

SUMMARY
There are multiple mechanisms of maintaining tolerance in the gut that protect the intestine from excessive inflammatory response to intestinal microorganisms. We report here that all four mammalian Peptidoglycan Recognition Proteins (PGRPs or Pglyrps) protect the host from colitis induced by dextran sulfate sodium (DSS). Pglyrp1−/−, Pglyrp2−/−, Pglyrp3−/−, and Pglyrp4−/− mice are all more sensitive than wild type (WT) mice to DSS-induced colitis due to changes to more inflammatory gut microflora, higher production of interferon-γ and interferon-inducible genes, and increase in NK cells in the colon upon initial exposure to DSS, which leads to severe hyperplasia of the lamina propria, loss of epithelial cells, and ulceration in the colon. Thus in WT mice PGRPs protect the colon from early inflammatory response and loss of the barrier function of intestinal epithelium by promoting normal bacterial flora and by preventing damaging production of interferon-γ by NK cells in response to injury.
INTRODUCTION
Mucosal surfaces, such as intestinal tract mucosa, rely both on innate and adaptive immunity for protection against invasion with commensals and pathogens. Much attention has been focused on pro-inflammatory effects of pattern recognition receptors, such as Toll-like receptors or Nod-like receptors, which recognize various microbial components. In the intestinal mucosa a single layer of mucus-coated epithelial cells separates billions of bacteria present in the colon from sterile tissues. This barrier must not only prevent entrance of these bacteria into the tissues, but it must also avoid mounting continuous inflammatory response to the highly pro-inflammatory microbial components present in the colon. Thus the balance of the immune responsiveness in the colon must be heavily tilted towards unresponsiveness (or inhibition of inflammation). However, the mechanisms of maintaining this unresponsiveness are not fully understood.
In this study we tested the role of Peptidoglycan Recognition Proteins (PGRPs or Pglyrps) in intestinal immunity. PGRPs are innate immunity proteins that are conserved from insects to mammals, recognize bacterial peptidoglycan, and function in antibacterial immunity. Mammals have four PGRPs, Pglyrp1, Pglyrp2, Pglyrp3, and Pglyrp4 (that were initially named PGRP-S, PGRP-L, PGRP-Iα, and PGRP-Iβ, respectively) (Kang et al., 1998; Liu et al., 2 001). Three PGRPs, Pglyrp1, Pglyrp3, and Pglyrp4 are directly bactericidal (Lu et al., 2006; Tydell et al., 2006; Wang et al., 2007), whereas Pglyrp2 is an N-acetylmuramoyl-L-alanine amidase that hydrolyzes peptidoglycan (Gelius et al., 2003; Wang et al., 2003). Pglyrp1 is highly expressed in PMN’s granules and is also expressed in other cells, e.g., intestinal M cells (Liu et al., 2000, 2001; Dziarski et al., 2003; Lo et al., 2003). Pglyrp3 and Pglyrp4 are expressed in the skin, salivary glands, throat, tongue, esophagus, stomach, intestine, and eyes (Mathur et al., 2004; Lu et al., 2006). Pglyrp2 is constitutively expressed in the liver and secreted into blood and its expression is induced in other cells, such as epithelial cells, including intestinal epithelium (Gelius et al., 2003; Wang et al., 2003; Lo et al., 2003; Xu et al., 2004; Wang et al., 2005; Zhang et al., 2005; Li et al., 2006). Mammalian PGRPs could influence host-parasite interactions through their anti-bacterial or peptidoglycan-hydrolytic properties, since normal bacterial flora is crucial for maintaining proper mucosal homeostatic balance. However, PGRPs also have immunomodulatory properties that are independent of their hydrolytic and anti-bacterial activities (Saha et al., 2009).
Based on these antibacterial and immunomodulatory properties of PGRPs and their expression in the intestine we hypothesized that mammalian PGRPs may play a role in the intestinal inflammation. One of the most frequent inflammatory diseases of unknown etiology in the intestinal tract is the inflammatory bowel disease (IBD). IBD affects one in 500 individuals and is characterized by chronic relapsing inflammation of the gastrointestinal tract likely due to dysregulated immune response to intestinal bacteria. It includes Crohn’s disease and ulcerative colitis. Crohn’s disease may affect the entire length of intestinal tract and usually includes chronic granulomatous inflammation, whereas ulcerative colitis affects primarily colon and lacks granulomatous inflammation (Podolsky, 2002; Sartor, 2003; Korzenik and Podolsky, 2006; Hanauer, 2006; Lakatos et al., 2006).
To test whether PGRPs play a role in IBD we selected dextran sulfate sodium (DSS)-induced colitis model. DSS-induced colitis is an established experimental mouse model of acute colitis often used to study the role of innate immunity in IBD, and especially ulcerative colitis. Oral administration of DSS in drinking water damages intestinal epithelium and induces inflammation and ulcerative colitis, most likely in response to enteric bacteria. The requirement for intestinal bacteria is evidenced by drastic reduction of the intestinal inflammatory response in this model by oral administration of antibiotics (Elson et al., 1995; Rath et al., 2001; Rakoff-Nahoum et al., 2004; Maeda et al., 2005).
We demonstrate that all four PGRPs protect mice from colitis, because Pglyrp1−/−, Pglyrp2−/−, Pglyrp3−/−, and Pglyrp4−/− mice are all more sensitive than wild type (WT) mice to DSS-induced colitis. We show that PGRPs protect mice from colitis by promoting normal intestinal flora that is less inflammatory than flora of PGRP-deficient mice, and by limiting production of interferon-γ (IFN-γ) by NK cells in the colon upon initial exposure to DSS. This allows WT mice to limit induction of IFN-inducible genes, early inflammatory response, and damage to the colon.
RESULTS
PGRP-deficient mice have higher susceptibility to DSS-induced colitis
Oral administration of 3% DSS in drinking water to WT BALB/c mice induced moderate colitis, manifested first by diarrhea and then some blood in the stools, ~15% weight loss, and ~27% mortality, when DSS was continued for 20 days (Figure 1). By contrast, Pglyrp1−/−, Pglyrp2−/−, Pglyrp3−/−, and Pglyrp4−/− mice showed significantly higher sensitivity to DSS-induced colitis than WT mice, manifested by extremely severe intestinal bleeding, accelerated and more severe weight loss (28–34%), and accelerated and higher mortality (60–95%) (Figure 1). Pglyrp2−/− and Pglyrp3−/− mice were the most sensitive, Pglyrp1−/− mice had intermediate sensitivity, and Pglyrp4−/− mice were the least sensitive, although still significantly more sensitive than WT mice (Figure 1 and Supplemental Results). These results are consistent with lower expression of Pglyrp4 than other PGRPs in most tissues, including colon (Liu et al., 2001; Mathur et al., 2004; Lu et al., 2006). PGRP-deficient mice were also similarly more sensitive than WT mice to colitis induced by 5% DSS, which induced more rapid and more severe colitis in all mice (Figure S1).
Figure 1. PGRP-deficient mice are more susceptible to DSS-induced colitis than WT mice.

(A) Gross rectal bleeding in Pglyrp3−/− mice but not in WT mice after treatment with 3% DSS in drinking water. (B) Survival, (C) body weight, and (D) stool and rectal bleeding scores of WT and Pglyrp1−/−, Pglyrp2−/−, Pglyrp3−/−, and Pglyrp4−/− mice after continuous treatment with 3% DSS in drinking water. N = 11–20 mice/group; significance of differences between PGRP-deficient and WT mice is indicated by asterisks; *, P<0.05; * *, P<0.001. In addition, Pglyrp2−/− and Pglyrp3−/− mice had significantly more severe colitis than Pglyrp4−/− mice (see Supplemental Results).
