

The crystal structure of human PI3K p85β iSH2 domain has been determined to 3.3 Å resolution. Comparison with the published structure of the bovine p85β iSH2 domain bound to the influenza A virus nonstructural protein 1 indicates that little or no structural change occurs upon complex formation. Structural analysis of human and bovine p85β iSH2 domains reveals conformational plasticity in the interhelical turn region of the coiled-coil motif.
Keywords: phosphatidylinositol 3-kinases, p85β unit, iSH2 domain, influenza virus NS1 protein binding
Abstract
Phosphatidylinositol 3-kinase (PI3K) proteins actively trigger signaling pathways leading to cell growth, proliferation and survival. These proteins have multiple isoforms and consist of a catalytic p110 subunit and a regulatory p85 subunit. The iSH2 domain of the p85β isoform has been implicated in the binding of nonstructural protein 1 (NS1) of influenza A viruses. Here, the crystal structure of human p85β iSH2 determined to 3.3 Å resolution is reported. The structure reveals that this domain mainly consists of a coiled-coil motif. Comparison with the published structure of the bovine p85β iSH2 domain bound to the influenza A virus nonstructural protein 1 indicates that little or no structural change occurs upon complex formation. By comparing this human p85β iSH2 structure with the bovine p85β iSH2 domain, which shares 99% sequence identity, and by comparing the multiple conformations observed within the asymmetric unit of the bovine iSH2 structure, it was found that this coiled-coil domain exhibits a certain degree of conformational variability or ‘plasticity’ in the interhelical turn region. It is speculated that this plasticity of p85β iSH2 may play a role in regulating its functional and molecular-recognition properties.
1. Introduction
Considering that some influenza A virus strains such as H5N1 have a mortality rate greater than 50% and the recent demonstration that a new influenza strain, namely the 2009 H1N1 strain, can so rapidly become a global threat, it is important to understand the mechanisms that regulate the influenza A virus life cycle. Influenza A has eight RNA fragments in its viral genome. One of these RNAs encodes a protein named nonstructural protein 1 (NS1), which has the ability to manipulate multiple pathways within the host cell (Nemeroff et al., 1998 ▶; Min et al., 2007 ▶; Gack et al., 2009 ▶). It has been shown that NS1 activates class IA phosphatidylinositol 3-kinase (PI3K) signaling, a pathway that is involved in cell proliferation and survival, by directly binding to the p85β regulatory subunit of PI3K (Hale et al., 2006 ▶).
Phosphatidylinositol 3-kinases can recruit a multitude of effectors with phosphoinositide-recognition domains by phosphorylating inositol lipids at the 3-position hydroxy group. These effectors then initiate multiple signaling cascades that are responsible for regulating cellular functions such as proliferation and cell-cycle regulation (Vanhaesebroeck et al., 2010 ▶). PI3K proteins are heterodimers that contain a catalytic p110 subunit and a regulatory p85 subunit (Cantley, 2002 ▶). The p110 catalytic subunit, of which there are multiple isoforms (p110α, p110β, p110δ and p110γ), is responsible for the kinase activity of the protein (Vanhaesebroeck et al., 2010 ▶). The multiple variants of the p85 regulatory subunit (p85α, p85β, p55α, p50α and p55γ) are responsible for stabilizing p110, inhibiting the catalytic activity of p110 and recruiting it to phosphorylated tyrosine residues of receptor or adaptor proteins (Vanhaesebroeck et al., 2010 ▶).
The p85β regulatory subunit of PI3K has five domains. The N-terminal SH3 domain is followed by a BH domain, along with two SH2 domains, aptly named nSH2 (N-terminal SH2) and cSH2 (C-terminal SH2), respectively. The fifth domain is the region between these two SH2 domains and is called the inter-SH2 or iSH2 domain.
The effector domain (ED) of the influenza A virus NS1 protein interacts directly with the iSH2 domain of p85β, the same domain responsible for binding the p110 catalytic subunit (Hale et al., 2008 ▶). NS1 can bind the iSH2 domain without displacing p110 (Hale et al., 2008 ▶). Additionally, reciprocal gain of function experiments identified the key role of Val573 of p85β in binding NS1 (Li et al., 2008 ▶). When Val573 of p85β is mutated to methionine, p85β loses its ability to bind to NS1; when Met583, the corresponding residue of p85α, is mutated to valine p85α gains the ability to bind NS1 (Li et al., 2008 ▶).
