

Abstract
The range of possible gene interactions in a multilocus model of a complex inherited disease is studied by exploring genotype-specific risks subject to the constraint that the allele frequencies and marginal risks are known. We quantify the effect of gene interactions by defining the interaction ratio, 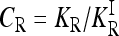 , where KR is the recurrence risk to relatives with relationship R for the true model and
, where KR is the recurrence risk to relatives with relationship R for the true model and 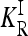 is the recurrence risk to relatives for a multiplicative model with the same marginal risks. We use a Markov chain Monte Carlo (MCMC) procedure to sample from the space of possible models. We find that the average of CR increases with the number of loci for both low frequency (p = 0.03) and higher frequency (p = 0.25) causative alleles. Furthermore, the probability that CR > 1 is nearly 1. Similar results are obtained when more weight is given to risk models that are closer to the comparable multiplicative model. These results imply that, in general, gene interactions will result in greater heritability of a complex inherited disease than is expected on the basis of a multiplicative model of interactions and hence may provide a partial explanation for the problem of missing heritability of complex diseases.
is the recurrence risk to relatives for a multiplicative model with the same marginal risks. We use a Markov chain Monte Carlo (MCMC) procedure to sample from the space of possible models. We find that the average of CR increases with the number of loci for both low frequency (p = 0.03) and higher frequency (p = 0.25) causative alleles. Furthermore, the probability that CR > 1 is nearly 1. Similar results are obtained when more weight is given to risk models that are closer to the comparable multiplicative model. These results imply that, in general, gene interactions will result in greater heritability of a complex inherited disease than is expected on the basis of a multiplicative model of interactions and hence may provide a partial explanation for the problem of missing heritability of complex diseases.
ALTHOUGH many genome-wide association studies (GWAS) have been performed and have found hundreds of SNPs associated with higher risk of complex inherited diseases, those SNPs so far account for only a small fraction of the inherited risk of those diseases (Altshuler et al. 2008). Several not mutually exclusive explanations have been proposed for the “missing heritability,” i.e., the heritability that is not yet accounted for by SNPs found in GWAS (Manolio et al. 2009): (i) common alleles of small effect that have not been found because GWAS done so far have been underpowered, (ii) low-frequency alleles of moderate effect that are difficult to find using HapMap SNPs, (iii) rare copy-number variants that are not in strong linkage disequilibrium (LD) with HapMap SNPs, (iv) inherited epigenetic factors that are not in strong LD with HapMap SNPs, and (v) interactions among causative alleles that conceal their true contribution to heritability. In this article we investigate the last possibility and determine the extent to which interactions may account for missing heritability.
Our analysis is in the same spirit as that of Culverhouse et al. (2002). We assume that the risk of being affected by a complex disease is determined by an individual's genotype at two or more loci and that the frequencies of causative alleles and the average risks for each one-locus genotype (the marginal risks) are known. Culverhouse et al. (2002) assumed the marginal risks were the same for all genotypes and all loci. In that case, causative alleles have odds ratios of 1; they contribute to risk only through their interactions. Culverhouse et al. found the risk function that maximized the heritability and showed that the maximum possible heritability attributable to interactions increased with the number of loci. They concluded that it is quite possible that interactions among loci that have no main effect could contribute substantially to the heritability of a complex disease and indeed could account for “virtually all the variation in affection status for diseases with any prevalence” (Culverhouse et al. 2002, p. 468).
We generalize the analysis of Culverhouse et al. in three ways. First, we allow causative alleles to have odds ratios >1. Second, we explore the entire space of models instead of focusing only on the risk model that maximizes heritability. Third, we examine how the importance of gene interactions depends on the “distance” between a risk model and a comparable multiplicative model. We show that gene interactions can substantially increase the heritability of risk as measured by recurrence risk, KR, and that the effect increases with the number of loci carrying causative alleles. Furthermore, we show that these results are true even if more weight is given to models that are closer to a comparable multiplicative model.
