

The structure of human vascular adhesion protein 1 in space group C2 with seven molecules in the asymmetric unit has been refined at a resolution of 2.9 Å.
Keywords: vascular adhesion protein 1, VAP-1, SSAO, copper amine oxidases, topaquinone, TPQ
Abstract
Human vascular adhesion protein 1 (VAP-1) is involved in lymphocyte–endothelial cell adhesion and has been implicated in many human inflammatory diseases. VAP-1 is a member of the copper amine oxidase family of enzymes with a trihydroxyphenylalanine quinone (TPQ) cofactor. Previously characterized crystals of VAP-1 suffered from anisotropy and contained disordered regions; in addition, one form was consistently twinned. In an effort to grow crystals that diffracted to higher resolution for inhibitor-binding studies, a construct with an N-terminal deletion was made and expressed in the Chinese hamster ovary (CHO) glycosylation mutant cell line Lec8. Screening produced crystals that displayed some anisotropy and contained seven molecules per asymmetric unit. These crystals belonged to space group C2, with unit-cell parameters a = 394.5, b = 115.8, c = 179.3 Å, β = 112.3°. The structure was refined to a resolution of 2.9 Å, with R cryst and R free values of 0.250 and 0.286, respectively.
1. Introduction
Inflammation is a normal consequence of an organism’s response to an infectious agent and is a required step in the process of tissue repair and wound healing. This disease-induced response often leads to excessive or aberrant stimulation, resulting in harmful inflammatory conditions (Butcher & Picker, 1996 ▶). Activation of the inflammatory cascade requires cells such as leukocytes to migrate from the circulation to the site of the insult (infection or wound) through a complex series of events. One possible approach to modulating nonproductive inflammation conditions is to inhibit the action of the adhesion molecules involved in the extravasation of leukocytes from the blood into the inflamed tissue (Luster et al., 2005 ▶). It has been recognized in recent years that vascular adhesion protein 1 (VAP-1) plays a significant role in this extravasation process (Salmi & Jalkanen, 1992 ▶, 2001 ▶). VAP-1 is a nonclassical leukocyte subtype-specific adhesion molecule which is upregulated at sites of inflammation. It mediates the attachment and rolling of leukocytes as well as aiding in the transmigration across the endothelial luminal wall in many tissue types (Salmi & Jalkanen, 2001 ▶). VAP-1 is a dual function protein, acting as both a cell-adhesion molecule and an enzyme with amine oxidase activity; both functions are required to facilitate cell migration.
VAP-1 was first identified as a lymphocyte–endothelial cell adhesion molecule expressed on the surface of endothelial cells in 1992 (Salmi & Jalkanen, 1992 ▶). Biochemical analysis later showed VAP-1 to be an extensively sialylated homodimeric glycoprotein of ∼90 kDa subunits (Salmi & Jalkanen, 1996 ▶) and revealed the leukocyte interaction to be carbohydrate-dependent. The subsequent cloning and purification revealed an unexpected significant sequence identity to the copper amine oxidase (CAO) family of enzymes (EC 1.4.3.6; Smith et al., 1998 ▶). In fact, VAP-1 is identical to the previously well known amine oxidase semicarbazide-sensitive amine oxidase (SSAO). CAOs catalyze the oxidative deamination of primary amines to the corresponding aldehydes, with the concomitant production of ammonia and hydrogen peroxide. CAOs are ubiquitous in nature (although noticeably absent from archaea) but do not seem to carry out a universal biological role. The CAO family of enzymes is distinguished by the presence of a type II copper ion and a quinone-type cofactor, which in VAP-1 is a protein-derived trihydroxyphenylalanine quinone (TPQ).
In a series of studies from many laboratories VAP-1 has been implicated in many human diseases and disorders, including lung inflammation, asthma, psoriasis, stroke, rheumatoid arthritis, colitis, uveitis, diabetes, metabolic disorders and vascular damage. Furthermore, recent research has established VAP-1 as a viable drug target. Both antibodies to the VAP-1 adhesion site and selective inhibitors of the amine oxidase activity have been shown to attenuate inflammation in multiple animal models of disease. Strong validation comes from the observation that VAP-1-deficient mice have clear defects in cell migration during inflammation (Stolen et al., 2005 ▶) and that mice overexpressing VAP-1 in the lung have an exaggerated response to lipopolysaccharide (LPS; Yu et al., 2006 ▶). Furthermore, this response can be attenuated with an amine oxidase inhibitor (Yu et al., 2006 ▶).
The formation of aldehyde products by VAP-1 has been shown to lead to cytotoxic damage in endothelial cells. The concomitant production of hydrogen peroxide may also have detrimental effects via free-radical formation and oxidative stress (Yu et al., 1994 ▶). Furthermore, it has been suggested that the hydrogen peroxide produced by VAP-1 has the potential to exacerbate the process of local lesion formation, as it is present in the vessel wall (Smith & Vainio, 2007 ▶). Supporting this proposal is the evidence that VAP-1 activity is elevated in patients with known risk factors associated with cardiovascular disease, including diabetes, obesity and hypertension.