Recently, the crystal structure of the complex between bovine p85β iSH2 and the ED of NS1 from an H1N1 strain of influenza A has been determined (Hale et al., 2010 ▶). This structure verifies the importance of many residues which had already been identified by immunoprecipitation and pull-down assays. For instance, Tyr89 of NS1, which has long been considered to play an important role in the interaction (Shin et al., 2007 ▶), is directly in the center of the complex with more buried surface area than any other residue (Hale et al., 2010 ▶). Also, this structure revealed the basis for the selectivity of NS1 for p85β and not p85α, in that replacement of Val573 in the structure with methionine would cause steric hindrance that would prevent the formation of the complex with NS1 (Hale et al., 2010 ▶).
A series of molecular-modeling experiments conducted using this crystal structure of the complex in conjunction with the p85α–p110 complex led to some interesting observations. Firstly, p110 could indeed bind to the iSH2 domain in conjunction with NS1 (Hale et al., 2010 ▶). Also, it was predicted that the ED of NS1 occupies a region in space that would directly overlap with the nSH2 domain (Hale et al., 2010 ▶). Thus, it was proposed that NS1 may inhibit the p110 catalytic subunit by mimicking the position of the nSH2 domain as well as by making some direct contacts with p110 (Hale et al., 2010 ▶).
Here, we report the three-dimensional structure of the iSH2 domain of human p85β in its unbound form. By comparison with the published structure of the NS1 ED-bound bovine p85β iSH2 domain (Hale et al., 2010 ▶), we conclude that the iSH2 domain of p85β does not undergo a large conformational change when binding NS1. However, there are some structural differences in the interhelical turn regions between bound and unbound iSH2 domains. Structural variation in these interhelical turn regions is also observed among iSH2 domains from the same asymmetric unit in the bovine p85β iSH2 structure. These observations suggest that this domain exhibits a significant degree of ‘conformational plasticity’ in the interhelical turn region which may play an important role in the function of p85β. This conformational plasticity in the interhelical turn is not observed in the published structures of the iSH2 domain of human p85α (Mandelker et al., 2009 ▶; Huang et al., 2007 ▶; Miled et al., 2007 ▶).
2. Materials and methods
2.1. Expression and purification
The iSH2 domain of the p85β subunit of human PI3K was cloned and purified using standard methods of the Northeast Structural Genomics Consortium (Acton et al., 2005 ▶; Xiao et al., 2010 ▶). The cDNA of human p85β was originally obtained from ATCC. A DNA insert containing the coding region for residues 433–610 of p85β was cloned into a pET-21c expression vector, sequence-verified and subsequently expressed in Escherichia coli strain BL21 (DE3) in LB broth using shaker flasks at 310 K. The cell pellet from 1 l fermentation was resuspended in 25 ml binding buffer consisting of 50 mM Tris–HCl pH 7.5, 500 mM NaCl, 40 mM imidazole, 1 mM TCEP, 0.02% NaN3. Cells were then disrupted by sonication on ice for 10 min. The sonicated mixture was spun down in the centrifuge at 50 000g for 40 min. The supernatant was then poured through a 3 ml Ni–NTA Superflow column (Qiagen) which was previously equilibrated in binding buffer. Next, the column was subjected to two 25 ml washes with NTA wash buffer consisting of 50 mM NaH2PO4 pH 8.0, 300 mM NaCl, 40 mM imidazole and 10 mM DTT. The protein was then eluted with 10 ml elution buffer consisting of 50 mM Tris–HCl pH 7.5, 500 mM NaCl, 500 mM imidazole, 1 mM TCEP, 0.02% NaN3 and 10 mM DTT. The sample was further purified using a Superdex 200 gel-filtration column (GE Healthcare) equilibrated in 10 mM Tris–HCl pH 7.5, 100 mM NaCl, 5 mM DTT and 0.02% NaN3. The purity of the fractions was visualized using SDS–PAGE gels and pure fractions were subsequently pooled and concentrated using Millipore Amicon Ultra centrifugal filter tubes.