Geometrically, the space of feasible genotype-specific risks subject to the aforementioned constraints (i.e., that the allele frequencies and marginal risks are known) corresponds to a high-dimensional convex polytope, and the computational problem of interest involves integrating a quadratic function over the polytope. The dimension of the polytope grows exponentially with the number of loci, and, therefore, analytic computation is intractable for more than two loci. Hence, we devise a Monte Carlo approach to tackle the problem. Note that, because of high dimensionality, rejection algorithms are not appropriate for this kind of problem. We instead employ a Markov chain Monte Carlo (MCMC) algorithm based on a random walk that always stays inside the polytope. We present empirical results for up to five loci and obtain a closed-form formula for the minimum of KR over the polytope; the latter result applies to an arbitrary number of loci. Interestingly, the minimum of KR decreases as the number L of loci increases, but the average of KR over the polytope increases with L.
MULTILOCUS MODEL OF RISK
Model constraints:
We assume that the risk of an individual being affected by a dichotomous complex disease depends on the genotype at L diallelic loci. We use f(k) to denote the probability that an individual with the L-locus genotype k = (k1, … , kL) is affected, where ki = 0, 1, 2 indicates the number of copies of the higher-risk allele (denoted by Ai) at locus i. Note that 0 ≤ f(k) ≤ 1 for all genotypes k. We assume that the frequency pi of Ai in a population is known. Further, we assume that the loci are unlinked and are in Hardy–Weinberg and linkage equilibrium.
The average risk in the population is
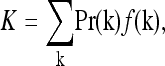 |
(1) |
where Pr(k) is the probability of genotype k in the population and the sum is over all genotypes. The marginal risks for each one-locus genotype are obtained by averaging over the other loci:
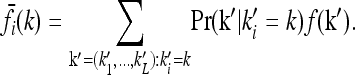 |
(2) |
In this article, we assume that these one-locus marginal risks 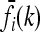 are known for each locus i and genotype k. Necessarily,
are known for each locus i and genotype k. Necessarily,
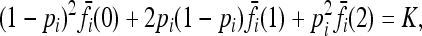 |
(3) |
for all loci i.
Recurrence risk and the induced multiplicative model:
To characterize a risk model, we begin with the recurrence risk, KR, which is the risk of the disease to a relative with relationship R of an individual that has the disease. Risch's (1990) recurrence risk ratio, λR, is defined as KR/K. The increase in KR over K indicates the effect of causative alleles shared by the two relatives because of identity by descent. In our notation, the recurrence risk corresponding to a risk model 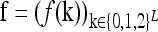 is given by
is given by
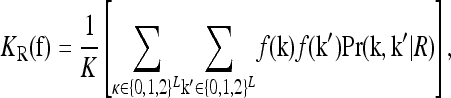 |
(4) |
where Pr(k, k′ | R) denotes the joint probability of the L-locus genotypes of two relatives with relationship R. In this article, we are concerned with the recurrence risk in full siblings, parents and offspring, half siblings, and first full cousins. Since we assume that all L loci are unlinked, the joint probability can be decomposed as
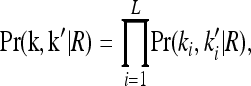 |
where 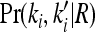 is the marginal joint probability for locus i. See Liu and Weir (2005) for details on computing
is the marginal joint probability for locus i. See Liu and Weir (2005) for details on computing 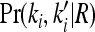 for various relationships.
for various relationships.
We define for any risk model the multiplicative model with the same average risk K and marginal risks 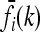 . We call this model the induced multiplicative model. It satisfies
. We call this model the induced multiplicative model. It satisfies
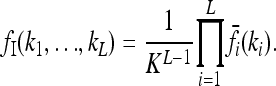 |
(5) |
The recurrence risk for the induced model 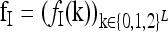 is denoted by
is denoted by
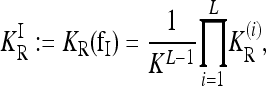 |
(6) |
where the one-locus recurrence risk 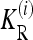 for locus i is defined as
for locus i is defined as
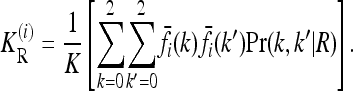 |
(7) |
The quantity we use to characterize the deviation of a risk model f from the induced multiplicative model is the interaction ratio
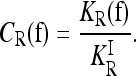 |
For a multiplicative model, CR = 1. If CR < 1, then KR is smaller than expected under a comparable multiplicative model. In that case, assuming a multiplicative model would overestimate the actual heritability of risk as measured by KR. If CR > 1, assuming a multiplicative model would underestimate the actual heritability of risk, in which case gene interactions are concealing some of the heritability.