Humans have three genes encoding CAOs: AOC1–3. AOC1 encodes a diamine oxidase (hDAO) that is involved in clearing both endogenous and exogenous histamine (Schwelberger, 2004 ▶). AOC2 encodes retina-specific amine oxidase (hRAO), which includes a putative transmembrane domain and has an enzymatic activity that is isolated primarily to the eye (Kaitaniemi et al., 2009 ▶). AOC3 encodes VAP-1. In most mammals another gene, AOC4, encodes a soluble form of VAP-1. However, a nonsense mutation in the AOC4 gene in humans translates to a nonfunctional truncated protein. In humans, soluble blood-borne VAP-1 (sVAP-1) results from proteolytic cleavage of membrane-bound VAP-1. Two human CAOs have been structurally characterized: VAP-1 (Jakobsson et al., 2005 ▶; Airenne et al., 2005 ▶) and hDAO (McGrath et al., 2009 ▶, 2010 ▶). Previous studies indicate broad yet largely separate substrate profiles for the two enzymes (Elmore et al., 2002 ▶; Marti et al., 2004 ▶). While the residues in close proximity to the TPQ are mostly conserved between VAP-1 and hDAO, the substrate channel of VAP-1 is wider than that in hDAO (McGrath et al., 2009 ▶). Apart from this general observation, it is difficult to rationalize the distinct substrate profiles.
CAOs are homodimers consisting of subunits of ∼700 residues (Guss et al., 2009 ▶), and crystal structures of CAOs from bacteria, yeasts, plants and mammals are available. The first structure of a CAO was that from Escherichia coli (ECAO; Parsons et al., 1995 ▶). ECAO was described as being shaped like a mushroom, with the D1 domain forming the stalk linked to a cap made up from the other three domains (D2–4). The availability of additional CAO structures from eubacteria (Arthrobacter globiformis), yeast (Hansenula polymorpha and Pichia pastoris), plants (Pisum sativum) and mammals (Homo sapiens and Bos taurus) revealed a common architecture (domains D2–4) showing a high degree of structural similarity despite pairwise sequence identities of ∼20–30%. All structures other than ECAO lack domain D1. The common architecture observed in CAOs hides the vast diversity shown in the substrate preferences and biological roles of these enzymes.
The native structure of human VAP-1 has previously been described in two crystal forms: P6522 (Airenne et al., 2005 ▶) and P43 (Jakobsson et al., 2005 ▶). In this paper, we describe the structure of a new crystal form of human VAP-1 refined to 2.9 Å resolution in space group C2 with seven molecules in the asymmetric unit. Overall, the structure is very similar to those previously characterized. However, a number of differences have been observed in loop regions, around disulfide bridges and in the active-site cavity around the TPQ. The large number of molecules in the asymmetric unit provides a unique opportunity to observe the effect of crystal packing and flexibility on the molecules of a CAO in one structure.
2. Materials and methods
2.1. Cloning, expression, purification and crystallization of VAP-1
A cloned DNA template corresponding to residues 34–763 of human VAP-1/SSAO and incorporating a mouse Ig κ signal sequence, an N-terminal FLAG epitope tag and a tobacco etch virus (TEV) cleavage site was assembled in a mammalian expression vector (pLO-CMV). Stably transfected pools of the CHO-K1 glycosylation mutant cell line Lec8 (Stanley, 1981 ▶) were derived following co-transfection of pLO-CMV VAP-1 together with a plasmid conferring resistance to the antibiotic puromycin. One positive pool was expanded into roller bottles and immunoaffinity chromatography resulted in the recovery of >20 mg enzymatically active protein from ∼5 l culture supernatant. The protein was purified using the following method. The pH of the 5.4 l fermentation harvest target protein was adjusted by the addition of 5 ml 3 M Tris–HCl pH 8.0 per litre. The medium was first pumped through an ∼50 ml Sephacryl S300 pre-filter column and then through two M2 anti-FLAG Minileak (Millipore) columns, each with a volume of ∼20 ml, at 300–350 ml h−1 in a 277 K cold room (the columns were run in series). The M2 anti-FLAG Minileak columns were washed with ∼1.0 l each of Tris-buffered saline solution (TBSA) at room temperature (RT). The protein was eluted by circulating 0.25 mg ml−1 FLAG peptide in TBSA through each column for 1 h at RT, followed by an additional low-pH recovery step using 5–10 column volumes of 0.5 M sodium citrate pH 3.0 (0.02% azide), the eluate dropping directly into an equivalent volume of 3 M Tris–HCl pH 8.0. Each eluant (at pH ∼8) was concentrated to ∼8 ml by ultrafiltration in a new 10 kDa 150 ml Omega stirred cell (Pall). After centrifugation to remove solids, the target protein was gel-filtered on a Superdex 200 column in TBSA buffer pH 8.0, pooling tubes (T24–34) in the main peak with an apparent mass of ∼190 kDa as estimated by comparison with the chromatographic profile of a standard set of size markers.