2.2. Crystallization and data collection
Crystallization screening of human p85β iSH2 was initially carried out using the Hauptmann–Woodward Institute high-throughput crystallization screening facility and was then further optimized using the hanging-drop vapor-diffusion method. 1 µl protein solution at a concentration of ∼6–8 mg ml−1 was mixed with 1 µl well solution. This protein could be crystallized under several different conditions and the crystals used for data collection were obtained in 0.5 M sodium malonate pH 7.6. The crystals formed at 277 K and were not stable at room temperature. The crystals were briefly soaked in a cryobuffer consisting of 2.0 M sodium malonate and were flash-frozen in liquid N2. These crystals only diffracted to around 3.5 Å resolution when measured on the home X-ray facility; a complete data set was subsequently collected to 3.3 Å resolution on beamline X29A at the National Synchrotron Light Source (Brookhaven, New York, USA). The X-ray diffraction data were processed with DENZO and scaled with SCALEPACK as implemented in HKL-2000 (Otwinowski & Minor, 1997 ▶).
2.3. Structure determination and refinement
The structure of the human p85β iSH2 domain was determined by molecular replacement with the program Phaser (McCoy et al., 2007 ▶) using the crystal structure of the bovine p85β iSH2 domain (chain D of PDB entry 3l4q; Hale et al., 2010 ▶) as the search probe. There is a clear molecular-replacement solution for the first molecule (TFZ = 13.1, LLG = 211). However, searching for more molecules in the asymmetric unit was not successful. Structure refinement confirmed that there is only one molecule in the asymmetric unit, although the solvent content is unusually high (81.4%). Structure refinement was carried out with the program PHENIX (Adams et al., 2002 ▶). For B-factor refinement, considering the data were only collected to 3.3 Å resolution, only group B factors were refined. Manual model building was performed using Coot (Emsley & Cowtan, 2004 ▶). The geometry of the model was examined with MolProbity (Chen et al., 2010 ▶). The final structure-refinement and geometry statistics are listed in Table 1 ▶.
Table 1. Data-collection and structure-refinement statistics.
Values in parentheses are for the highest resolution shell.
| Data collection | |
| Resolution () | 3.30 (3.423.30) |
| Space group | R32 |
| Wavelength () | 1.075 |
| Unit-cell parameters (, ) | a = b = 174.59, c = 102.70, = = 90, = 120 |
| Unique reflections | 9161 (897) |
| Multiplicity | 22.1 (22.5) |
| Completeness (%) | 99.9 (100) |
| R merge † (%) | 8.8 (56.7) |
| I/(I) | 39.0 (6.83) |
| Structure refinement | |
| Resolution () | 3.3 |
| R cryst ‡ (%) | 24.87 |
| R free § (%) | 27.73 |
| No. of reflections used | 8955 |
| No. of reflections in R free set | 427 |
| No. of non-H protein atoms | 1385 |
| No. of water molecules | 14 |
| R.m.s. deviation, bond lengths () | 0.625 |
| R.m.s. deviation, bond angles () | 0.003 |
| Ramachandran plot statistics | |
| Most favored (%) | 98.7 |
| Additional allowed (%) | 1.3 |
| Generously allowed (%) | 0 |
| MolProbity all-atom clash score | 18.04 (97th percentile¶ at >3) |
| MolProbity geometry score | 2.25 (99th percentile¶ at 3.30 0.25) |
R
merge = 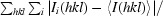
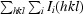 , where I
i(hkl) is the intensity of an individual reflection and I(hkl) is the average intensity of that reflection.
, where I
i(hkl) is the intensity of an individual reflection and I(hkl) is the average intensity of that reflection.
R
cryst = 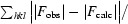
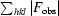 , where F
calc is the calculated structure factor.
, where F
calc is the calculated structure factor.
R free is as R cryst but calculated for a randomly selected 5.0% of reflections that were not included in the refinement.
The 100th percentile is the best among structures of comparable resolution; the 0th percentile is the worst.
3. Results and discussion
The DNA fragment encoding the iSH2 domain of human p85β consisting of residues 433–610 was cloned into a pET-21c vector and the protein was expressed in E. coli BL21 (DE3) cells. Ni2+-affinity column purification followed by gel-filtration chromatography resulted in ∼20 mg protein from 1 l cell culture with high purity as determined by SDS–PAGE (Fig. 1 ▶ a) and mass spectrometry. The purified p85β iSH2 sample was crystallized under different conditions and the best diffracting crystals were grown from sodium malonate (Fig. 1 ▶ b) and were used for data collection on beamline X29A at NSLS. A complete data set was collected to 3.3 Å resolution.