To determine the pathologic basis of higher sensitivity of PGRP-deficient mice to colitis, we compared the histology of intestinal tract in WT and PGRP-deficient mice treated with DSS. The most prominent changes were found in the colon, with the most severe pathology in the distal colon. In both proximal and distal colon PGRP-deficient mice had extensive hyperplasia of the lamina propria, thickened submucosa, loss of morphology and crypt architecture, loss of epithelial layer, decrease in goblet cells, and severe ulceration. By contrast, WT mice after the same length of DSS intake did not show these changes (Figure 2). Within 4 days of administration of 5% DSS hyperplasia of the lamina propria was visible in all PGRP-deficient mice. However, the extent of hyperplasia was different in different strains; it was extensive in Pglyrp2−/− and Pglyrp3−/− mice, moderate in Pglyrp1−/− mice, and mild in Pglyrp4−/− and WT mice. There was loss of epithelial cells in small sections in Pglyrp2−/− and Pglyrp3−/− mice. By day 7 there was extensive loss of epithelial layer, few remaining goblet cells, and ulceration in all PGRP-deficient mice, with most severe changes in Pglyrp2−/− and Pglyrp3−/− mice. Frank blood was also present in the lumen in distal colon in all PGRP-deficient mice. Similar, but milder, morphological changes were also seen in the cecum in DSS-treated PGRP-deficient mice, whereas few changes were seen in the small intestine (data not shown). The progressive complete loss of colonic epithelium in PGRP-deficient mice was the most likely cause of their mortality as it resulted in dehydration, because one of the important functions of colonic epithelium is re-absorption of water (see Supplemental Results).
Figure 2. DSS-treated PGRP-deficient mice have colon ulceration, hyperplasia of the lamina propria, and extensive loss of colonic epithelium.
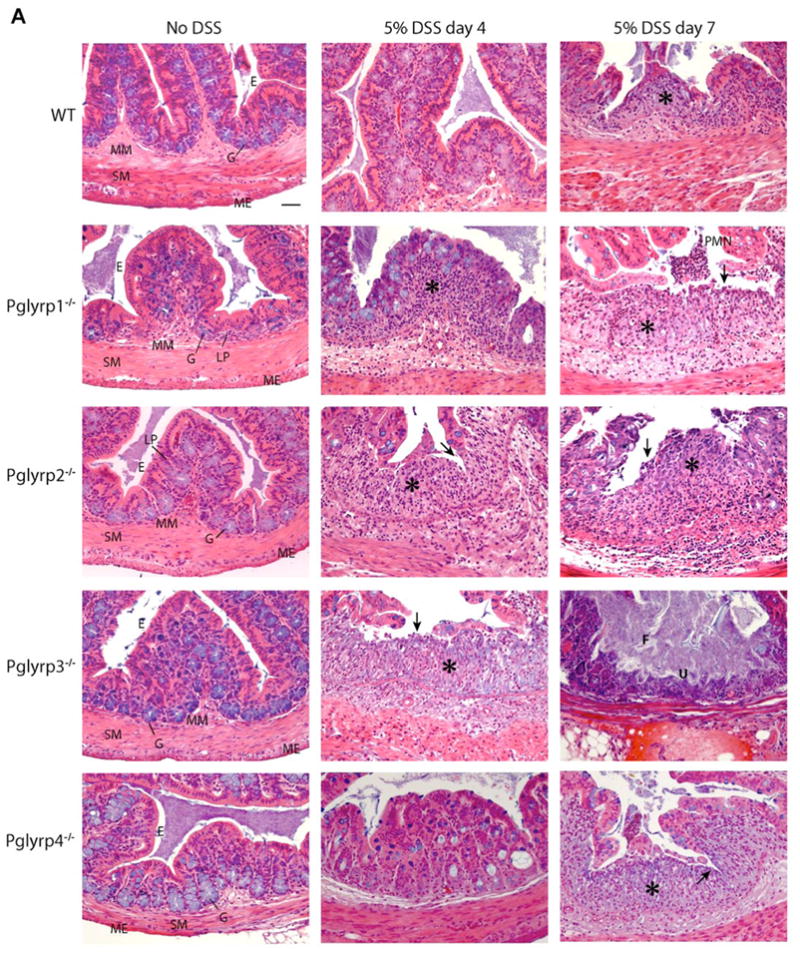

H&E stained cross-sections of the proximal (A) and distal (B) colon show normal histology in all untreated mice. Epithelial cells (E), lamina propria (LP), goblet cells (G), muscularis mucosa (MM), submucosa (SM), and muscularis externa (ME) are indicated. By day 4 of DSS treatment, PGRP-deficient mice showed hyperplasia of the lamina propria (*) and early stages in the loss of crypt architecture. By day 7 these changes were more pronounced and were accompanied by a decrease in the number of goblet cells, extensive loss of epithelial cells (→), ulceration (U), and infiltration with PMNs (PMN). Frank blood in the lumen (Bl) was present in distal colon in DSS-treated PGRP-deficient mice (B). Fecal matter in the lumen is indicated (F). Representative results from 6–12 mice/group are shown. All panels are at the same magnification. Bar = 50 μm.
In PGRP-deficient mice inflammatory cells (mainly PMNs and lymphocytes) were seen scattered throughout the intestinal wall and often migrating through the intestinal wall, and in later stages (day 7) pus was present in the lumen of the colon (Figure 2). However, overall the numbers of inflammatory cells were not very high in light of full exposure of subepithelial layers to the bacteria-laden contents of colonic lumen. Thus, the main pathologic finding in PGRP-deficient mice was the loss of normal colon morphology and architecture due to hyperplasia of the lamina propria and loss of colonic epithelium with moderate inflammation. These results demonstrate that all four PGRPs play an important role in protecting mice against DSS-induced tissue damage and colitis.
PGRPs protect mice from induction of IFN-γ and activation of IFN-inducible genes in the colon
To determine the mechanism responsible for the higher sensitivity of PGRP-deficient mice to DSS-induced colitis, we then looked for the genes whose expression was differentially induced in PGRP-deficient mice, compared to WT mice, following exposure to DSS. We first compared induction of expression of 84 pro-inflammatory genes in WT and Pglyrp3−/− mice 1, 2, 3, or 4 days after treatment with 4% or 5 % DSS, using qRT-PCR. We looked for the earliest and biggest difference in induction of gene expression between Pglyrp3−/− and WT mice to identify the initial events that make PGRP-deficient mice more susceptible to colitis. Treatment with both 4% and 5% DSS induced higher expression of pro-inflammatory genes in Pglyrp3−/− than WT mice. The biggest and the earliest difference was seen on day 2 of 5% DSS treatment: expression of 10 out of 84 pro-inflammatory genes was induced at least 2 times higher in Pglyrp3−/− mice than in WT mice. In particular, chemokines CXCL-9, CXCL-10, CXCL-11, and CCL-8 and IFN-γ were induced 5 to 16 times higher in Pglyrp3−/− than in WT mice (highlighted in Table S1).
To perform unbiased analysis of gene expression in WT and PGRP-deficient mice, we then used whole genome expression arrays to compare gene expression in WT mice and Pglyrp1−/−, Pglyrp2−/−, Pglyrp3−/−, and Pglyrp4−/− mice that were untreated or treated for 2 days with 5% DSS. Out of 28,853 genes analyzed 20,751 were expressed, and 1883 to 2728 genes were expressed significantly higher in PGRP-deficient than in WT mice following DSS treatment, whereas 2191 to 3751 genes were expressed significantly lower. The expression of 32 to 115 genes was increased more than 2 times higher in PGRP-deficient than in WT mice, whereas the expression of only 6 to 14 genes was decreased more than 2 times in PGRP-deficient than in WT mice (Table S2 and NCBI GEO accession number GSE18859). These results indicate overall higher level of gene activation following DSS treatment in PGRP-deficient than in WT mice.