The protein was concentrated to 5.7 mg ml−1 and set up in sitting-drop crystallization experiments at the Bio21 Collaborative Crystallization Centre. Droplets consisting of 200 nl protein solution and 200 nl reservoir solution were equilibrated against 50 µl reservoir solution at 293 K in SD-2 sitting-drop plates (IDEX Corp). Initial screens were performed with three in-house polyethylene glycol-rich screens (288 conditions) and yielded crystals overnight in over 30 conditions. The crystals grew to full size within 5 d. A number of these crystals were tested for diffraction in situ (PXScanner) and none showed diffraction beyond low resolution (7–8 Å). More extensive tests on the MX1 beamline of the Australian Synchrotron (AS) did not yield a significant improvement in the observed diffraction limits. Treatment of the protein with the endoglycosidases EndoF1 and PNGaseF was attempted, but made no obvious difference to the quality of the crystals. Adding a further step of protein purification using ion-exchange chromatography (MonoQ) separated the protein preparation into two fractions. The first, larger fraction produced crystals that looked similar and behaved very similarly to the initial crystals, but the second fraction produced some crystals that were morphologically distinct (long thin rods rather than chunky, sometimes ragged-ended, prisms) and showed anisotropic diffraction to 2.2 and 3 Å resolution in the strong and weaker directions, respectively. These crystals were grown using VAP-1 at 3.5 mg ml−1 in 0.2 M diammonium hydrogen citrate, 20% PEG 3350. A remarkable difference in diffraction was seen by ‘stepping along’ crystals on the MX2 (microfocus) beamline at the AS: the resolution varied from no diffraction to 2.2 Å within a single crystal.
2.2. X-ray data collection and structure solution
Data were recorded on the MX2 beamline at the AS from a crystal that had been flash-cooled in a cold nitrogen stream. Immediately before harvesting and cooling the crystal, 1 µl of a cryosolution (the reservoir components with the addition of 10% ethylene glycol and 10% glycerol) was added to the droplet containing the crystal. The data were integrated with MOSFLM (Leslie, 1992 ▶) and scaled using SCALA (Collaborative Computational Project, Number 4, 1994 ▶). Molecular replacement was carried out using a model of VAP-1 (PDB code 2c10; Jakobsson et al., 2005 ▶) without solvent molecules, metal ions and the TPQ. A clear molecular-replacement solution was found with the program Phaser (McCoy, 2007 ▶) in space group C2 (Z score = 80.49, log-likelihood gain = 22334.8). Refinement was carried out using REFMAC5 (Murshudov et al., 1997 ▶) within the CCP4 program suite (Collaborative Computational Project, Number 4, 1994 ▶). In between the rounds of refinement the model was inspected manually and rebuilt using Coot (Emsley & Cowtan, 2004 ▶). Initially, strict NCS restraints were applied to the seven monomers within the asymmetric unit. The NCS restraints were kept between the different chains throughout the refinement process in order to keep the refinement residuals within an acceptable criterion and were gradually relaxed in areas where differences between the various monomers became apparent. Solvent molecules were added progressively as the electron-density maps improved. Isotropic B factors were refined after having been set to the Wilson B (43.1 Å2) and resulted in a lowering of the R cryst and R free values by ∼1.0%. Thereafter, the B factors for the individual water molecules were set to 43.1 Å2 and only an overall B factor was refined. This reflects the relatively low resolution of the data.
3. Results and discussion
3.1. VAP-1 in space group C2
The data-processing and refinement statistics for VAP-1 in space group C2 are listed in Table 1 ▶. There are seven molecules in the asymmetric unit (Fig. 1 ▶). Six of these monomers form three biological units (A/B, C/D and F/G), whereas molecule E forms a dimer with the E chain of the crystallographic twofold symmetry-related molecule (−x, y, −z + 1). The final structure includes residues A39–41, A57–202, A206–761, B39–43, B57–202, B206–501, B504–761, C35–43, C57–202, C206–501, C505–762, D36–42, D57–202, D206–762, E39–41, E58–202, E206–502, E505–761, F41, F57–202, F206–501, F504–761, G37–42, G57–202 and G206–761. The N-terminal residues A/B/E34–38, C34, D34–35, G34–36, F34–40, A42–56, B/C44–56, D/G43–56, E42–57 and F42–56 have been excluded from the model as well as the last one or two residues at the C-terminus of each chain as they were not located in significant electron density and were most likely to be disordered. Two surface loops are also disordered with little or no visible electron density: residues 203–205 are omitted from the structure and residues 502–504 are disordered for most chains, in which case they too are absent from the final model.
Table 1. Diffraction data and refinement statistics.