Figure 1.

Purification and crystallization of human p85β iSH2 domain. (a) SDS–PAGE shows a single band for the purified human p85β iSH2 domain. Lane 1 contains markers and lane 2 contains human p85β iSH2. The molecular weights (kDa) of the markers are labeled. (b) Crystals of p85β iSH2 domain grown in sodium malonate buffer.
The crystal structure of human p85β iSH2 was determined by the molecular-replacement method using the bovine iSH2 structure as the search model (chain D of PDB entry 3l4q; Hale et al., 2010 ▶). These crystals of human p85β iSH2 belonged to space group R32 and there was only one molecule in the asymmetric unit, indicating an extremely high solvent content of 81.4% in the crystals. The refined final model of p85β iSH2 contains residues 436–598 and 14 water molecules. The final R and R free factors are 24.9% and 27.7%, respectively, indicating a well refined structure at 3.3 Å resolution. The detailed refinement statistics are listed in Table 1 ▶. The coordinates and structure factors have been deposited in the PDB with accession code 3mtt.
The overall structure of p85β iSH2 is a coiled-coil motif (Fig. 2 ▶ a), like other homologous iSH2 domains from human p85α (Mandelker et al., 2009 ▶; Huang et al., 2007 ▶; Miled et al., 2007 ▶) and bovine p85β (Hale et al., 2010 ▶). Residues 439–511 and 515–583 form the two major α-helices and there is a small α-helix (residues 588–594) at the C-terminus.
Figure 2.

Overall structure of p85β iSH2 domain and comparison with other iSH2 domain structures. (a) Overall structure of p85β iSH2 domain, colored by temperature factors of the Cα atoms. Colors from blue to red indicate temperature factors from low to high. The N- and C-termini are labeled. (b) Human p85β iSH2 domain (green) superimposed on bovine p85β iSH2 domain (PDB entry 3l4q chain D; cyan) determined in complex with influenza A NS1 effector domain (yellow). The primary structural differences are in the interhelical turn region between the coiled coils, distant from the NS1A-binding site. (c) Structure superposition of human p85β iSH2 domain (green) and bovine p85β iSH2 domain in 3l4q chains C (magenta) and D (cyan). The conformation of the turn linking the two long α-helices shifts in these three structures, indicating a certain degree of ‘conformational plasticity’ in the interhelical turn region of this coiled-coil domain. This region is highlighted by a black box with dashed-line borders.
As outlined in §1, the crystal structure of the complex between the ED of influenza A PR8 NS1 and bovine p85β iSH2 has been determined to 2.3 Å resolution (PDB code 3l4q; Hale et al., 2010 ▶). The region of sequence overlap between the structures of this bovine iSH2 and our human iSH2 contains 99% sequence identity, allowing a direct comparison between the free and NS1-bound states of the iSH2 domain of p85β. Even with this high sequence similarity, only one of the two bovine p85β iSH2 chains in PDB entry 3l4q, the D chain, yielded a molecular-replacement solution for human p85β iSH2; the C chain was sufficiently different in structure that it could not provide a molecular-replacement solution. When superimposing human p85β iSH2 on the two bovine p85β iSH2 chains in 3l4q with the program LSQKAB from the CCP4 program suite (Collaborative Computational Project, Number 4, 1994 ▶), the r.m.s. values over 116 Cα atoms in the helices (no residues in the turn region linking the two helices were included) are 2.63 and 1.65 Å for the C and D chains, respectively. Although the observed r.m.s.d. is high considering the level of sequence identity that these two proteins share, no drastic overall conformational change is observed. This is consistent with the current idea that displacement of the nSH2 domain would activate the kinase activity of p110 rather than the activation stemming from some large change in the iSH2 upon binding NS1. Fig. 2 ▶(b) depicts the superimposition of NS1-bound bovine iSH2 (Hale et al., 2010 ▶) with the human iSH2 structure determined in this work, demonstrating that no major conformational changes result from NS1 binding.