We then further analyzed the array data to identify the common genes that were induced higher in PGRP-deficient mice than in WT mice by DSS treatment. We first compared the induction of gene expression in the colon after 2 days of treatment with 5% DSS in each knockout strain individually (Pglyrp1−/−, Pglyrp2−/−, Pglyrp3−/−, and Pglyrp4−/−) to the induction of gene expression in WT mice using the whole genome arrays. For each data set comparing Pglyrp−/− mice to WT mice we arranged all the genes on the array in descending order from the highest to the lowest ratio of gene induction in Pglyrp−/− mice versus WT mice (these data sets have been deposited in NCBI GEO, accession number GSE18859). In this study we focused on the common mechanism of increased sensitivity to colitis in all four PGRP-deficient mice. Therefore, we then merged the above four whole genome array data sets for Pglyrp1−/−, Pglyrp2−/−, Pglyrp3−/−, and Pglyrp4−/− mice (each compared to WT mice) to identify the top common genes differentially expressed in all PGRP-deficient versus WT mice.
The majority of genes that were induced higher in all four PGRP-deficient strains of mice than in WT mice after DSS treatment were IFN-inducible genes (90% of top 50 genes and 83% of top 75 genes). The expression of these genes was increased in the colon in PGRP-deficient mice by DSS, whereas it was reduced or unchanged in WT mice, as demonstrated by the whole genome array (Table S3). We confirmed this higher induction of top 75 IFN-inducible genes in all PGRP-deficient than in WT mice using qRT-PCR (Figure 3 and Table S3).
Figure 3. Higher expression of IFN-inducible genes in the colon in PGRP-deficient than in WT mice following DSS treatment.

A. Top 75 genes expressed higher in the colon of PGRP-deficient than WT mice after 2 days of oral treatment with 5% DSS were identified by whole genome expression array as mostly IFN-inducible genes. Their expression is shown here measured by qRT-PCR. The results are the ratio of the amount of mRNA in DSS-treated to untreated mice. The gene symbols are (IFN-inducible genes are in bold face): A1-12: Iigp1, Tgtp, Indo1, Ifit2, H28, Ifi44, Igtp, Upp1, Cxcl10, Gbp1, Cxcl9, Mpa2l; B1-12: Ifi47, Gbp2, Usp18, Slfn4, Gbp5, Gbp4, Zbp1, Trim30, Oas3, Irgm1, Apol9b, Gbp6; C1-12: Oas2, Ifit3, Tap1, Oasl2, Stat1, Herc5, Ifit1, Tnfsf10, Mx2, Ccl8, Ly6a, Psmb8; D1-12: Rsad2, Phf11, Cmpk2, Cxcl11, Ms4a1, Nlrc5, Ddx60, Ly6e, Nos2, Isg15, Mx1, Slfn2; E1-12: Psmb9, Stat2, Reg3b, Gzmb, Insig1, Klhl6, Rtp4, Ube2l6, Xdh, Cd274, Cd72, Rnf213; F1-12: Dhx58, Ly6c1, Fcgr4, Trim15, Art2a, Cd19, Batf2, Parp9, Pbef1, Parp14, Ifi27, Cxcr5; G1-3: Mlkl, Lgals9, Cd74. Gene descriptions and detailed results are shown in Table S3.
B and C. IFN-γ and IFN-inducible chemokines are preferentially induced by DSS treatment in the colon in PGRP-deficient but not in WT mice. B. Top 6 inflammatory genes expressed higher in the colon of PGRP-deficient than WT mice after 2 days of oral treatment with 5% DSS out of a panel of 84 cytokines, chemokines, and other marker genes for various immune cell types were identified by qRT-PCR as IFN-γ and IFN-inducible genes (CXCL-9, CXCL-10, CXCL-11, CCL-8, and iNOS-2), which are characteristic of an NK cell profile. The results are the ratio of the amount of mRNA in DSS-treated to untreated mice. Detailed results for all 84 genes are shown in Table S4. C. Kinetics of induction of IFN-γ and IFN-inducible chemokines in the colon in DSS-treated Pglyrp3−/− and WT mice; means of 3 assays on 5 mice/group ± SEM.
Because mammals have many IFN genes, we next determined which IFNs were induced by DSS treatment in the colon of PGRP-deficient mice. After 2 days of treatment with 5% DSS, WT mice did not increase the expression of any of the 16 IFN genes in the colon. By contrast, the same treatment in PGRP-deficient mice significantly increased the expression of IFN-γ (Ifng) in the colon, but not any of the other 15 IFN genes (Figure 4A). These results indicate that the genes most highly and preferentially induced in all PGRP-deficient mice (Figure 3 and Table S3) are genes that are known to be IFN-inducible (Borden et al., 2007; Sadler and Williams, 2008) are most likely induced by IFN-γ.
Figure 4. IFN-γ is preferentially induced by DSS treatment in the colon in PGRP-deficient but not in WT mice (A) and IFN- γ is required for full manifestation of colitis in PGRP-deficient mice (B).

A. Expression of 16 IFN genes was measured by qRT-PCR in the colons of PGRP-deficient and WT mice after 2 days of oral treatment with 5% DSS. The results are the ratio of the amount of mRNA in DSS-treated to untreated mice; means of 3 assays on 5 mice/group ± SEM; significance of differences between PGRP-deficient and WT mice is indicated by an asterisk, *, P<0.01. B. Higher survival of Pglyrp3−/− mice treated with neutralizing anti-IFN-γ mAbs than IgG-injected control mice following treatment with 5% DSS in drinking water for 11 days; 6 mice/group; *, P = 0.01.
IFN-γ is required for full manifestation of colitis in PGRP-deficient mice
To determine whether IFN-γ is required for the development of colitis in PGRP-deficient mice in vivo, we determined the sensitivity to DSS-induced colitis of Pglyrp3−/− mice in which IFN-γ activity was inhibited with neutralizing anti-IFN-γ mAb. These mice had significantly higher survival (Figure 4B, similar to the survival of WT mice, Figure S1) and significantly lower weight loss and stool scores (data not shown) than mice treated with isotype control IgG following exposure to DSS. These results demonstrate that IFN-γ is required for full manifestation of severe colitis in PGRP-deficient mice and show that induction of IFN-γ is not merely a consequence of mucosal damage.
PGRPs protect mice from DSS-induced increase in NK cells, but do not influence Th1, Th2, and Th17 responses in the colon
Two main cells that produce IFN-γ are Th1 cells and NK cells. To find the source of IFN-γ we first tested which cells were preferentially increased in the colons of DSS-treated PGRP-deficient mice, compared to WT mice. We measured the amounts of RNA for marker genes characteristic of various immune and inflammatory cell types in the colon. After 2 days of 5% DSS treatment, NK cells and PMNs were significantly increased in the colons of PGRP-deficient mice but not WT mice (Figure 5A). At that time B cells, T cells (total T cells and CD4+ and CD8+ T cells), monocytes and macrophages were not significantly increased in the colons of any DSS-treated strains. These results suggest that NK cells may be the source of INF-γ in PGRP-deficient mice and are consistent with higher induction of chemokines in PGRP-deficient mice and the overall moderate inflammatory response in the colon.
Figure 5. DSS treatment results in early increase in NK cells and PMNs in the colon in PGRP-deficient but not in WT mice (A) and NK cells are required for full manifestation of colitis in PGRP-deficient mice (B).