The space of disease risk models:
In what follows, we provide a geometric description of the space of disease risk models that are consistent with given constraints. Since f(k) ∈ [0,1] for all k, the 3L-tuple 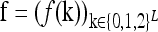 takes value in the 3L-dimensional unit hypercube, denoted Y. Note that different points in Y correspond to different disease risk models.
takes value in the 3L-dimensional unit hypercube, denoted Y. Note that different points in Y correspond to different disease risk models.
For each locus i ∈ {1, …, L} and genotype k ∈ {0, 1, 2}, Equation 2 relates the disease risk function f to the marginal risk 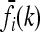 . More explicitly, we have
. More explicitly, we have
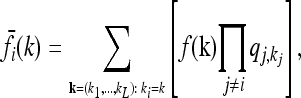 |
(8) |
where 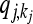 denotes the equilibrium frequency of genotype kj at locus j, given by
denotes the equilibrium frequency of genotype kj at locus j, given by
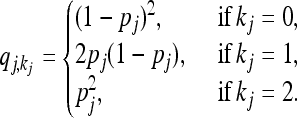 |
Note that the 3L equations in (8) are not all independent, since, for each i ∈ {1, …, L},
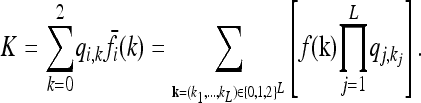 |
(9) |
Now, suppose that K, pi, and 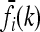 are given, for each locus i and genotype k. Then, (8) and (9) define 2L + 1 linearly independent affine hyperplanes H1, …, H2L+1 in the unit hypercube Y, and the intersection of those hyperplanes restricted to Y defines the space Δ of disease risk models consistent with the given information; i.e.,
are given, for each locus i and genotype k. Then, (8) and (9) define 2L + 1 linearly independent affine hyperplanes H1, …, H2L+1 in the unit hypercube Y, and the intersection of those hyperplanes restricted to Y defines the space Δ of disease risk models consistent with the given information; i.e.,
 |
Note that Δ is a convex polytope of dimension 3L − 2L − 1 embedded in the 3L-dimensional Y. The induced model with the risk function fI(k) shown in (5) is consistent with the given constraints (8) and (9), and it corresponds to a particular point in the polytope Δ.
In our study, instead of specifying the marginal risks 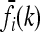 directly, we assume a multiplicative model within each locus and specify the genotype relative risk GRRi for each locus i. Under this assumption, the marginal risks are related by
directly, we assume a multiplicative model within each locus and specify the genotype relative risk GRRi for each locus i. Under this assumption, the marginal risks are related by 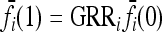 and
and 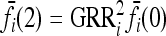 , and the first equality in (9) implies
, and the first equality in (9) implies
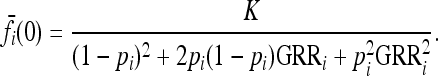 |
(10) |
Since 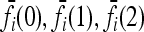 are linear in K, note that
are linear in K, note that 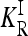 defined in (6) is proportional to K.
defined in (6) is proportional to K.
Sampling from the polytope:
The main computational problem involved in our work concerns computing the expectation 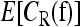 with respect to the uniform distribution over the polytope Δ. The dimension of the polytope grows exponentially with the number of loci, rendering analytic integration intractable for more than two loci. Hence, we employ a Markov chain Monte Carlo algorithm to estimate the expectation.
with respect to the uniform distribution over the polytope Δ. The dimension of the polytope grows exponentially with the number of loci, rendering analytic integration intractable for more than two loci. Hence, we employ a Markov chain Monte Carlo algorithm to estimate the expectation.