Values in parentheses are for the highest resolution shell.
| Space group | C2 |
| X-ray source | AS MX2 |
| Wavelength () | 0.97946 |
| Unit-cell parameters (, ) | a = 394.5, b = 115.8, c = 179.3, = 90, = 112.3, = 90 |
| No. of molecules in asymmetric unit | 7 |
| Resolution range () | 110.432.90 (3.062.90) |
| No. of unique reflections | 163097 (23606) |
| Completeness (%) | 98.6 (98.1) |
| Multiplicity | 3.7 (3.8) |
| I/(I) | 9.4 (1.9) |
| R merge † | 0.192 (0.88) |
| R p.i.m. ‡ | 0.115 (0.523) |
| No. of reflections for R cryst | 154651 (11137) |
| No. of reflections for R free | 8187 (584) |
| R cryst | 0.250 (0.317) |
| R free | 0.286 (0.356) |
| Model content | |
| Total atoms (non-H) | 39103 |
| Protein residues | 4939 |
| Carbohydrate residues | 50 |
| Water molecules | 230 |
| Copper ions | 7 |
| Calcium ions | 14 |
| Ramachandran data | |
| Allowed (%) | 100 |
| Favoured (%) | 94.9 |
| Outliers (%) | 0 |
| R.m.s.d. bond lengths () | 0.016 |
| R.m.s.d. bond angles () | 1.531 |
| PDB code | 3ala |
R
merge = 
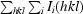 .
.
R
p.i.m. = 
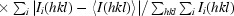 (Weiss, 2001 ▶).
(Weiss, 2001 ▶).
Figure 1.

A molecular-surface representation depicting the arrangement of the seven molecules in the asymmetric unit: A (green), B (cyan), C (magenta), D (yellow), E (pink), F (grey) and G (blue). The carbohydrates attached to the surface are represented as sticks.
When comparing this new crystal form of VAP-1 with the two previously available structures, the Cα atoms of the individual chains align with a root-mean-square deviation (r.m.s.d.) of 0.33–0.40 Å for PDB entry 2c10 (all chains; Jakobsson et al., 2005 ▶) and 0.40–0.44 Å for PDB entry 1us1 (all chains; Airenne et al., 2005 ▶). The major differences in the Cα trace (>0.6 Å) can be found in various surface loops, including the areas around residues 79–85, 160–162, 268–290, 293, 319–321, 501–505, 551–558, 580–583, 665–666 and 740–752. Some of these residues participate directly in the formation of crystal contacts.
Pairwise secondary-structural alignment of the seven protein chains in the asymmetric unit yields r.m.s.d.s for Cα atoms of 0.15–0.22 Å. The small r.m.s.d. values are in part a result of the use of tight NCS restraints. However, despite refining the structure with tight NCS restraints there are some apparent differences between the molecules in the asymmetric unit and as a result the NCS restraints were relaxed for these regions. They include the N-terminal residues around Cys41, for which the Cα r.m.s.d.s fluctuate by up to 2.2 Å. These residues have not been observed in previous structures and it is likely that this part of the molecule provides a flexible connection to the transmembrane region defined by residues 6–26. Residues 80–82 (Gly-Pro-Gly) in chain F differ in their conformation when overlaid with other chains, with Pro81 being displaced by up to 3.4 Å.
Jakobsson et al. (2005 ▶) suggested that the C-terminal residues 762–763, although not observed in their structure (PDB code 2c10), might form part of the entrance to the active site. The electron density in the current structure allowed His762 of chains C and D to be modelled. As predicted, the imidazole ring of His762 lies over the mouth of the active-site channel, about 4–5 Å away from the phenyl ring of Phe173. In this position it limits access to the active site and possibly provides some discrimination between different substrates (Fig. 2 ▶). In 2c10 the phenyl ring of Phe173 forms part of the wall of the active-site channel, whereas in the structure determined by Airenne and coworkers (PDB code 1us1; Airenne et al., 2005 ▶) it protrudes out across the opening. In the present structure, Phe173 is in the same position as modelled by Airenne and coworkers. In this conformation the phenyl group of Phe173 is rotated about the Cα—Cβ bond when compared with the structure determined by Jakobsson and coworkers (PDB code 2c10; Jakobsson et al., 2005 ▶) and in effect Phe173 and His762 form a ‘bridge’ across the substrate-channel opening. The absence of His762 in the electron density of other chains in the present structure and in structures 2c10 and 1us1 hints at the apparent flexibility of this residue. Furthermore, Ser763, although not observed in the electron density, may also serve to further delimit the substrate-channel opening.
Figure 2.

A surface model of the mouth of the active-site cavity is shown in (a) as modelled in chain D, where His762 could be modelled, and in (b) with His762 removed from the structure. Phe173 is on the opposite side of the opening and effectively delimits the active-site channel opening with His762. The residues forming the active-site cavity are shown in cyan (chain D) and magenta (chain C). TPQ471 and the proposed active-site gate residue Leu469 can be seen at the bottom of the funnel, as well as the active-site copper, which is depicted as a sphere.