A. The amounts of mRNA for the indicated marker genes characteristic of B cells, T cells, NK cells, macrophages (MΦ) and PMNs were measured by qRT-PCR in the colons of PGRP-deficient and WT mice after 2 days of oral treatment with 5% DSS. The results are the ratio of the amount of mRNA in DSS-treated to untreated mice; means of 3 assays on 5 mice/group ± SEM; significance of differences between PGRP-deficient and WT mice is indicated by an asterisk, *, P<0.01. B. Higher survival of Pglyrp3−/− mice depleted of NK cells with anti-AsiloGM1 Ab than IgG-injected control mice following treatment with 5% DSS in drinking water for 11 days; 6 mice/group; *, P = 0.01.
To further investigate the source of IFN-γ we measured the expression of an extended panel of cytokines, chemokines, and other marker genes characteristic of Th1, Th2, Th17, and NK cells to determine which of these genes were differentially induced in the colons of PGRP-deficient mice, compared to WT mice, following exposure to DSS. We included Th1, Th2, and Th17 marker genes because Th1 cells are the major source of IFN-γ and enhanced Th1 and Th17 responses are often associated with IBD and with several experimental models of colitis (Watanabe et al., 2004, 2006; Yen et al., 2006).
In this extended Th1, Th2, Th17, and NK cell profile of 84 genes, the only genes preferentially induced by DSS in all PGRP-deficient mice, compared to WT mice, were Ifng and IFN-inducible genes, Cxcl9, Cxcl10, Cxcl11, Ccl8, and Nos2 (Figure 3B and Table S4). Kinetic analysis of the expression of IFN-γ and four IFN-inducible chemokines revealed preferential activation of all these genes in Pglyrp3−/− mice but not in WT mice on days 2, 3, and 4 of DSS treatment (Figure 3C). Our results suggest that IFN-γ originated from NK cells, because the induction of the above genes is a typical NK cell profile and because there was an increase in the number of NK cells in DSS-treated PGRP-deficient, but not WT mice (Figure 5A). It was unlikely that IFN-γ originated from Th1 cells, because expression of other Th1 marker genes (such as Il2, Tnf, Il18, Il18bp, Irf1) was not higher in PGRP-deficient than in WT mice (Table S4). Similar pattern of higher expression of Ifng and IFN-inducible genes, Cxcl9, Cxcl10, Cxcl11, and Ccl8 was also seen in colonic epithelial cells isolated from DSS-treated Pglyrp1−/−, Pglyrp2−/−, Pglyrp3−/−, and Pglyrp4−/− (but not WT) mice (data not shown), indicating that IFN-γ was produced at or near the epithelial cell layer.
We also tested whether higher expression of these IFN-inducible chemokines in PGRP-deficient than in WT mice was solely due to the higher production of IFN in these mice, or also due to higher responsiveness of cells from PGRP-deficient mice to IFN. We used cultured primary colonic fibroblasts, which are one of the target cells for IFN and which are known to be high producers of pro-inflammatory chemokines. Primary colonic fibroblasts from PGRP-deficient mice produced the same amounts of Cxcl9, Cxcl10, Cxcl11, and Ccl8 mRNA as fibroblasts from WT mice following stimulation with IFN-γ (Figure S2). These results confirm that the difference in the responses of PGRP-deficient and WT mice is due to the higher IFN-γ production and not due to increased responsiveness to IFN-γ.
The genes characteristic of Th17 (Il17a, Il17f, Il21, Il22, Il23a, Cxcl1, Cxcl2, Cxcl5, Tgfb1, Sele, Vcam1) or Th2 (Il3, Il4, Il5, Il9, Il10, Il13) responses were not preferentially induced in the colons of DSS-treated PGRP-deficient mice compared to WT mice (Table S4). Our results, therefore, indicate that there is preferential increase in NK cells and production of IFN-γ (likely by NK cells) and no preferential shift to Th1, Th17 or Th2 responses in the colon of DSS-treated PGRP-deficient mice compared to WT mice. Thus, PGRPs protect the colon from excessive damage by limiting IFN-γ production and infiltration with NK cells.
NK cells are required for full manifestation of colitis in PGRP-deficient mice
To determine the role of NK cells in the development of colitis in PGRP-deficient mice in vivo, we compared the sensitivity of NK-cell-depleted and not depleted Pglyrp3−/− mice to DSS-induced colitis. Pglyrp3−/− mice depleted of NK cells with anti-AsiloGM1 Abs had significantly higher survival (Figure 5B) and significantly lower stool scores (data not shown) than mice injected with control IgG following treatment with DSS. These results demonstrate that NK cells are required for full manifestation of severe colitis in PGRP-deficient mice and show that recruitment of NK cells is not merely a consequence of mucosal damage.
PGRPs are differentially expressed in various parts of oral cavity and gastrointestinal tract
To gain further insight how PGRPs cause greater sensitivity to colitis, we compared expression of all PGRPs in various sites in the oral cavity and gastrointestinal tract. All PGRPs were expressed in most sites in the oral cavity and gastrointestinal tract, but their level of expression varied. Pglyrp1 was highly expressed in most parts of the gastrointestinal tract and its expression was the highest in the oral cavity, ileum, and cecum (Figure S3). Pglyrp2 was highly expressed in the esophagus and throughout the small and large intestine. Pglyrp3 was highly expressed in the esophagus, tongue and throat, whereas Pglyrp4 was primarily expressed in the esophagus, salivary glands, tongue, throat, and lactating mammary glands, but the overall expression of Pglyrp4 in these tissues was much lower than of other PGRPs. Pglyrp3 and Pglyrp4 had very low expression in other parts of the gastrointestinal tract (Figure S3). These results confirm and extend previous results in humans (Liu et al., 2001; Lu et al., 2006) and mice (Mathur et al., 2004). One possibility how low expression of Pglyrp3 and Pglyrp4 in the colon could influence colitis could be through up-regulation of Pglyrp3 and Pglyrp4 expression during early stages of colitis. Indeed, expression of all PGRPs in the colon was significantly increased by the treatment with DSS, and expression of Pglyrp2, Pglyrp3, and Pglyrp4 in cultured primary colonic fibroblasts was also significantly increased by the treatment with bacteria or cytokines (Figure S4).
PGRPs change bacterial flora in the intestine
Since PGRPs have antibacterial or peptidoglycan-lytic activities, one possibility how expression of PGRPs in the intestinal tract could affect the sensitivity of the host to colitis could be through influencing the composition of the gastrointestinal bacterial flora. This mechanism could be especially applicable to Pglyrp3 and Pglyrp4, which are highly expressed in the oral cavity, salivary glands, upper gastrointestinal tract, and even lactating mammary glands. To test this hypothesis, we compared the composition of bacterial flora in the stools of WT and PGRP deficient mice, by measuring the amounts of DNA for all Eubacteria and for eight major groups of mouse intestinal bacteria using previously validated qPCR methods (Barman et al., 2008; Salzman et al., 2010). PGRP-deficient mice had significant changes in the composition of their bacterial flora in their stools. The amounts of bacteria from the following bacterial groups were significantly reduced in the following knockout mice compared to WT mice: Lactobacillus/Lactococcus group in Pglyrp2−/−, Pglyrp3−/− and Pglyrp4−/− mice, Segmented Filamentous bacteria group in Pglyrp1−/−, Pglyrp2−/− and Pglyrp4−/− mice, Clostridium perfringens group in Pglyrp2−/− and Pglyrp3−/− mice, and Enterobacteriaceae and Eubacterium rectale/Clostridium coccoides groups in Pglyrp3−/− mice (Figure 6A). Mouse Intestinal Bacteroides group was also significantly decreased in Pglyrp2−/− mice and increased in Pglyrp4−/− mice. These results indicate that all PGRPs are important for maintaining the balance of normal flora in the gut and that deficiencies in individual PGRPs can cause significant, but not identical changes in gut normal flora.
