transcript level quantification confirmed these results. VEGF mRNA expression in the fat-1 mice was only 0.78 fold as high as compared to the WT mice, while it was 2.55 fold higher in the DKO than in the WT group (Fig. 3B).
In vitro Human RPE Cells
1. PPAR γ Expression
ARPE-19 cells expressed all three subtypes of PPARs. Baseline expression of PPAR γ in these cells is seen in Fig. (4). After assessing cell viability at different time points and concentrations of H2O2, we found that 70% viability at 0.5mM H2O2 for 2 hours was sufficient. At these conditions it was found that PPAR protein expression mildly increased (Fig. 4). Transcript levels of PPAR γ were again determined. Levels were 8.4 fold higher than in unexposed control cells.
Fig. (4).
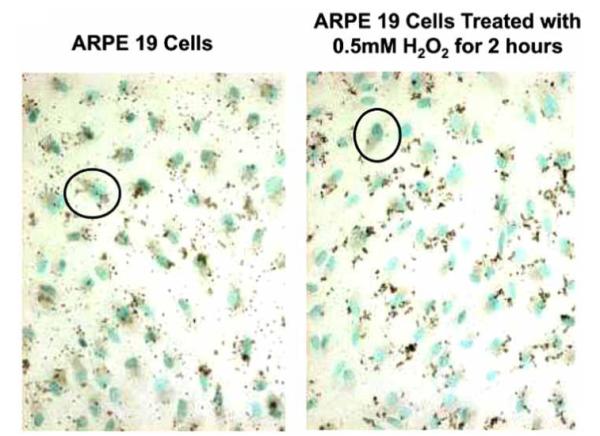
PPAR γ expression. ARPE19 cells exposed to 0.5 mM H2O2 for 2 hours show an increase in PPAR γ expression (black color) as compared to unexposed cells. Positive stain is brown. Nuclei are counterstained with methyl green. (original magnification, 100x).
2. Downstream Effects of PPAR γ
Downstream PPAR γ activity was measured indirectly by assessing VEGF, MMP-9 and HO-1 levels in the H2O2 exposed and unexposed ARPE 19 cells. VEGF protein levels increased after H2O2 exposure (Fig. 5A). For transcript expression assessment, the cells were exposed to lower levels of H2O2 to insure that the compensatory change in the transcript levels could be easily evaluated. A concentration of 0.2 mM H2O2 was chosen based on the positive response. Transcript levels for PPAR γ, VEGF, MMP-9 and HO-1 were all measured (Fig. 5B). At 0.1mM H2O2, compared to unexposed ARPE 19 cells, PPAR γ levels increased slightly (1.2 fold). Consistent with protein findings, VEGF levels increased 3.5 times. MMP-9 levels were 1.7 fold higher than in the exposed cells. HO-1 levels were significantly inc-reased, as expected, to about 23 times higher than in unexposed control cells.
Fig. (5).

VEGF expression. (A) ARPE 19 cells exposed to 0.5 mM H2O2 for 2 hours show an increase in VEGF protein expression (black color) as compared to unexposed cells. Positive is in brown. Nuclei are counterstained with methyl green. (original magnification, 100x). (B) Transcript expression of PPAR γ, VEGF, MMP-9 and HO -1 in ARPE 19 cells exposed to 0.1 mM H2O2 for 2 hours. PPAR γ is 1.2 fold increased. VEGF is 3.5 times higher while MMP-9 is 1.7 times higher than unexposed cells. HO-1 is 23 fold increased over unexposed ARPE19 cells.
In vivo Human Eyes
Positive immunoreactivity against PPAR γ was found in normal human retina (Fig. 6). Retinas from humans with both wet and dry AMD show an increased expression of PPAR γ as compared to retinas from age-matched normal eyes (Fig. 6). PPAR γ expression within the retina illustrates the same distribution as seen in the mouse models.
Fig. (6).

Retinas from humans with both dry and wet AMD express PPAR γ (black color) at a higher level than retinas taken from age-matched controls. (ABC stain, original magnification, 100x).
DISCUSSION
We found that PPAR α, β and γ were diffusely expressed in normal neuroretina and RPE of humans and mice. Interestingly, PPAR γ was higher in the Ccl2-/-/Cx3cr1-/- mice with retinal AMD-like lesions as well as in the fat-1 mouse with high endogenous n-3 fatty acids. Although VEGF was significantly higher in the Ccl2-/-/Cx3cr1-/- mice, it was lower in the fat-1 mice as compared to normal WT controls. In contrast, VEGFR1 and VEGFR2 levels were relatively stable in the fat-1 and Ccl2-/-/Cx3cr1-/- mice.