3.2. Crystal contacts
Apart from the dimer interface, in which the D4 domains of each monomer closely interact with each other (as described by Airenne et al., 2005 ▶; Jakobsson et al., 2005 ▶), there are very few unambiguous contacts between the seven chains in the asymmetric unit (see Table 2 ▶ for a summary). Dimer A/B sits in the middle of the asymmetric unit, participating in the formation of the most contacts with the other chains. Chain A forms contacts with chain C through residues residing at the beginning of domain D2 of both monomers. Only one hydrogen bond is measurable (A/Gln88 N∊2⋯C/Arg78 O, 3.5 Å), but there are most likely to also be interactions involving A/Gln88 O∊1⋯C/Arg78 N∊/Nη1/Nη2, A/Arg90 Nη2⋯C/Gln77 O∊1/N∊2 and A/Gln181 O∊1⋯C/Arg73 N∊/Nη1/Nη2, for which there is no visible electron density for the side chains. Monomer A also interacts with monomers E (A/Thr296 Oγ⋯E/His148 N∊2) and F (A/Asp730 Oδ1⋯F/Ala421 N, A/Asp730 Oδ2⋯F/Asp85 Oδ2, A/Gly732 O⋯F/Gln88 N∊2 and A/Cys734 N⋯F/Gln88 O∊1), forming most of the interactions in the asymmetric unit. The A/F interactions occur just after the surface-exposed RGD motif on chain A (residues 726–728) that has been suggested to be important for cell–cell adhesion and the liberation of soluble VAP-1 into the bloodstream (Smith et al., 1998 ▶; Airenne et al., 2005 ▶; Jakobsson et al., 2005 ▶). Chains D and G are located so that they are not in contact with any other molecules in the asymmetric unit apart from their respective dimer partners.
Table 2. Observed contacts between the monomers in the asymmetric unit, excluding dimer-interface interactions.
| Atom X | Atom Y | Distance () |
|---|---|---|
| A/Gln88N2 | C/Arg78O | 3.4 |
| A/Thr296O | E/His148N2 | 3.1 |
| A/Asp730O1 | F/Ala421N | 3.0 |
| A/Asp730O2 | F/Gln420N | 3.0 |
| A/Gly732O | F/Gln88N2 | 3.3 |
| A/Cys734N | F/Gln88O1 | 2.7 |
Crystal contacts are present between the asymmetric unit and eight symmetry-related molecules (−x, y, −z; x, y − 1, z; −x, y − 1, −z + 1; −x + 1/2, y −1/2, −z + 1; −x, y, −z + 1; −x + 1/2, y + 1/2, −z + 1; x, y + 1, z; −x, y + 1, −z + 1), one of which dimerizes with monomer E (−x, y, −z + 1). Owing to the crystallographic symmetry operations, the interactions between the molecules in the asymmetric unit and the symmetry molecules are the same for six of them: x, y − 1, z = x, y + 1, z; −x, y − 1, −z + 1 = x, y + 1, −z + 1; −x + 1/2, y − 1/2, −z + 1 = −x + 1/2, y + 1/2, −z + 1. The equivalent residues in the different chains are often involved in forming the crystal contacts and interactions between A, D, E and F and C, B, G and E, respectively (Table 3 ▶). Persistent crystal contacts include Gln276 O∊1/N∊2⋯Leu568 N/O and Gly284 O⋯Ser594 Oγ. Residues 276–284 are located at the end of domain D3 and the beginning of the linker between D3 and D4. The backbone Cα positions for residues 282–284 differ from those in structures 2c10 and 1us1 and the side chain of residue 276 has adopted a different conformation, flipping by around 90° to make crystal packing possible. Where density for loop residues 501–505 is visible it appears that these residues are involved in hydrogen bonding to nearby symmetry molecules. This loop has been modelled differently when compared with 2c10 and 1us1, but the electron density of this part of the molecule is weak. Another interesting feature is the visibility of the long stretch of amino-acid residues at the N-terminus of chains C and D around the Cys41–Cys748 disulfide bridge. These residues are involved in interactions with a symmetry molecule (C/Ser36 O⋯A/Leu82 N and C/Ser36 N⋯A/Gly83 O) and the crystal contacts have probably contributed to stabilizing them. In structure 2c10, in which the disulfide was modelled in a similar orientation, these residues are not visible.
Table 3. Proteinprotein contacts between the symmetry-related molecules in the crystal.