Over the past 20 years, there have been a series of theoretical developments (e.g., see Dyer et al. 1991; Kannan et al. 1997; Lovász and Vempala 2006) on fully polynomial-time randomized approximation schemes for computing the volume of convex bodies in 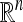 . The key component of these algorithms concerns the problem of sampling points uniformly at random from convex bodies. The convexity property implies that one can devise an MCMC algorithm with acceptance rate 1. In our work, we employed the hit-and-run sampling algorithm (Smith 1984; Lovász 1999), which goes as follows: Suppose that the current state in the Markov chain is xt ∈ Δ. To sample the next state, choose a direction uniformly at random and move uniformly along that direction, restricted to Δ. The point after the move is the next state xt+1 ∈ Δ.
. The key component of these algorithms concerns the problem of sampling points uniformly at random from convex bodies. The convexity property implies that one can devise an MCMC algorithm with acceptance rate 1. In our work, we employed the hit-and-run sampling algorithm (Smith 1984; Lovász 1999), which goes as follows: Suppose that the current state in the Markov chain is xt ∈ Δ. To sample the next state, choose a direction uniformly at random and move uniformly along that direction, restricted to Δ. The point after the move is the next state xt+1 ∈ Δ.
The mixing time of the hit-and-run algorithm has been shown to be O*(n3), after some appropriate preprocessing (Lovász 1999), where n denotes the dimension of the ambient space. We remark that there are at least two sources of complication in applying this complexity result in practice. First, it is an asymptotic result and the O* notation actually hides a large constant, as well as the dependence on error parameters. Second, in the application we are considering, the dimension n grows exponentially with the number L of loci. Hence, to check convergence, we took an empirical approach, by keeping track of the running average of CR (f).
For L = 2, 3, 4, we ran our sampler for 20 million iterations, taking samples every 20 iterations to compute the expectation 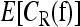 . For L = 5, we used 30 million iterations, again taking samples every 20 iterations to compute
. For L = 5, we used 30 million iterations, again taking samples every 20 iterations to compute 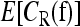 . For a few parameter settings, we tried using 100 million iterations and obtained results very close to that for 30 million iterations. In all runs, we started the chain at the induced multiplicative model fI. The computation was done using a single core of a Mac Pro with two 3.0-GHz Quad-Core Intel Xeon processors, and the running time for a given parameter setting was 88 sec for L = 2, 822 sec for L = 3, 2.2 hr for L = 4, and 28.3 hr for L = 5.
. For a few parameter settings, we tried using 100 million iterations and obtained results very close to that for 30 million iterations. In all runs, we started the chain at the induced multiplicative model fI. The computation was done using a single core of a Mac Pro with two 3.0-GHz Quad-Core Intel Xeon processors, and the running time for a given parameter setting was 88 sec for L = 2, 822 sec for L = 3, 2.2 hr for L = 4, and 28.3 hr for L = 5.
RESULTS
Random sampling of risk models:
With L diallelic loci, there are 3L genotypes and hence 3L d.f. for an arbitrary risk function. If we fix the marginal risks at each locus, the risk function is subject to 2L + 1 constraints (Culverhouse et al. 2002). This can be seen by noting first that there are 3L marginal risks but that (3) implies that only two of those marginal risks are independent. The average risk, K, must be specified also, adding one more constraint. Even for small L, there is a large range of risk functions subject to these constraints. There are 4 d.f. in the risk function for L = 2, 20 for L = 3, 72 for L = 4, and 232 for L = 5. To explore efficiently the space of feasible risk functions subject to these constraints, we implemented an MCMC algorithm described in the previous section. That algorithm allowed us to randomly sample risk functions from the space of feasible risk functions subject to the given constraints. In our empirical study, instead of specifying the marginal risks directly, we assumed a multiplicative model within each locus and specified the genotype relative risk GRRi for each locus i. Recall that specifying K, pi, and GRRi completely fixes the marginal risks for locus i; see (10).