Under normal condition, there is a low level of PPAR γ expression in ARPE19 cells. PPAR γ and its downstream pathway products of VEGF, MMP9 and HO-1 transcript levels were also increased in H2O2 stressed ARPE19 cells as compared to the unstressed cells. Findings in human AMD retinas and controls exhibited high levels of PPAR supporting the above results.
PPARs are members of the steroid/thyroid nuclear receptor superfamily of ligand-activated transcription factors [9, 10]. They can be activated either exogenously by fatty acid derivatives or endogenously by prostaglandin derivatives that complex with the PPARs and translocate to the nucleus where they form heterodimers with retinoid-X receptors (RXR). The entire complex is activated to bind the target DNA at a specific region called the PPAR Response Elements (PREs). This area is usually located in an enhancer or promoter sequence of the target gene. Once bound, transcription is enabled [10]. PPARγ and RXRα form a non-symmetric complex, allowing the ligand-binding domain (LBD) of PPARγ to contact multiple domains in both proteins. The PPARγ LBD cooperates with both DNA-binding domains to enhance response-element binding. Recently, crystal structures of two related complexes have demonstrated that distinct ligands can modulate the multiple domain-domain interfaces [30].
PPAR α is encoded on chromosome 22q12 and expressed in brown adipose tissue, liver, kidney, heart, central nervous system (CNS), and skeletal muscle. The gene for PPAR β, also known as PPAR δ and NUC1, is on chromosome 6p12. The protein is expressed in the intestines, kidney, liver and CNS. The PPAR γ gene is found on chromosome 3p25. PPAR γ is expressed in adipocytes, colon, renal epithelial cells, monocytes, macrophages, CNS, and RPE cells [10, 11]. All three subtypes share structural and functional similarities. PPAR α and γ are involved in lipid and glucidic metabolism, immune regulation, cell differentiation and apoptosis.
Since PPAR is known to have an anti-inflammatory effect and plays a role in down regulating factors that increase damage associated with AMD [8], PPAR γ may be a protective factor, qualifying its upregulation in the diseased mouse model as a compensatory mechanism. Upregulation was seen in the Ccl2-/-/Cx3cr1-/- mice that are under oxidative stress [31], the ARPE 19 cells exposed to H2O2, and in the retinas from humans with AMD. Indeed, inflammatory markers (chemokines and macrophages) have been found in the Ccl2-/-/Cx3cr1-/- mice [22] and human AMD eyes [1, 32, 33].
Long chain polyunsaturated fatty acids, LCPUFAs, specifically DHA, are major dietary fatty acids. Importantly these omega-3 fatty acids are a major structural lipid of the retinal photoreceptor outer segment and play a role in neovascularization and inflammation [4]. Though the amount of dietary intake is the limiting resource for turnover of the photoreceptor, PPARs promote lipid mobilization for the restructuring of these cell membranes. This decreases lasting damage from oxidative stress. PPARs are known to upregulate antioxidant genes, such as HO-1, and further inhibit damage to RPE [34].
Transgenic fat-1 mice produce endogenous n-3 PUFA [35], which increase PPAR expression and enhance the protective effects on their downstream molecules. LCPUFAs are present in high concentrations in the retina and cell membrane integrity is dependant on the ability to turnover these lipids, thus, counteracting environmental damage. Humans are dependent on dietary intake of these LCPUFAs since we are unable to produce them de novo [36-38]. The fat-1 model serves to demonstrate what diet modification with supplementation of n-3 fatty acids can achieve. AMD patients on n-3 fatty acids rich diet are reported to slow their disease progression in 6 well- established clinical studies [4]. Moreover, PPAR γ can regulate lipid mobilization, and with the endogenous n-3 fatty acids, we have found active PPAR γ in this model.
An increase in PPAR γ should decrease VEGF expression in the retina. Indeed, increased levels of PPAR γ correlate with decreased VEGF protein and transcript levels in the fat-1 mice. However, the comparatively mild compensatory increase in the PPAR γ in Ccl2-/-/Cx3cr1-/- mice was insufficient to suppress the elevated levels of VEGF. PPAR γ is not the only factor regulating levels of VEGF. The findings that the Ccl2-/-/Cx3cr1-/- were unable while fat-1 mice were able to normalize levels of VEGF suggest that normal PPAR pathway regulation of VEGF levels may be overwhelmed in diseased conditions.