Figure 6. PGRP-deficient mice have changes in intestinal bacterial flora that induce higher cytokine and chemokine production.

A. Changes in the composition of bacterial flora in the stools of WT and PGRP-deficient mice in all Eubacteria and the following bacterial groups: Mouse Intestinal (MI) Bacteroides, Bacteroides sp., Eubacterium rectale/Clostridium coccoides, Clostridium leptum, Lactobacillus/Lactococcus, Segmented filamentous bacteria (SFB), Enterobacteriaceae, and Clostridium perfringens, measured by qPCR; means ± SEM of 12 mice per group; *, P ≤ 0.05 versus WT (t-test). B. Higher induction of IL-6 and CXCL-1 production in cultures of primary colonic WT mouse fibroblasts by stools from PGRP-deficient than from WT mice; means of 5 experiments ± SEM; *, P ≤ 0.05 versus WT (paired t-test).
Intestinal flora from PGRP-deficient mice increases sensitivity to colitis
We next considered whether the above changes in normal gut flora in PGRP-deficient mice have any functional consequences. First we tested whether it differentially induces cytokine production by intestinal cells. Indeed, stools from PGRP-deficient mice have significantly higher capacity to induce cytokine (IL-6) and chemokine (CXCL-1) production in cultured primary mouse colonic fibroblasts (Figure 6B) and peritoneal macrophages (Figure S5) than stools from WT mice. Of note, Pglyrp3−/− and Pglyrp2−/− mice, which were the most sensitive to colitis, had the most changes in their gut flora and their stools had the highest cell-activating capacity, whereas Pglyrp1−/− and Pglyrp4−/− mice, which were less sensitive to colitis, had fewer changes in the gut flora and their stools had lower cell-activating capacity.
Second, we tested whether the imbalance in normal flora caused by the lack of individual PGRPs and a shift to microflora with higher capacity to stimulate proinflammatory cytokines and chemokines in host intestinal cells predisposed mice to the higher sensitivity to colitis. We treated germ-free mice with daily gavages of stools from WT or PGRP-deficient mice and oral 4% DSS in drinking water. Germ-free mice receiving stools from Pglyrp2−/− mice or Pglyrp3−/− mice were more sensitive to DSS colitis than mice receiving stools from WT mice, as manifested by significantly higher mortality, significantly higher weight loss, and significantly higher colitis scores (Figures 7A–C and S6). Therefore, a shift in intestinal flora to more damaging pro-inflammatory microbiota is an important cause of increased sensitivity to colitis in PGRP-deficient mice. Thus, PGRPs promote normal intestinal flora that protects WT mice from severe colitis.
Figure 7. Intestinal flora from PGRP-deficient mice renders germ-free mice more susceptible to DSS-induced colitis than intestinal flora from WT mice (A–C).

(A) Survival, (B) body weight, and (C) stool and rectal bleeding scores of WT germ-free mice continuously treated for 18 days with 4% DSS in drinking water and gavaged daily with stool homogenates from conventionally raised WT, Pglyrp2−/−, or Pglyrp3−/− mice. N = 10 mice/group (from 2 experiments with 5 mice/group in each experiment); significance of differences between mice gavaged with stools from PGRP-deficient and WT mice is indicated by asterisks; *, P<0.05; * *, P<0.005.
(D) Sequence of events leading to more severe colitis in PGRP-deficient mice.
PGRPs do not induce signal transduction inhibitors in the colon
We also tested a possibility that the protective effect of PGRPs could be exerted through induction of inhibitors of inflammatory pathways. If this was the mechanism of PGRP action, deleting PGRPs would result in lower production of inhibitors, which would cause higher cytokine responses and increased inflammation. Since IFN can be induced through activation of Toll-like receptors, we tested whether several inhibitors known to regulate TLR signaling (Rothlin et al., 2007; Watanabe et al., 2008) are differentially induced in DSS-treated PGRP-deficient and WT mice. However, none of the 12 known inhibitors of inflammatory pathways that we have tested by qRT-PCR (Atf3, Bcl3, Il18bp, Inpp5d, Irak3, Irf1, Irf4, Irf7, Rnf216, Socs1, Socs3, Tollip) was expressed lower in PGRP-deficient than in WT mice (Table S5). The expression of these inhibitors was also not decreased in PGRP-deficient mice in the whole genome arrays (data not shown; see NCBI GEO accession number GSE18859). Expression of other relevant inhibitors, such as TAM receptors (Tyro3, Axl, Mertk) (Rothlin et al., 2007), Irf2 (IFN-regulatory factor-2), and pyrimidinergic and purinergic receptors (P2ry4, P2ry6, P2ry14) that modulate IFN responses (Hida et al., 2000; Shin et al., 2008) was also not changed after treatment with DSS in PGRP-deficient mice compared to WT mice, as determined by whole genome arrays (data not shown; see NCBI GEO accession number GSE18859). There were no known IFN inhibitors in the 100 most down regulated genes in PGRP-deficient mice compared to WT mice following DSS treatment determined by the whole genome arrays (data not shown; see NCBI GEO accession number GSE18859). Some inhibitors, e.g., Socs1 (Suppressor of cytokine signaling 1) and Zbp1 (Z-DNA binding protein 1, which is an IFN-regulatory factor) were increased in DSS-treated PGRP-deficient mice compared to WT mice (Tables S3 and S5), which indicates activation of inhibitory feedback mechanisms by higher induction of cytokines in PGRP-deficient compared to WT mice. These results indicate that PGRPs do not exert their anti-inflammatory effect through induction of higher expression of the above inhibitors.
DISCUSSION
There are multiple mechanisms of maintaining tolerance in the gut that protect the intestine from excessive inflammatory response to intestinal microorganisms and damage to the intestinal wall. We report here a previously unknown mechanism in which all four mammalian PGRPs protect the host from DSS-induced colitis. We show that Pglyrp1−/−, Pglyrp2−/−, Pglyrp3−/−, and Pglyrp4−/− mice are all more sensitive than WT mice to DSS-induced colitis. The key events controlled by PGRPs are maintaining less inflammatory normal intestinal flora, resistance to production of IFN-γ, and resistance to infiltration with NK cells in the colon upon initial exposure to DSS. This allows WT mice to avoid induction of early inflammatory response and damage in the colon. In contrast to WT mice, all PGRP-deficient mice have intestinal flora with higher pro-inflammatory activity and have high induction of IFN-γ in the colon followed by robust activation of IFN-inducible genes, including CXCL-9, CXCL-10, and CXCL-11, which attract more IFN-γ-producing NK cells. This cycle then leads to the early changes in colon morphology, loss of epithelial lining in the colon, inflammation, and increased mortality (Figure 7D).
Microbial flora in the gut has a great influence on the development of the immune system and changes in the intestinal flora often determine the sensitivity to inflammatory diseases and the outcomes of host-pathogen interactions (Sansonetti and Medzhitov, 2009). Our results demonstrate that PGRPs (which have bactericidal or bacteriolytic activities) significantly influence normal flora in the gut. Deficiencies in individual PGRPs result in significant changes in the normal bacterial flora in the colon to the extent that the stools from PGRP-deficient mice have higher immunostimulatory activity than the stools from WT mice, and when transferred to germ-free mice induce higher sensitivity to colitis than stools from WT mice. Such a change could be, for example, due to a decrease in the number of Lactobacilli, which are usually considered as beneficial probiotics, although detailed analysis which bacteria are responsible for this effect will require further study. Nevertheless, our results offer one possible explanation how a deficiency in a single PGRP increases sensitivity to colitis, and also how relatively high expression of PGRPs in the oral cavity, salivary glands, and the upper segments of the gastrointestinal tract influence the outcome of colitis through induction of changes in the intestinal flora.