In the structure of NS1-bound bovine p85β iSH2, the two iSH2 chains (the C and D chains in 3l4q) are identical molecules in the same crystal structure and we would expect their structures to be very similar to each other. However, when superimposed they show some differences, especially in the turn region linking the two long α-helices; these conformational differences result in a ‘shift’ in the position of this turn (Fig. 2 ▶ c). The NS1–p85β iSH2 interactions, which are at the other end of the coiled coil (Fig. 2 ▶), are not influenced by this ‘shift’ in the coiled-coil interhelical turn region. We looked further into this plasticity by aligning human p85β iSH2 with the C and D chains of 3l4q (also shown in Fig. 2 ▶ c) and observed that there is a ‘shift’ in the turn regions in all three p85β iSH2 chains: i.e. there is conformational variability in this interhelical turn region even among the molecules of the asymmetric unit. These differences may reflect the ability of the p85β iSH2 coiled-coil domain to ‘flex’. The B factors in this interhelical turn are not particularly high, as shown in Fig. 2 ▶(a). The evidence for plasticity is that different interhelical turn conformations are stabilized in different crystal structures.
We also investigated whether similar conformational variability is observed among the crystal structures of human p85α iSH2. To date, the human p85α iSH2 domain structure has been determined in various environments (PDB codes 2rd0, 2v1y, 3hhm and 3hiz; Mandelker et al., 2009 ▶; Huang et al., 2007 ▶; Miled et al., 2007 ▶). When these structures of different crystal forms are superimposed they fit to each other very well, with r.m.s. values of around 1.0 Å. Accordingly, the structural variability that we observe in the coiled-coil interhelical turn region of the p85β iSH2 domain does not appear to occur in the human p85α iSH2 domain.
Although p85α and p85β share 73% sequence identity, their functions are quite different. Until recently, the focus of much research has been on p85α because of its clear link to many cancers. Knowing that NS1 selectively activates PI3K by binding to p85β but not p85α and that NS1-activated PI3K does not exhibit the predicted anti-apoptotic effect illustrates that the two isoforms play different roles in the cell (Jackson et al., 2009 ▶). While the exact biological function of p85β still not very clear, and with little insight into the structural basis underlying the functional differences of isoforms p85β and p85α, we speculate that the structural plasticity of the p85β iSH2 domain could have a contributing role in distinguishing the functional properties of these two otherwise highly homologous proteins.
The crystal packing in human p85β iSH2 crystals is unique. According to the Matthews coefficient calculation (Matthews, 1968 ▶; Kantardjieff & Rupp, 2003 ▶), three molecules in the asymmetric unit will give a V M of 2.21 Å3 Da−1 (44.3% solvent content) and two molecules in the asymmetric unit will give a V M of 3.31 Å3 Da−1 (62.87% solvent content). However, molecular replacement could only find one molecule in the asymmetric unit, which corresponds to a V M of 6.63 Å3 Da−1 and a solvent content of 81.4%. This extremely high solvent content could explain why these crystals, which are large in size and sharp in shape, only diffracted to 3.3 Å even using synchrotron X-ray radiation.
In conclusion, we have determined the crystal structure of the human p85β iSH2 domain, which mainly consists of a coiled-coil motif. Compared with the NS1-bound bovine p85β iSH2 domain structure, we do not observe significant conformational changes upon NS1 binding. However, we observed some degree of conformational plasticity in the interhelical turn region of iSH2, distant from the NS1-binding region. Although it is possible that structural variants in this region may be stabilized by crystal packing, such structural variations in different crystal forms are an indication of inherent conformational plasticity. Although this conformational variability has not been observed in the p85α isoform, all of the reported structures of p85α iSH2 are complexes with p110α domains, which may stabilize particular conformations of the iSH2 interhelical turn region. We speculate that this conformational plasticity observed among the different crystal forms and complexes of p85β iSH2 may contribute to determining the molecular-recognition properties of p85β.
Supplementary Material
PDB reference: iSH2 domain of human phosphatidylinositol 3-kinase p85β subunit, 3mtt
Acknowledgments
X-ray diffraction data for this study were measured on beamline X29A of the National Synchrotron Light Source, where the financial support comes principally from the Offices of Biological and Environmental Research and of Basic Energy Sciences of the US Department of Energy and from the National Center for Research Resources of the National Institutes of Health. We thank Dr Howard Robinson for his help with data collection and the HWI high-throughput crystallization screening facility for initial crystallization screening. This work was supported in part by NIGMS grant U54-GM074958 (GTM).