Figure 1 summarizes the results for full and half siblings. The results for parents and offspring are similar to those for full siblings. For full first cousins, the overall effect of gene interactions is weaker than for half siblings but the average CR still increases with L. The qualitative pattern for K = 0.005 is the same for both low-frequency (p = 0.03) and higher-frequency (p = 0.25) causative alleles but the average of CR is larger for rare alleles. Similar results were found for K = 0.02. The average CR increases with L and the probability that CR > 1 is nearly 1 even for L = 2. That is, gene interactions will tend to make the true similarity of relatives larger than expected if the multiplicative model is assumed to be correct, and the effect is more pronounced when causative alleles are rarer.
Figure 1.—

Expected increase in the interaction ratio (CR) and the fraction of models for which CR > 1 as functions of the number of causative loci, for low- and high-frequency alleles. The average CR increases with the number of loci and the probability that CR > 1 is nearly 1 for all loci. The effects of gene interactions are more pronounced when causative alleles are rarer. We used K = 0.005 and pi = p for all loci i. Within each locus, we assumed a multiplicative model and set the genotype relative risk (GRR) to 1.25. For all loci i, the corresponding marginal risks are 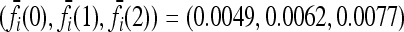 for p = 0.03 and
for p = 0.03 and 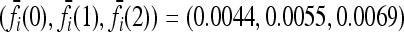 for p = 0.25. See (10).
for p = 0.25. See (10).
The results in Figure 1 were based on the assumption that all loci have the same genotype relative risks and allele frequencies. When there are both rare and common alleles, the increase in average CR increases with the proportion of rare alleles, except when L = 2. See Figure 2.
Figure 2.—

Expected increase in the interaction ratio as a function of the number of loci with rare causative alleles, given the total number of loci is L. Except for the case of L = 2, when there are both rare and common alleles, the increase in average CR increases with the proportion of rare alleles. The same parameter values as in Figure 1 are assumed here. In particular, rare risk alleles are assumed to have frequency p = 0.03, while common risk alleles are assumed to have frequency p = 0.25.
Nonrandom sampling of risk models:
The above results were obtained by sampling all feasible risk functions with equal probability. On intuitive grounds, however, it is reasonable to assume that risk functions that are closer in some sense to the induced multiplicative model are more likely alternatives than risk functions that are more unlike the induced multiplicative model. This assumption embodies the intuition that, although genes are not completely independent in their effect on risk, they are more likely to be partially independent than they are to interact in a completely arbitrary way. To test whether taking account of the similarity to a multiplicative model affects the above conclusions, we define a distance between a risk function f and the induced multiplicative risk function fI to be
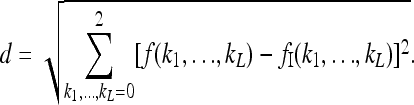 |
(11) |
This is a Euclidean distance in the ambient space with 3L dimensions.
In exploring the space of feasible models, we computed the d for each model. The left side of Figure 3 shows the cumulative distribution of d for one of the sets of parameter values. Other results are similar. The right side of Figure 3 shows the conditional average of the interaction ratio CR for full siblings, given that the distance d from the induced multiplicative model is within the yth percentile. The graph shows that, except for risk models that are very close to the multiplicative model, the average of CR is nearly the average obtained by randomly sampling risk models. In other words, even models of risk that are similar to the induced multiplicative model conceal on average substantial amounts of heritability.
Figure 3.—

Effects of nonrandom sampling of risk models for L = 4. The left plot shows the cumulative probability that a randomly sampled risk model has a Euclidean distance d (measured as a fraction of the empirical maximum possible distance dmax) from the induced multiplicative model for low- and high-frequency causative alleles. The right plot shows the conditional expectation of the interaction ratio CR for full siblings, given that the distance from the induced multiplicative model is within the yth percentile. This result implies that even models of risk that are similar to the induced multiplicative model conceal on average substantial amounts of heritability. The same parameter values as in Figure 1 are assumed here.