| Atom X | Atom Y | Distance () | Symmetry operation | Translation† |
|---|---|---|---|---|
| A/Leu568N | B/Gln276O1 | 3.0 | x,y, z | |
| A/Leu568O | B/Gln276N2 | 2.4 | x,y, z | |
| F/Gln276N2 | E/Leu568N | 3.4 | x,y 1, z | x,y + 1, z |
| F/Gln276N2 | E/Leu568O | 2.7 | x,y 1, z | x,y + 1, z |
| F/Arg269N | E/Gly505N | 3.3 | x,y 1, z + 1 | x,y + 1, z + 1 |
| F/Arg269N2 | E/Gly502O | 2.8 | x,y 1, z + 1 | x,y + 1, z + 1 |
| G/Leu568N | E/Gln276N2 | 2.8 | x,y 1, z + 1 | x,y + 1, z + 1 |
| G/Leu568O | E/Gln276N2 | 2.9 | x,y 1, z + 1 | x,y + 1, z + 1 |
| G/Ser594O | E/Gly284O | 3.5 | x,y 1, z + 1 | x,y + 1, z + 1 |
| A/Gly82N | C/Ser36O | 3.4 | x + 1/2, y 1/2, z + 1 | x + 1/2, y + 1/2, z + 1 |
| A/His148N2 | C/Asp730O2 | 3.3 | x + 1/2, y 1/2, z + 1 | x + 1/2, y + 1/2, z + 1 |
| C/Leu568N | A/Gln276O1 | 3.1 | x + 1/2, y 1/2, z + 1 | x + 1/2, y + 1/2, z + 1 |
| C/Leu568O | A/Gln276N2 | 2.4 | x + 1/2, y 1/2, z + 1 | x + 1/2, y + 1/2, z + 1 |
| D/Gln276N2 | B/Leu568N | 2.9 | x + 1/2, y 1/2, z + 1 | x + 1/2, y + 1/2, z + 1 |
| D/Gln276N2 | B/Leu568O | 3.4 | x + 1/2, y 1/2, z + 1 | x + 1/2, y + 1/2, z + 1 |
| D/Ala503O | A/Arg269N | 3.0 | x + 1/2, y 1/2, z + 1 | x + 1/2, y + 1/2, z + 1 |
| A/Lys567N | B/Gln276N2 | 3.2 |
Indicating a translational symmetry operation in one axis, 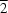 , i.e. if atom a
x,y,z in the asymmetric unit interacts with b
x,y 1, z then b
x,y,z will also interact with a
x,y + 1, z.
, i.e. if atom a
x,y,z in the asymmetric unit interacts with b
x,y 1, z then b
x,y,z will also interact with a
x,y + 1, z.
3.3. Cysteine residues and disulfides
Four disulfide bonds have been modelled in each molecule: Cys41–Cys748, Cys198–Cys199, Cys404–Cys430 and Cys734–Cys741, with the exception of Cys734–Cys741 in chain G, in which the side chain of Cys734 appears to be partially disordered. However, since this disulfide bond is unambiguously present in all other monomers it was retained in the model for completeness. In all seven molecules there is clear evidence of Cys748 forming an intramolecular disulfide bond with the N-terminal residue Cys41. This has previously been described for another truncated version of VAP-1 (residues 29–763; PDB code 2c10; Jakobsson et al., 2005 ▶), but is in contrast to the full-length protein structure (PDB code 1us1; Airenne et al., 2005 ▶), in which this cysteine forms an intermolecular disulfide bond with Cys748 of the other molecule in the dimer. The observation of the disulfide bond between Cys748 and Cys41 is therefore most likely to be an artefact of the expression of a truncated form of VAP-1. The observation of an intermolecular disulfide bond in 1us1 is consistent with an intermolecular disulfide bond formed by the corresponding cysteines in the structure of the related human CAO hDAO (McGrath et al., 2009 ▶). There is some residual electron density between F/Cys748 and G/Cys748 to suggest an intermolecular disulfide bridge in the dimer formed by chains F and G. However, this electron density has been left unmodelled owing to the large amount of noise in the electron-density maps as well as the presence of clearly interpretable electron density for the Cys41–Cys748 disulfide bond present in the other chains.
3.4. Active site
The active site of VAP-1 includes the TPQ cofactor and a copper ion (Cu2+) that is coordinated by the N∊2 atoms of the imidazole side chains of His520 and His522 and the Nδ1 atom of His684. The TPQ has been found to be present in one or both of two distinct conformations: an inactive ‘on-copper’ conformation in which the TPQ is directly coordinated to the copper ion through the O4 atom and an active ‘off-copper’ conformation in which the TPQ is indirectly bound to the copper through a hydrogen bond between O2 and the axially coordinated water molecule Wa. A second equatorially positioned water molecule, We, forms a square-pyramidal arrangement with Wa and the active-site histidine residues, but We is labile and has only been observed occasionally (reviewed by Dawkes & Phillips, 2001 ▶; Brazeau et al., 2004 ▶). In the present structure, the TPQ is in an active ‘off-copper’ conformation in all seven monomers, although the electron density is not entirely unambiguous. The axial water is visible in chains B and C and forms a 2.6–3 Å long hydrogen bond to TPQ O2. The positions of the O4 and O5 atoms vary in the different subunits (see Fig. 3 ▶). In chains A, B, D, E and G TPQ O4 sits at a distance of 3.1–3.5 Å from the hydroxyl group of Tyr372, a conserved residue in all known CAO structures that is believed to stabilize an ‘off-copper’ TPQ, and TPQ O5 is between 2.9 and 3.4 Å away from the carboxylate group of the catalytic base Asp386. This arrangement has been observed in most other CAOs in which the TPQ is ‘off-copper’ (Wilce et al., 1997 ▶; Li et al., 1998 ▶; Lunelli et al., 2005 ▶; Mure et al., 2005 ▶). However, in monomers C and F the distance from TPQ O4 to Tyr372 is longer (4.4 and 4.9 Å, respectively). This conformation of the TPQ resembles the previously solved VAP-1 structure in the active form (Jakobsson et al., 2005 ▶). The different conformations of the TPQ cause the position of the Cβ atom to vary by up to 2.0 Å between the different subunits.