Changes in intestinal flora have been also observed in other models of IBD. For example, NOD2 is one of the strongest genetic risk factors for Crohn’s disease, and NOD2-deficient mice have changes in intestinal flora that result in diminished ability to prevent intestinal colonization with pathogenic bacteria and to effectively kill these bacteria in the intestine (Petnicki-Ocwieja et al., 2009). Moreover, bacterial flora in the gut has also systemic influence on inflammatory diseases not only in the intestine, but also in other organs, such as joints and lungs (Maslowski et al., 2009). Thus, effects of PGRPs on the intestinal flora could have systemic consequences in other inflammatory diseases.
The involvement of NK cells in colitis is consistent with the current notion of NK cells being “conductors of inflammatory responses” (Di Santo, 2008). The proposed involvement of IFN-γ-producing NK cells rather than Th1 cells in DSS-induced colitis is also consistent with the notion that this form of colitis is due to direct damage of epithelial cells in the colon, that it more closely resembles human ulcerative colitis, and that it is based on innate (rather than T cell-mediated) response (Xavier and Podolsky, 2007; Zenewicz et al., 2008). Ulcerative colitis and its DSS model contrast Crohn’s disease, which involves granulomatous infiltrations of both small intestine and colon, which is primarily Th1-dependent, and for which IL-22-producing NK cells are protective (Watanabe et al., 2004; Rakoff-Nahoum et al., 2006; Zenewicz et al., 2008). Our results demonstrate protective role of PGRPs and pro-inflammatory role of IFN-γ and IFN-γ-producing NK cells in DSS-induced colitis, and further confirm pro-inflammatory role of IFN-γ and IFN-induced chemokines in colitis.
IFN production can lead to colitis, as evidenced by spontaneous development of intestinal inflammation similar to ulcerative colitis as a side effect in patients treated with IFN for viral infections (Wenner and Piccoli, 1997; Mavrogiannis et al., 2001; Sprenger et al., 2005). Moreover, NK cells from IBD patients produce increased amounts of IFN-γ (Liu et al., 2009). Our results are consistent with the above reports and with other findings of pivotal role of IFN-γ in DSS colitis through induction of apoptosis and inhibition of proliferation of colonic epithelial cells (Ito et al., 2006; Nava et al., 2010), as well as with previous reports demonstrating the role of IFN and IFN-induced NK cell-attracting chemokines, CXCL-9, CXCL-10, and CXCL-11, and their receptor CXCR3, in various models of colitis (Watanabe et al., 2004, 2006; Rakoff-Nahoum et al., 2006; Louis et al., 2008) and in patients with IBD (Parronchi et al., 1997; Singh et al., 2007; Strober et al., 2007; Budarf et al., 2009). Their role in the pathogenesis of IBD is also supported by the effectiveness of anti-CXCL-10 antibodies or blocking CXCR3 in the treatment of colitis (Singh et al., 2007).
Our results reveal a previously unknown in vivo role of PGRPs as anti-inflammatory proteins that prevent damaging production of IFN-γ in response to injury. PGRPs have an indirect effect on colitis through modulation of intestinal flora, as discussed above. PGRPs, however, could also directly modulate inflammation, as previously shown in an arthritis model (Saha et al., 2009). All PGRPs are expressed along the entire length of the gastrointestinal tract, with the highest expression in the upper segments (oral cavity, esophagus, and salivary glands), including intestinal epithelial cells, and their expression is increased following treatment with DSS or stimulation with bacteria or cytokines. However, PGRPs are usually not expressed in immune cells, such as lymphocytes, monocytes, and macrophages (Liu et al., 2000, 2001; Wang et al., 2005; Lu et al., 2006; Li et al., 2006). Thus, PGRPs are likely to exert their effects on local non-immune cells in the gut, such as epithelial cells and fibroblasts, which are rich sources of chemokines, including NK cell-attracting CXCL-9, CXCL-10, and CXCL-11 (Figure S2).
We recently observed an opposite effect of Pglyrp2 in a model of peptidoglycan-induced arthritis, i.e., Pglyrp2 has a pro-inflammatory effect and is required for the induction of arthritis by peptidoglycan in BALB/c mice (Saha et al., 2009). By contrast, other PGRPs have no pro-inflammatory effect in this arthritis model and Pglyrp1 is anti-inflammatory, similar to its effect in the colon observed in the current study. Such anti-inflammatory and pro-inflammatory effects in various models of inflammation are not unique to PGRPs, as some cytokines also have dual pro- and anti-inflammatory effects depending on the local milieu and the types of effector and target cells involved in the inflammatory process. For example, IL-22 targets non-immune cells, such as epithelial cells, endothelial cells, keratinocytes, and fibroblasts, and has a pro-inflammatory effect in the skin (Zheng et al., 2007; Ma et al., 2008) and anti-inflammatory effect in the gastrointestinal tract (Sugimoto et al., 2008; Zenewicz et al., 2008). Similarly, local cells that produce cytokines and are targets for cytokines, e.g., fibroblasts, are heterogenous in various organs and tissues and show different responsiveness to various stimulatory signals (Koumas et al., 2002; Smith, 2005).
Mammalian Pglyrp2 is an N-acetylmuramoyl-L-alanine amidase that hydrolyzes bacterial cell wall peptidoglycan (Gelius et al., 2003; Wang et al., 2003). However, its amidase activity is not required for its pro-inflammatory effect in the arthritis model (Saha et al., 2009). The amidase activity is also likely not required for the anti-inflammatory effect of PGRPs reported here, because all four PGRPs have this anti-inflammatory effect, and only one of these PGRPs (Pglyrp2) has amidase activity (Wang et al., 2003). These results are consistent with the emerging view that many proteins possess multiple functions in vivo.
IBD is a complex disease of unknown etiology that is determined both by genetic predisposition and environmental factors. Initial genetic analysis identified nine genetic loci that predispose patients to IBD (Lakatos et al., 2006). The first gene identified in one of these loci that predisposes patients to the Crohn’s disease form of IBD is a mutated NOD2 (Hugot et al., 2001; Ogura et al., 2001). More recent large-scale genome-wide association screenings identified 34 loci, in which more than 20 genes have proven significant association with IBD, including IL23R, ATG16L1, IL12B, and IL10 (Cho, 2008; Budarf et al., 2009). These findings opened up a possibility of still many more so far unidentified genes to be associated with IBD, especially that specific genes still have not been identified in all of the above IBD susceptibility loci. Of interest to our results is the susceptibility locus 19p13 (originally named IBD6 locus) (Rioux et al., 2000; Lakatos et al., 2006), for which the susceptibility gene still has not been identified (Budarf et al., 2009) and which harbors human PGLYRP2 at position 19p13.12. Thus, PGLYRP2 could be one of the human IBD susceptibility genes in this locus. Although other human PGRPs do not map to any of the above 34 IBD susceptibility loci, it is still possible that mutations in these PGRPs could also predispose patients to IBD, since many more genes are associated with IBD than initially suspected. Genotyping of IBD patients is underway to test these possibilities.