The minimum of CR:
The extrema of CR over the space of feasible disease models can be computed numerically using quadratic programming methods. Surprisingly, it turns out that we can actually obtain a closed-form formula for the minimum of CR by considering the following interpretation of the recurrence risk (4): Given an individual a in a population, let Xa denote an indicator random variable such that Xa = 0 (respectively, Xa = 1) if the individual is unaffected (respectively, affected) by a given complex disease. Then, for two relatives a and b with relationship R, the recurrence risk (4) can be written as
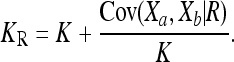 |
Since the recurrence risk 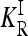 for the induced multiplicative model is completely determined by the given constraints, minimizing
for the induced multiplicative model is completely determined by the given constraints, minimizing 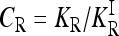 is equivalent to minimizing KR, which in turn translates to minimizing the covariance term Cov(Xa, Xb | R). Now, since Cov(Xa, Xb | R) can be partitioned into additive, dominance, and interaction variance components (James 1971)—which are all nonnegative—we conclude that the minimum Cov(Xa, Xb | R) is attained by a model with vanishing interaction variances. In such a model, Cov(Xa, Xb | R) is given by a sum of one-locus variance components over the L loci:
is equivalent to minimizing KR, which in turn translates to minimizing the covariance term Cov(Xa, Xb | R). Now, since Cov(Xa, Xb | R) can be partitioned into additive, dominance, and interaction variance components (James 1971)—which are all nonnegative—we conclude that the minimum Cov(Xa, Xb | R) is attained by a model with vanishing interaction variances. In such a model, Cov(Xa, Xb | R) is given by a sum of one-locus variance components over the L loci:
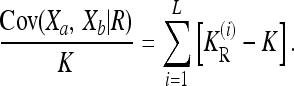 |
In summary,
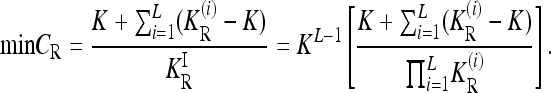 |
(12) |
In the symmetric case with pi = p and 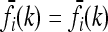 , for all loci i, the risk function f(k) that minimizes CR is given by
, for all loci i, the risk function f(k) that minimizes CR is given by
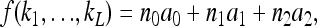 |
where nk is the number of loci with genotype k and 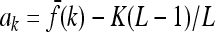 .
.
It can be checked that numerical optimization based on the method of Lagrange multipliers produces results that coincide with (12). In the cases we considered in this article, the marginal risks 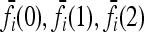 are linear in K. Therefore, the
are linear in K. Therefore, the 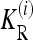 are proportional to K, and hence the minimum of CR is independent of K. Shown in Figure 4 are plots of the minimum of CR for pi = p and GRRi = 1.25 for all loci i. Note that the minimum of CR decreases as the number of loci increases and that the rate of decrease is more rapid for larger values of p. It is interesting that the average of CR increases with the number L of loci, despite the fact that the minimum of CR decreases as L increases. Computing the maximum of CR is a more difficult problem, both numerically and analytically, and we do not address it in this article. For L ≤ 4 and special values of K and p, Culverhouse et al. (2002) estimated the maximum of CR for purely epistatic models, in which case
are proportional to K, and hence the minimum of CR is independent of K. Shown in Figure 4 are plots of the minimum of CR for pi = p and GRRi = 1.25 for all loci i. Note that the minimum of CR decreases as the number of loci increases and that the rate of decrease is more rapid for larger values of p. It is interesting that the average of CR increases with the number L of loci, despite the fact that the minimum of CR decreases as L increases. Computing the maximum of CR is a more difficult problem, both numerically and analytically, and we do not address it in this article. For L ≤ 4 and special values of K and p, Culverhouse et al. (2002) estimated the maximum of CR for purely epistatic models, in which case 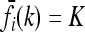 for all i = 1,…, L and k = 0, 1, 2.
for all i = 1,…, L and k = 0, 1, 2.
Figure 4.—

A graph of the minimum interaction ratio CR for full siblings plotted against the number of causative loci. The minimum of CR does not depend on K. Also, it decreases as the number of loci increases, and the rate of decrease is more rapid for larger values of p. It is interesting that, although the minimum of CR decreases as the number L of loci increases, the average of CR over the polytope increases with L.