References
- Acton, T. B. et al. (2005). Methods Enzymol. 394, 210–243. [DOI] [PubMed]
- Adams, P. D., Grosse-Kunstleve, R. W., Hung, L.-W., Ioerger, T. R., McCoy, A. J., Moriarty, N. W., Read, R. J., Sacchettini, J. C., Sauter, N. K. & Terwilliger, T. C. (2002). Acta Cryst. D58, 1948–1954. [DOI] [PubMed]
- Cantley, L. C. (2002). Science, 296, 1655–1657. [DOI] [PubMed]
- Chen, V. B., Arendall, W. B., Headd, J. J., Keedy, D. A., Immormino, R. M., Kapral, G. J., Murray, L. W., Richardson, J. S. & Richardson, D. C. (2010). Acta Cryst. D66, 12–21. [DOI] [PMC free article] [PubMed]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763.
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Gack, M. U., Albrecht, R. A., Urano, T., Inn, K.-S., Huang, I.-C., Carnero, E., Farzan, M., Inoue, S., Jung, J. U. & García-Sastre, A. (2009). Cell Host Microbe, 8, 439–449. [DOI] [PMC free article] [PubMed]
- Hale, B. G., Batty, I. H., Downes, C. P. & Randall, R. E. (2008). J. Biol. Chem. 283, 1372–1380. [DOI] [PubMed]
- Hale, B. G., Jackson, D., Chen, Y.-H., Lamb, R. A. & Randall, R. E. (2006). Proc. Natl Acad. Sci. USA, 103, 14194–14199. [DOI] [PMC free article] [PubMed]
- Hale, B. G., Kerry, P. S., Jackson, D., Precious, B. L., Gray, A., Killip, M. J., Randall, R. E. & Russell, R. J. (2010). Proc. Natl Acad. Sci. USA, 107, 1954–1959. [DOI] [PMC free article] [PubMed]
- Huang, C.-H., Mandelker, D., Schmidt-Kittler, O., Samuels, Y., Velculescu, V. E., Kinzler, K. W, Vogelstein, B., Gabelli, S. B. & Amzel, L. M. (2007). Science, 318, 1744–1748. [DOI] [PubMed]
- Jackson, D., Killip, M. J., Galloway, C. S., Russell, R. J. & Randall, R. E. (2009). Virology, 396, 94–105. [DOI] [PubMed]
- Kantardjieff, K. A. & Rupp, B. (2003). Protein Sci. 12, 1865–1871. [DOI] [PMC free article] [PubMed]
- Li, Y., Anderson, D. H., Liu, Q. & Zhou, Y. (2008). J. Biol. Chem. 283, 23397–23409. [DOI] [PubMed]
- Mandelker, D., Gabelli, S. B., Schmidt-Kittler, O., Zhu, J., Cheong, I., Huang, C.-H., Kinzler, K. W., Vogelstein, B. & Amzel, L. M. (2009). Proc. Natl Acad. Sci. USA, 106, 16996–17001. [DOI] [PMC free article] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol. 33, 491–497. [DOI] [PubMed]
- McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C. & Read, R. J. (2007). J. Appl. Cryst. 40, 658–674. [DOI] [PMC free article] [PubMed]
- Miled, N., Yan, Y., Hon, W. C., Perisic, O., Zvelebil, M., Inbar, Y., Schneidman-Duhovny, D., Wolfson, H. J., Backer, J. M. & Williams, R. L. (2007). Science, 317, 239–242. [DOI] [PubMed]
- Min, J.-Y., Li, S., Sen, G. C. & Krug, R. M. (2007). Virology, 363, 236–243. [DOI] [PubMed]
- Nemeroff, M., Barabino, S. M. L., Keller, W. & Krug, R. M. (1998). Mol. Cell, 1, 991–1000. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol. 276, 307–326. [DOI] [PubMed]
- Shin, Y.-K., Li, Y., Liu, Q., Anderson, D. H., Babiuk, L. A. & Zhou, Y. (2007). J. Virol. 81, 12730–12739. [DOI] [PMC free article] [PubMed]
- Vanhaesebroeck, B., Guillermet-Guibert, J., Graupera, M. & Bilanges, B. (2010). Nature Rev. Mol. Cell Biol. 11, 329–341. [DOI] [PubMed]
- Xiao, R. et al. (2010). J. Struct. Biol. 172, 21–33. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: iSH2 domain of human phosphatidylinositol 3-kinase p85β subunit, 3mtt