MMPs, zinc-dependent endopeptidases, are important in the resorption of extracellular matrixes in both physiological and pathological processes [39]. MMP-9, a specific MMP, is thought to degrade the fibrinous cap found on atherosclerotic plaques, destabilizing the plaque, and priming it for rupture [40]. Since AMD is associated with sustained chronic inflammation and loss of integrity of Bruch’s membrane, it has been hypothesized that MMPs may play a role in the pathogenesis of the disease [41-44]. Moreover, it is reported that natural as well as synthetic PPARγ ligands act to inhibit the expression of AP-1, STAT-1, and NF-κB thereby indirectly decreasing levels of MMP-9 [45]. Consistent with findings for VEGF, ARPE19 cells produce higher level of MMP-9 under H2O2 exposure.
HO-1 transcript levels were markedly elevated in the ARPE19 cells exposed to hydrogen peroxide. This is expected since this is an antioxidant gene, upregulated by PPAR, as well as by the stress induced from the H2O2. The increase should then aid in the protective effects of PPAR γ.
Through inhibition of VEGF expression, downregulation of MMP-9, induction of antioxidant genes and increased lipid mobilization, PPARs can be hypothesized to decrease the impetus for progression of AMD. Here we found that PPARs are constitutively expressed throughout the retina. PPAR γ is mildly increased in the Ccl2-/-Cx3cr1-/- mouse AMD model as a possible protective mechanism. High PPAR γ expression down-regulates VEGF in the fat-1 mice, which are capable of endogenous production of n-3 fatty acids. Findings in human AMD and normal retinas also depict the same patterns.
ACKNOWLEDGEMENT
The fat-1 mice were obtained from Jing X. Kang, PhD., Department of Medicine, Massachusetts General Hospital & Harvard Medical School, Boston, MA. The NEI Intramural Research Program and American Health Assistance Foundation funded this study.
REFERENCES
- [1].Coleman HR, Chan CC, Ferris FL, et al. Age-related macular degeneration. Lancet. 2008;372(9652):1835–45. doi: 10.1016/S0140-6736(08)61759-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Friedman DS, O’Colmain BJ, Muñoz B, et al. Prevalence of age-related macular degeneration in the United States. Arch Ophthalmol. 2004;122(4):564–72. doi: 10.1001/archopht.122.4.564. [DOI] [PubMed] [Google Scholar]
- [3].De Jong PTVM. Age-related macular degeneration. N Engl J Med. 2006;355(14):1474–85. doi: 10.1056/NEJMra062326. [DOI] [PubMed] [Google Scholar]
- [4].SanGiovanni JP, Chew EY. The role of omega-3 long-chain polyunsaturated fatty acids in health and disease of the retina. Prog Retia Eye Res. 2005;24(1):87–138. doi: 10.1016/j.preteyeres.2004.06.002. [DOI] [PubMed] [Google Scholar]
- [5].Winkler BS, Boulton ME, Gottsch JD, Sternberg P. Oxidative damage and age-related macular degeneration. Mol Vis. 1999;5(1):32–4. [PMC free article] [PubMed] [Google Scholar]
- [6].Strauss O. The retinal pigment epithelium in visual function. Physiol Rev. 2005;85(1):845–81. doi: 10.1152/physrev.00021.2004. [DOI] [PubMed] [Google Scholar]
- [7].Ding X, Patel M, Chan CC. Molecular pathology of age-related macular degeneration. Prog Retia Eye Res. 2009;28(1):1–18. doi: 10.1016/j.preteyeres.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Herzlich AA, Tuo J, Chan CC. The role of PPARs in AMD. PPAR Res. 2008:389507. doi: 10.1155/2008/389507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Kersten S, Desvergne B, Wahli W. Roles of PPARs in health and disease. Nature. 2000;405(6785):421–4. doi: 10.1038/35013000. [DOI] [PubMed] [Google Scholar]
- [10].Auwerx J. PPAR γ, the ultimate thrifty gene. Diabetologia. 1999;42(9):1033–49. doi: 10.1007/s001250051268. [DOI] [PubMed] [Google Scholar]
- [11].Reiss AB, Vagell ME. PPAR γ activity in the vessel wall: anti-atherogenic properties. Curr Med Chem. 2006;13(26):3227–38. doi: 10.2174/092986706778742909. [DOI] [PubMed] [Google Scholar]