DISCUSSION
In human genetics, gene interactions are regarded with ambivalence. On one hand, what has been learned about metabolic pathways and gene-regulatory networks makes clear that genes and gene products may interact in so many ways that independent effects of individual genes are probably the exception rather than the norm (Cordell 2002; Carlborg and Haley 2004; Moore and Williams 2005; Phillips 2008). On the other hand, tradition, convenience, and a large body of evidence from quantitative genetic studies encourage the use of models of disease risk that assume causative loci are independent in their effects or are additive on an underlying scale of disease liability (Risch 1990; Hill et al. 2008; Wray and Goddard 2010). Although many statistical methods have been developed to detect interacting genes in GWAS data sets (Marchini et al. 2005; Zhao et al. 2006; Zhang and Liu 2007; Gayán et al. 2008), those and similar methods are intended to test for specific pairs of loci that interact and not the overall importance of gene interactions to the average risk and heritability of complex diseases.
In this article, we quantified the potential effects of gene interactions. We showed that, whether or not there are detectable marginal effects of individual loci, gene interactions will tend to increase the heritability of disease risk as measured by the recurrence risk, KR, from what is expected on the basis of the assumption of no gene interactions. Furthermore, a greater increase in heritability is expected if there are more causative loci and when causative alleles are rare rather than common, and the pattern is found even with models of disease risk that are closer than average to a model that assumes no gene interactions. Therefore, relatively weak interactions among multiple causative loci could conceal substantial heritability.
The magnitude of the effect of interactions depends on the frequencies of causative alleles. To illustrate what our results mean, consider the case with low-frequency causative alleles (p = 0.03), each of which increases disease risk by a factor of 1.25. If there are five such loci, the interaction ratio, CR for full siblings is ∼1.57 (see Figure 2) for K = 0.005. If these five loci were identified and the GRRs estimated and a multiplicative model of interactions is assumed, we could conclude that these loci together would cause the recurrence risk KR to exceed K by a factor of 1.01. As a consequence, we would conclude that these five loci contribute very little to the increased risk to full siblings of an affected individual. However, our results indicate that, if the interaction model is sampled randomly from the space of models with the same GRRs, these loci would cause KR to exceed K by an average of 1.59. Furthermore, this would be true even if the risk model were chosen to be close to a multiplicative model in the sense we defined above. In this example, then, most of the increase in the risk to close relatives would be concealed if the true model were not multiplicative. The effect is weaker for higher-frequency risk alleles. If p = 0.25, CR for full siblings is ∼1.14. The increase in KR = 1.05 if a multiplicative model is assumed. The expected increase for a randomly chosen model of interactions is ∼1.20.
Untyped rare causative alleles may enhance the potential role of epistasis in explaining missing heritability. As our results show, the effect of epistasis on heritability is more pronounced for rare causative alleles than for common ones. Suppose c common causative alleles were typed in a GWAS, while r rare causative alleles were not typed. Our results (see Figure 2) suggest that the recurrence risk KR for the c + r loci can be substantially greater than that for the c common causative alleles only and that this difference in general grows with the number r of untyped rare causative alleles. In contrast, the recurrence risk 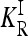 in the induced multiplicative model will be similar in the two cases.
in the induced multiplicative model will be similar in the two cases.
Our analysis cannot tell us whether gene interactions actually contribute to disease risk and the heritability of complex disease. Only empirical studies can do that. But our results indicate that there is ample room for gene interactions to have a strong effect on disease heritability. It is not necessary to assume extreme or bizarre types of gene interaction. Most deviations from independent gene action have an important effect on heritability, so the possibility of extensive interactions should be kept in mind when interpreting the results of GWAS and family studies of complex inherited diseases.
Acknowledgments
This research was supported in part by National Institutes of Health (NIH) grant R00-GM080099, by an Alfred P. Sloan Research Fellowship, by a Packard Fellowship for Science and Engineering (to Y.S.S.), and by NIH grant R01-GM40282 (to M.S.).