The observation that mice deficient in single PGRP genes are all more sensitive to colitis indicates that the functions of four mammalian PGRPs are not identical and non-redundant, and suggests that the mechanisms through which individual PGRPs protect mice from colitis are at least partially different. In this report we focused on the similarities among all four PGRPs and we designed our analysis of gene expression to identify common genes whose expression is similarly changed in colitis in all four PGRP-deficient strains. The rationale for this approach was to identify steps that are common in the pathogenesis of colitis in all four PGRP-deficient strains, because all PGRPs recognize peptidoglycan and are anti-bacterial. We reveal that the common steps in the pathogenesis of colitis in all PGRP-deficient mice are changes in the intestinal microflora, induction of NK cell-attracting chemokines, NK cell infiltration, IFN-γ production by NK cells, and activation IFN-inducible genes, followed by loss of colon morphology and epithelial layer (Figure 7D). However, the colitis phenotype in all four PGRP-deficient strains is not the same. The severity of colitis and kinetics of its development, as well as some details of pathology are not the same in all four strains. Similarly, gene expression profiles in whole genome expression arrays showed both similarities (on which we focused in this study) and differences (on which we will focus in future studies). This is not surprising, because all four PGRPs were preserved in evolution in all mammals, because each PGRP has a different expression pattern, and because deficiencies in each PGRP cause non-identical changes in gut flora. This possibility could thus explain non-redundancy of PGRPs in increased sensitivity to colitis, if changes in the gut flora in each PGRP-deficient strain are sufficiently similar to increase sensitivity to colitis, but not identical. This would result in some similarities and some differences between pathogenesis and manifestations of colitis in each PGRP-deficient strain. Colitis is a complex disease with several interacting pathogenic mechanisms determined by many genetic, microbial, and other environmental factors. Our results offer a possible link between the previously observed antibacterial activities of PGRPs and their immunomodulatory effects in vivo.
EXPERIMENTAL PROCEDURES
Mice
We generated Pglyrp1−/−, Pglyrp2−/−, Pglyrp3−/−, and Pglyrp4−/− mice by deleting exons 1, 1–4, 2–5, or 3–4, respectively, followed by backcrossing to BALB/c background, as described in Supplemental Data and Figures S7 and S8.
Colitis model, neutralization of IFN-γ, and depletion of NK cells
To induce experimental colitis female 8–10 week-old mice were given 3%, 4%, or 5% DSS (dextran sulfate sodium, MP Biomedicals) in drinking water (Elson et al., 1995). The development of colitis was evaluated as described in Supplemental Data using: (a) severity of colitis based on stool and rectal bleeding scores of 0–16; (b) weight loss; (c) mortality; and (d) histological analysis. The significance of differences in colitis scores and weight loss were determined using two-sample t-test and mortality using Chi-square test. IFN-γ was neutralized in Pglyrp3−/− mice by intravenous injections of anti-IFN-γ mAb (rat clone XMG1.2, endotoxin-free from Southern Biotech, 100 μg on day 0 and 50 μg on days 2, 4, 6, and 8 of 5% DSS treatment). Control mice were similarly treated with isotype control rat IgG1κ anti-KLH mAb (rat clone KLH/G1-2-2, endotoxin-free from Southern Biotech). NK cells were depleted from Pglyrp3−/− mice by intravenous injections of 40 μl anti-Asilo-GM1 Abs (Wako Chemicals) (Kasai et al., 1981) on days 0, 3, and 7 of 5% DSS treatment, which resulted in sustained >96% decrease in NK cells (CD49b+ with Dx5 mAb and CD335+ with NKp46 29A1.4 mAb) and <10% change in CD3+, CD4+, CD8+, and CD19+ cells, measured by flow cytometry using Miltenyi MACSQuant flow cytometer. Control group received three injections of normal rabbit IgG.
Whole genome arrays and quantitative real-time reverse transcription PCR (qRT-PCR)
Whole genome arrays (Affymetrix Mouse GeneChip® Gene 1.0 ST Array) were used to measure gene expression in untreated and DSS-treated (5%, 48 hrs) WT and Pglyrp1−/−, Pglyrp2−/−, Pglyrp3−/−, and Pglyrp4−/− mice (5 per group, 50 mice total). RNA from each mouse (isolated as described in Supplemental Data) was individually processed and hybridized. The cDNA synthesis and labeling, hybridization, scanning, and data extraction was done as described in Supplemental Data. We first compared induction of gene expression in the colon after 2 days of 5% DSS treatment in each knockout strain individually (Pglyrp1−/−, Pglyrp2−/−, Pglyrp3−/−, and Pglyrp4−/−) to the induction of gene expression by DSS in WT mice. We compared the fold induction of mRNA in the colon of Pglyrp−/− mice treated with DSS to untreated Pglyrp−/− mice, the fold induction of mRNA in the colon of WT mice treated with DSS to untreated WT mice, the significance of differences between the fold induction of mRNA in Pglyrp−/− versus WT mice, and the ratio of the fold induction of mRNA in the colon of Pglyrp−/− mice treated with DSS to WT mice treated with DSS for each gene, which were calculated as described in Supplemental Data. For each data set comparing Pglyrp−/− mice to WT mice we arranged all the genes on the array in descending order from the highest to the lowest ratio of gene induction in Pglyrp−/− mice versus WT mice. The entire array data sets including these calculations have been deposited in NCBI GEO (accession number GSE18859).
We then merged the above four whole genome array data sets for Pglyrp1−/−, Pglyrp2−/−, Pglyrp3−/−, and Pglyrp4−/− mice (each compared to WT mice) and identified the top common 75 genes differentially expressed in all PGRP-deficient versus WT mice. Predicted genes, gene models, unidentified genes, and ESTs were omitted from this top 75 genes list. The results with these top 75 genes are presented here in Figure 3A and Table S3.
Quantitative reverse transcription real-time PCR (qRT-PCR) was used to quantify the amounts of mRNA in colon, colonic epithelial cells, or colonic fibroblasts using Mouse Inflammatory Cytokines and Receptors RT2 Profiler PCR Array, or for other genes using custom RT2 Profiler PCR Arrays designed by us, or individual primer sets (all from SA Biosciences), as described previously (Saha et al., 2009) and in Supplemental Data. The lists of genes are provided in Tables S1 and S3–S5. The results were calculated as described in Supplemental Data and reported as mean fold increases after DSS treatment (DSS-treated/untreated) for each strain, or ratios of fold increases in PGRP-deficient to WT mice.
Cell isolation, culture, and cytokine production
Mouse primary colonic epithelial cells, colonic fibroblasts, and peritoneal macrophages were isolated as described in Supplemental Data. Colonic fibroblasts or macrophages were then seeded into 10-cm plates for RNA isolation or into 24-well plates for cytokine secretion. Details of cell culture, stimulation, and qRT-PCR and ELISA assays for IL-6 and CXCL-1 are given in Supplemental Data.
qPCR for stool flora analysis
The abundance of specific bacterial groups in mouse stools was measured by qPCR using group-specific 16s rDNA primers (Barman et al., 2008; Salzman et al., 2010) as described in Supplemental Data.
Germ-free mice experiments
Germ-free mice (Swiss Webster female, 4–5-week-old, from Taconic Farms), maintained under sterile conditions, were treated with 4% DSS in drinking water and gavaged daily into the stomach with 12 mg stools from WT, Pglyrp2−/−, or Pglyrp3−/− mice prepared as described in Supplemental Data. The development of colitis was evaluated as described above.
Supplementary Material
Acknowledgments
We are grateful to Patrick Bankston for help in interpreting histology slides, Robert Rukavina and Julie Cook for maintaining and breeding our mice, and Jeanette McClintick for help with whole genome micro-arrays, which were done at the Center for Medical Genomics (supported in part by grants from the Indiana 21st Century Research and Technology Fund, Indiana Genomics Initiative, and Lilly Endowment, Inc). This work was supported by USPHS Grants AI028797 and AI073290 from NIH.
Footnotes
The authors have no conflict of interest.
Supplemental Data includes Supplemental Results, Supplemental Experimental Procedures, Supplemental References, eight figures and five tables and can be found on line.