Figure 3.

(a) The variation of the active-site cofactor TPQ471. (b) The arrangement of the TPQ in chains B (beige) and C (blue). The active-site residues Tyr372 and Asp386 are shown to interact differently with the TPQ. In chain B (as well as chains A, D, E and G), TPQ O5 interacts with the Asp386 Oδ1/Oδ2 atoms and TPQ O4 interacts with Tyr372. However, in chain C (which is also representative of chain F), both TPQ atoms O4 and O5 are within hydrogen-bonding distance of Asp386 Oδ1/Oδ2. The copper is coordinated by the three conserved histidines, His520, His522 and His684, as well as TPQ O2 through the axial water molecule Wa in chains B and C.
The backbone and side-chain positions of the other active-site residues resemble those in previously described VAP-1 structures. For the seven NCS molecules in the asymmetric unit the variations between the active sites are small, even though the NCS restraints were removed for a number of these residues during refinement. Leu469 has previously been proposed to act as a potential gating residue in VAP-1, controlling the substrate accessibility to the active site. This residue also appears to be well ordered in all monomers, except in chain G, and overlays very closely when the different molecules are superposed.
3.5. Glycosylation
VAP-1 is extensively glycosylated, with four putative O-linked glycosylation sites (Ser43, Ser47, Thr212 and Thr679) and six N-linked glycosylation sites (Asn137, Asn232, Asn294, Asn592, Asn618 and Asn666) (Smith et al., 1998 ▶; Airenne et al., 2005 ▶). Glycosidase digestion and mutational studies have shown that all of the N-linked glycosylation sites are utilized, but have found little or no evidence of O-linked glycosylation (Salmi & Jalkanen, 1996 ▶; Maula et al., 2005 ▶). It is clear that glycosylation is imperative for the function of VAP-1 as an adhesion molecule (Salmi & Jalkanen, 1996 ▶) and it has also been proposed to be important for enzyme activity (Maula et al., 2005 ▶). In the two previously solved native structures of VAP-1 none of the potential O-linked carbohydrates were modelled, although there was some evidence for Thr212 being glycosylated in 1us1. However, structural evidence for N-linked sugars has been found in both structures, with up to five of the six potential sites being occupied. In the structure reported here, there is only clear evidence of N-linked glycosylation at Asn137, Asn232 and Asn592 in all seven monomers, as well as Asn666 in chain D. There is some weak residual electron density indicating the possible presence of glycosylation at sites E/Asn294, A/Asn618 and F/Asn618, but this was insufficient to confidently model a glycan at these positions.
4. Conclusions
We have reported a new crystal form of VAP-1 at 2.9 Å resolution in space group C2 with seven molecules in the asymmetric unit. Overall, the structure is very similar to the two previously reported structures of VAP-1. However, the large number of molecules in the asymmetric unit highlights regions of molecular flexibility. It also provides the opportunity to observe conformational variability in the positions of active-site residues in the productive ‘off-copper’ conformation.
Supplementary Material
PDB reference: human vascular adhesion protein 1, 3ala
Acknowledgments
This research was supported by an ARC Linkage Project (LP0669658) to CC, IM and JMG and an associated Australian Postgraduate Award for Industry to APM. This work was also supported by Pharmaxis Ltd. We would like to thank the staff of the Bio21 Collaborative Crystallization Centre, the CSIRO fermentation facility and the Australian Synchrotron beamline scientists Drs Caradoc-Davies and Cowieson for their assistance.
References
- Airenne, T. T., Nymalm, Y., Kidron, H., Smith, D. J., Pihlavisto, M., Salmi, M., Jalkanen, S., Johnson, M. S. & Salminen, T. A. (2005). Protein Sci. 14, 1964–1974. [DOI] [PMC free article] [PubMed]
- Brazeau, B. J., Johnson, B. J. & Wilmot, C. M. (2004). Arch. Biochem. Biophys. 428, 22–31. [DOI] [PubMed]
- Butcher, E. C. & Picker, L. J. (1996). Science, 272, 60–66. [DOI] [PubMed]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763.
- Dawkes, H. C. & Phillips, S. E. (2001). Curr. Opin. Struct. Biol. 11, 666–673. [DOI] [PubMed]
- Elmore, B. O., Bollinger, J. A. & Dooley, D. M. (2002). J. Biol. Inorg. Chem. 7, 565–579. [DOI] [PubMed]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Guss, J. M., Zanotti, G. & Salminen, T. (2009). Copper Amine Oxidases: Structures, Catalytic Mechanisms and Role in Pathophysiology, edited by G. Floris & B. Mondovi, pp. 119–141. Boca Raton: Taylor & Francis.