References
- Altshuler, D., M. Daly and E. Lander, 2008. Genetic mapping in human disease. Science 322(5903): 881–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlborg, Ö., and C. Haley, 2004. Epistasis: Too often neglected in complex trait studies? Nat. Rev. Genet. 5(8): 618–625. [DOI] [PubMed] [Google Scholar]
- Cordell, H., 2002. Epistasis: what it means, what it doesn't mean, and statistical methods to detect it in humans. Hum. Mol. Genet. 11(20): 2463–2468. [DOI] [PubMed] [Google Scholar]
- Culverhouse, R., B. Suarez, J. Lin and T. Reich, 2002. A perspective on epistasis: limits of models displaying no main effect. Am. J. Hum. Genet. 70(2): 461–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyer, M., A. Frieze and R. Kannan, 1991. A random polynomial-time algorithm for approximating the volume of convex bodies. J. Assoc. Comput. Mach. 38(1): 1–17. [Google Scholar]
- Gayán, J., A. González-Pérez, F. Bermudo, M. Sáez, J. Royo et al., 2008. A method for detecting epistasis in genome-wide studies using case-control multi-locus association analysis. BMC Genomics 9(1): 360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill, W., M. Goddard and P. Visscher, 2008. Data and theory point to mainly additive genetic variance for complex traits. PLoS Genet. 4(2): e1000008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James, J. W., 1971. Frequency in relatives for an all-or-none trait. Ann. Hum. Genet. 35(1): 47–49. [DOI] [PubMed] [Google Scholar]
- Kannan, R., L. Lovász and M. Simonovits, 1997. Random walks and an O*(n5) volume algorithm for convex bodies. Random Struct. Algorithms 11(1): 1–50. [Google Scholar]
- Liu, W., and B. S. Weir, 2005. Genotypic probabilities for pairs of inbred relatives. Philos. Trans. R. Soc. B 360 1379–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovász, L., 1999. Hit-and-run mixes fast. Math. Program. Ser. A 86(3): 443–461. [Google Scholar]
- Lovász, L., and S. Vempala, 2006. Simulated annealing in convex bodies and an O*(n4) volume algorithm. J. Comput. Syst. Sci. 72(2): 392–417. [Google Scholar]
- Manolio, T. A., F. S. Collins, N. J. Cox, D. B. Goldstein, L. A. Hindorff et al., 2009. Finding the missing heritability of complex diseases. Nature 461(7265): 747–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchini, J., P. Donnelly and L. R. Cardon, 2005. Genome-wide strategies for detecting multiple loci that influence complex diseases. Nat. Genet. 37(4): 413–417. [DOI] [PubMed] [Google Scholar]
- Moore, J. H., and S. M. Williams, 2005. Traversing the conceptual divide between biological and statistical epistasis: systems biology and a more modern synthesis. BioEssays 27(6): 637–646. [DOI] [PubMed] [Google Scholar]
- Phillips, P. C., 2008. Epistasis—the essential role of gene interactions in the structure and evolution of genetic systems. Nat. Rev. Genet. 9(11): 855–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risch, N., 1990. Linkage strategies for genetically complex traits. I. Multilocus models. Am. J. Hum. Genet. 46(2): 222–228. [PMC free article] [PubMed] [Google Scholar]
- Smith, R. L., 1984. Efficient Monte-Carlo procedures for generating points uniformly distributed over bounded regions. Oper. Res. 32 1296–1308. [Google Scholar]
- Wray, N. R., and M. E. Goddard, 2010. Multi-locus models of genetic risk of disease. Genome Med. 2(2): 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Y., and J. S. Liu, 2007. Bayesian inference of epistatic interactions in case-control studies. Nat. Genet. 39(9): 1167–1173. [DOI] [PubMed] [Google Scholar]
- Zhao, J., L. Jin and M. Xiong, 2006. Test for interaction between two unlinked loci. Am. J. Hum. Genet. 79(5): 831–845. [DOI] [PMC free article] [PubMed] [Google Scholar]