- [12].Kapadia R, Yi JH, Vemuganti R. Mechanisms of anti-inflammatory and neuroprotective actions of PPAR-gamma agonists. Front Biosci. 2008;1(13):1813–26. doi: 10.2741/2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Perl DP. Neuropathology of Alzheimer’s disease and related disorders. Neurol Clin. 2000;18(4):847–64. doi: 10.1016/s0733-8619(05)70229-2. [DOI] [PubMed] [Google Scholar]
- [14].Forno LS. Neuropathology of Parkinson’s disease. J Neuropathol Exp Neurol. 1996;55(3):259–72. doi: 10.1097/00005072-199603000-00001. [DOI] [PubMed] [Google Scholar]
- [15].Ferrara N, Kerbel RS. Angiogenesis as a therapeutic target. Nature. 2005;438(7070):967–74. doi: 10.1038/nature04483. [DOI] [PubMed] [Google Scholar]
- [16].Vinores SA, Xiao W-H, Aslam S, et al. Implication of the hypoxia response element of the VEGF promoter in mouse models of retinal and choroidal neovascularization, but not retinal vascular development. J Cell Physiol. 2006;206(3):749–58. doi: 10.1002/jcp.20525. [DOI] [PubMed] [Google Scholar]
- [17].Mustonen T, Alitalo K. Endothelial receptor tyrosine kinases involved in angiogenesis. J Cell Biol. 1995;129(4):895–8. doi: 10.1083/jcb.129.4.895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Kroll J, Waltenberger J. The vascular endothelial growth factor receptor KDR activates multiple signal transduction pathways in porcine aortic endothelial cells. J Biol Chem. 1997;272(51):32521–7. doi: 10.1074/jbc.272.51.32521. [DOI] [PubMed] [Google Scholar]
- [19].Parenti A, Morbidelli L, Cui XL, et al. Nitric oxide is an upstream signal of vascular endothelial growth factor-induced extracellular signal-regulated kinase (1/4) activation in postcapillary endothelium. J Biol Chem. 1998;273(7):4220–6. doi: 10.1074/jbc.273.7.4220. [DOI] [PubMed] [Google Scholar]
- [20].Kang JX, Wang J, Wu L, Kang ZB. Transgenic mice: fat-1 mice convert n-6 to n-3 fatty acids. Nature. 2004;427(6974):504. doi: 10.1038/427504a. [DOI] [PubMed] [Google Scholar]
- [21].Tuo J, Bojanowski CM, Zhou M, et al. Murine Ccl2/Cx3cr1 deficiency results in retinal lesions mimicking human age-related macular degeneration. Invest Ophthalmol Vis Sci. 2007;48(1):3827–36. doi: 10.1167/iovs.07-0051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Ross RJ, Zhou M, Shen D, et al. Immunological protein expression profile in ccl2/cxcr1 deficient nice with lesions similar to age-related macular degeneration. Exp Eye Res. 2008;86(4):675–83. doi: 10.1016/j.exer.2008.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Huet O, Petit JM, Ratinaud MH, Julien R. NADH-dependent dehydrogenase activity estimation by flow cytometric analysis of 3-(4,5-dimethylthiazolyl-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reduction. Cytometry. 1992;13(1):532–9. doi: 10.1002/cyto.990130513. [DOI] [PubMed] [Google Scholar]
- [24].Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Meth. 1983;65(1):55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- [25].Beatty S, Koh H, Henson D, Boulton M. The role of oxidative stress in the pathogenesis of age-related macular degeneration. Surv Ophthalmol. 2000;45(2):115–34. doi: 10.1016/s0039-6257(00)00140-5. [DOI] [PubMed] [Google Scholar]
- [26].Chan CC, Ross RJ, Shen D, et al. Ccl2/Cx3cr1-deficient mice: an animal model for age-related macular degeneration. Ophthalmic Res. 2008;40(1):124–8. doi: 10.1159/000119862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Chan CC, Tuo J, Bojanowski CM, Csaky KG, Green WR. Detection of CX3CR1 single nucleotide polymorphism and expression on archived eyes with age-related macular degeneration. Histol Histopathol. 2005;20(1):857–63. doi: 10.14670/hh-20.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Tuo J, Ning B, Bojanowski CM, et al. Synergic effect of polymorphisms in ERCC6 5′ flanking region and complement factor H on age-related macular degeneration predisposition. Proc Natl Acad Sci USA. 2006;103(1):9256–61. doi: 10.1073/pnas.0603485103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Tuo J, Ross RJ, Herzlich AA, et al. A high omega-3 fatty acid diet reduces lesions in a murine model of macular degeneration. Am J Pathol. 