- Jakobsson, E., Nilsson, J., Ogg, D. & Kleywegt, G. J. (2005). Acta Cryst. D61, 1550–1562. [DOI] [PubMed]
- Kaitaniemi, S., Elovaara, H., Gron, K., Kidron, H., Liukkonen, J., Salminen, T., Salmi, M., Jalkanen, S. & Elima, K. (2009). Cell. Mol. Life Sci. 66, 2743–2757. [DOI] [PMC free article] [PubMed]
- Leslie, A. G. W. (1992). Jnt CCP4/ESF–EACBM Newsl. Protein Crystallogr. 26
- Li, R., Klinman, J. P. & Mathews, F. S. (1998). Structure, 6, 293–307. [DOI] [PubMed]
- Lunelli, M., Di Paolo, M. L., Biadene, M., Calderone, V., Battistutta, R., Scarpa, M., Rigo, A. & Zanotti, G. (2005). J. Mol. Biol. 346, 991–1004. [DOI] [PubMed]
- Luster, A. D., Alon, R. & von Andrian, U. H. (2005). Nature Immunol. 6, 1182–1190. [DOI] [PubMed]
- Marti, L., Abella, A., De La Cruz, X., Garcia-Vicente, S., Unzeta, M., Carpene, C., Palacin, M., Testar, X., Orozco, M. & Zorzano, A. (2004). J. Med. Chem. 47, 4865–4874. [DOI] [PubMed]
- Maula, S. M., Salminen, T., Kaitaniemi, S., Nymalm, Y., Smith, D. J. & Jalkanen, S. (2005). Eur. J. Immunol. 35, 2718–2727. [DOI] [PubMed]
- McCoy, A. J. (2007). Acta Cryst. D63, 32–41. [DOI] [PMC free article] [PubMed]
- McGrath, A. P., Hilmer, K. M., Collyer, C. A., Dooley, D. M. & Guss, J. M. (2010). Acta Cryst. F66, 137–142. [DOI] [PMC free article] [PubMed]
- McGrath, A. P., Hilmer, K. M., Collyer, C. A., Shepard, E. M., Elmore, B. O., Brown, D. E., Dooley, D. M. & Guss, J. M. (2009). Biochemistry, 48, 9810–9822. [DOI] [PMC free article] [PubMed]
- Mure, M., Kurtis, C. R., Brown, D. E., Rogers, M. S., Tambyrajah, W. S., Saysell, C., Wilmot, C. M., Phillips, S. E., Knowles, P. F., Dooley, D. M. & McPherson, M. J. (2005). Biochemistry, 44, 1583–1594. [DOI] [PubMed]
- Murshudov, G. N., Vagin, A. A. & Dodson, E. J. (1997). Acta Cryst. D53, 240–255. [DOI] [PubMed]
- Parsons, M. R., Convery, M. A., Wilmot, C. M., Yadav, K. D., Blakeley, V., Corner, A. S., Phillips, S. E., McPherson, M. J. & Knowles, P. F. (1995). Structure, 3, 1171–1184. [DOI] [PubMed]
- Salmi, M. & Jalkanen, S. (1992). Science, 257, 1407–1409. [DOI] [PubMed]
- Salmi, M. & Jalkanen, S. (1996). J. Exp. Med. 183, 569–579. [DOI] [PMC free article] [PubMed]
- Salmi, M. & Jalkanen, S. (2001). Trends Immunol. 22, 211–216. [DOI] [PubMed]
- Schwelberger, H. G. (2004). Histamine: Biology and Medical Aspects, edited by A. Falus, pp. 43–52. Budapest: SpringMed Publishing.
- Smith, D. J., Salmi, M., Bono, P., Hellman, J., Leu, T. & Jalkanen, S. (1998). J. Exp. Med. 188, 17–27. [DOI] [PMC free article] [PubMed]
- Smith, D. J. & Vainio, P. J. (2007). Ann. N. Y. Acad. Sci. 1110, 382–388. [DOI] [PubMed]
- Stanley, P. (1981). Mol. Cell. Biol. 1, 687–696. [DOI] [PMC free article] [PubMed]
- Stolen, C. M., Marttila-Ichihara, F., Koskinen, K., Yegutkin, G. G., Turja, R., Bono, P., Skurnik, M., Hanninen, A., Jallkanen, S. & Salmi, M. (2005). Immunity, 22, 105–115. [DOI] [PubMed]
- Weiss, M. S. (2001). J. Appl. Cryst. 34, 130–135.
- Wilce, M. C., Dooley, D. M., Freeman, H. C., Guss, J. M., Matsunami, H., McIntire, W. S., Ruggiero, C. E., Tanizawa, K. & Yamaguchi, H. (1997). Biochemistry, 36, 16116–16133. [DOI] [PubMed]
- Yu, P. H., Davis, B. A., Boulton, A. A. & Zuo, D. M. (1994). J. Neural. Transm. Suppl. 41, 397–406. [DOI] [PubMed]
- Yu, P. H., Lu, L.-X., Fan, H., Kazachkov, M., Jiang, Z.-J., Jalkanen, S. & Stolen, C. (2006). Am. J. Pathol. 168, 718–726. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: human vascular adhesion protein 1, 3ala