2009;175(1):799–807. doi: 10.2353/ajpath.2009.090089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Chandra V, Huang P, Hamuro Y, et al. Structure of the intact PPAR-gamma-RXR-nuclear receptor on DNA. Nature. 2008;456(7220):350–56. doi: 10.1038/nature07413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Ding X, Patel M, Shen D, et al. Enhanced HtrA2/Omi expression in oxidative injury to retinal pigment epithelial cells and murine models of neurodegeneration. Invest Ophthalmol Vis Sci. 2009;50:4957–66. doi: 10.1167/iovs.09-3381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Dastgheib K, Green WR. Granulomatous reaction to Bruch’s membrane in age-related macular degeneration. Arch Ophthalmol. 1994;112(6):813–8. doi: 10.1001/archopht.1994.01090180111045. [DOI] [PubMed] [Google Scholar]
- [33].Penfold PL, Killingsworth MC, Sarks SH. Senile macular degeneration: the involvement of immunocompetent cells. Graefes Arch Clin Exp Ophthalmol. 1985;223(2):69–76. doi: 10.1007/BF02150948. [DOI] [PubMed] [Google Scholar]
- [34].Qin S, McLaughlin AP, De Vries GW. Protection of RPE cells from oxidative injury by 15-deoxy-Δ-12,14-prostaglandin J2 by augmenting GSH and activating MAPK. Invest Ophthalmol Vis Sci. 2006;47(11):5098–105. doi: 10.1167/iovs.06-0318. [DOI] [PubMed] [Google Scholar]
- [35].Kang JX. Fat-1 transgenic mice: a new model for omega-3 research. Prostaglandins Leukot Essent Fatty Acids. 2007;77(5-6):263–7. doi: 10.1016/j.plefa.2007.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Lands WM. Dietary fat and health: the evidence and the politics of prevention: careful use of dietary fats can improve life and prevent disease. Ann NY Acad Sci. 2005;1055(1):179–92. doi: 10.1196/annals.1323.028. [DOI] [PubMed] [Google Scholar]
- [37].Hibbeln JR. Healthy intakes of n—3 and n—6 fatty acids: estimations considering worldwide diversity. Am J Clin Nutr. 2006;83(6):1483S–93S. doi: 10.1093/ajcn/83.6.1483S. [DOI] [PubMed] [Google Scholar]
- [38].Okuyama H, Ichikawa Y, Sun Y, Hamazaki T, Lands WM. ω3 fatty acids effectively prevent coronary heart disease and other late-onset diseases: the excessive linoleic acid syndrome. World Rev Nutr Diet. 2007;96(1):83–103. doi: 10.1159/000097809. [DOI] [PubMed] [Google Scholar]
- [39].Folgueras AR, Pendás AM, Sánchez LM, López-Otín C. Matrix metalloproteinases in cancer: from new functions to improved inhibition strategies. Int J Dev Biol. 2004;48(5-6):411–24. doi: 10.1387/ijdb.041811af. [DOI] [PubMed] [Google Scholar]
- [40].Marx N, Froehlich J, Siam L, et al. Antidiabetic PPAR γ-activator rosiglitazone reduces MMP-9 serum levels in type 2 diabetic patients with coronary artery disease. Arterioscler Thromb Vasc Biol. 2003;23(2):283–8. doi: 10.1161/01.atv.0000054195.35121.5e. [DOI] [PubMed] [Google Scholar]
- [41].Collins AR, Meehan WP, Kintscher U, et al. Troglitazone inhibits formation of early atherosclerotic lesions in diabetic and nondiabetic low density lipoprotein receptor-deficient mice. Arterioscler Thromb Vasc Biol. 2001;21(3):365–71. doi: 10.1161/01.atv.21.3.365. [DOI] [PubMed] [Google Scholar]
- [42].Chau KY, Patel N. Plasma levels of matrix metalloproteinase - 2 and -9 in age-related macular degeneration. Eye. 2007;21(12):1511–5. doi: 10.1038/sj.eye.6702722. [DOI] [PubMed] [Google Scholar]
- [43].Guo L, Hussain AA. Age-dependent variation in metalloproteinase activity of isolated human Bruch’s membrane and choroid. Invest Ophthalmol Vis Sci. 1999;40(1):2676–82. [PubMed] [Google Scholar]
- [44].Lambert V, Wielockx B. MMP-2 and MMP-9 synergize in promoting choroidal neovascularization. FASEB J. 2003;17(1):2290–2. doi: 10.1096/fj.03-0113fje. [DOI] [PubMed] [Google Scholar]
- [45].Ricote M, Li A. The peroxisome proliferator-activated receptor γ is a negative regulator of macrophage activation. Nature. 1998;391(1):79–82. doi: 10.1038/34178. [DOI] [PubMed] [Google Scholar]