References
- Barman M, Unold D, Shifley K, Amir E, Hung K, Bos N, Salzman N. Enteric salmonellosis disrupts the microbial ecology of the murine gastrointestinal tract. Infect Immun. 2008;76:907–915. doi: 10.1128/IAI.01432-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borden EC, Sen GC, Uze G, Silverman RH, Ransohoff RM, Foster GR, Stark GR. Interferons at age 50: past, current and future impact on biomedicine. Nat Rev Drug Discov. 2007;6:975–990. doi: 10.1038/nrd2422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budarf ML, Labbé C, David G, Rioux JD. GWA studies: rewriting the story of IBD. Trends Genet. 2009;25:137–146. doi: 10.1016/j.tig.2009.01.001. [DOI] [PubMed] [Google Scholar]
- Cho JH. The genetics and immunopathogenesis of inflammatory bowel disease. Nat Rev Immunol. 2008;8:458–466. doi: 10.1038/nri2340. [DOI] [PubMed] [Google Scholar]
- Di Santo JP. Natural killer cells: diversity in search of a niche. Nat Immunol. 2008;9:473–475. doi: 10.1038/ni.f.201. [DOI] [PubMed] [Google Scholar]
- Dziarski R, Platt KA, Gelius E, Steiner H, Gupta D. Defect in neutrophil killing and increased susceptibility to infection with non-pathogenic Gram-positive bacteria in peptidoglycan recognition protein-S (PGRP-S)-deficient mice. Blood. 2003;102:689–697. doi: 10.1182/blood-2002-12-3853. [DOI] [PubMed] [Google Scholar]
- Elson CO, Sartor RB, Tennyson GS, Riddell RH. Experimental models of inflammatory bowel disease. Gastroenterology. 1995;109:1344–1367. doi: 10.1016/0016-5085(95)90599-5. [DOI] [PubMed] [Google Scholar]
- Gelius E, Persson C, Karlsson J, Steiner H. A mammalian peptidoglycan recognition protein with N-acetylmuramoyl-L-alanine amidase activity. Biochem Biophys Res Commun. 2003;306:988–994. doi: 10.1016/s0006-291x(03)01096-9. [DOI] [PubMed] [Google Scholar]
- Hanauer SB. Inflammatory bowel disease: epidemiology, pathogenesis, and therapeutic opportunities. Inflamm Bowel Dis Suppl. 2006;1:S3–9. doi: 10.1097/01.mib.0000195385.19268.68. [DOI] [PubMed] [Google Scholar]
- Hida S, Ogasawara K, Sato K, Abe M, Takayanagi H, Yokochi T, Sato T, Hirose S, Shirai T, Taki S, Taniguchi T. CD8(+) T cell-mediated skin disease in mice lacking IRF-2, the transcriptional attenuator of interferon-α/β signaling. Immunity. 2000;13:643–655. doi: 10.1016/s1074-7613(00)00064-9. [DOI] [PubMed] [Google Scholar]
- Hugot JP, Chamaillard M, Zouali H, Lesage S, Cezard JP, Belaiche J, Almer S, Tysk C, O’Morain CA, Gassull M, et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature. 2001;411:599–603. doi: 10.1038/35079107. [DOI] [PubMed] [Google Scholar]
- Ito R, Shin-Ya M, Kishida T, Urano A, Takada R, Sakagami J, Imanishi J, Kita M, Ueda Y, Iwakura Y, et al. Interferon-gamma is causatively involved in experimental inflammatory bowel disease in mice. Clin Exp Immunol. 2006;146:330–338. doi: 10.1111/j.1365-2249.2006.03214.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang D, Liu G, Lundstrom A, Gelius E, Steiner H. A peptidoglycan recognition protein in innate immunity conserved from insects to humans. Proc Natl Acad Sci USA. 1998;95:10078–10082. doi: 10.1073/pnas.95.17.10078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasai M, Yoneda T, Habu S, Maruyama Y, Okumura K, Tokunaga T. In vivo effect of anti-asialo GM1 antibody on natural killer activity. Nature. 1981;291:334–335. doi: 10.1038/291334a0. [DOI] [PubMed] [Google Scholar]
- Korzenik JR, Podolsky DK. Evolving knowledge and therapy of inflammatory bowel disease. Nat Rev Drug Discov. 2006;5:197–209. doi: 10.1038/nrd1986. [DOI] [PubMed] [Google Scholar]
- Koumas L, Smith TJ, Phipps RP. Fibroblast subsets in the human orbit: Thy-1+ and Thy-1− subpopulations exhibit distinct phenotypes. Eur J Immunol. 2002;32:477–485. doi: 10.1002/1521-4141(200202)32:2<477::AID-IMMU477>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- Lakatos PL, Fischer S, Lakatos L, Gal I, Papp J. Current concept on the pathogenesis of inflammatory bowel disease-crosstalk between genetic and microbial factors: pathogenic bacteria and altered bacterial sensing or changes in mucosal integrity take “toll”? World J Gastroenterol. 2006;12:1829–1841. doi: 10.3748/wjg.v12.i12.1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Wang S, Wang H, Gupta D. Differential expression of peptidoglycan recognition protein 2 in the skin and liver requires different transcription factors. J Biol Chem. 2006;281:20738–20748. doi: 10.1074/jbc.M601017200. [DOI] [PubMed] [Google Scholar]
- Liu C, Gelius E, Liu G, Steiner H, Dziarski R. Mammalian peptidoglycan recognition protein binds peptidoglycan with high affinity, is expressed in neutrophils, and inhibits bacterial growth. J Biol Chem. 2000;275:24490–24499. doi: 10.1074/jbc.M001239200. [DOI] [PubMed] [Google Scholar]
- Liu C, Xu Z, Gupta D, Dziarski R. Peptidoglycan recognition proteins: a novel family of four human innate immunity pattern recognition molecules. J Biol Chem. 2001;276:34686–34694. doi: 10.1074/jbc.M105566200. [DOI] [PubMed] [Google Scholar]
- Liu Z, Yang L, Cui Y, Wang X, Guo C, Huang Z, Kan Q, Liu Z, Liu Y. Il-21 enhances NK cell activation and cytolytic activity and induces Th17 cell differentiation in inflammatory bowel disease. Inflamm Bowel Dis. 2009;15:1133–1144. doi: 10.1002/ibd.20923. [DOI] [PubMed] [Google Scholar]
- Lo D, Tynan W, Dickerson J, Mendy J, Chang HW, Scharf M, Byrne D, Brayden D, Higgins L, Evans C, et al. Peptidoglycan recognition protein expression in mouse Peyer’s Patch follicle associated epithelium suggests functional specialization. Cell Immunol. 2003;224:8–16. doi: 10.1016/s0008-8749(03)00155-2. [DOI] [PubMed] [Google Scholar]
- Louis NA, Robinson AM, MacManus CF, Karhausen J, Scully M, Colgan SP. Control of IFN-αA by CD73: implications for mucosal inflammation. J Immunol. 2008;180:4246–4255. doi: 10.4049/jimmunol.180.6.4246. [DOI] [PubMed] [Google Scholar]
- Lu X, Wang M, Qi J, Wang H, Li X, Gupta D, Dziarski R. Peptidoglycan recognition proteins are a new class of human bactericidal proteins. J Biol Chem. 2006;281:5895–5907. doi: 10.1074/jbc.M511631200. [DOI] [PubMed] [Google Scholar]
- Ma HL, Liang S, Li J, Napierata L, Brown T, Benoit S, Senices M, Gill D, Dunussi-Joannopoulos K, Collins M, et al. IL-22 is required for Th17 cell-mediated pathology in a mouse model of psoriasis-like skin inflammation. J Clin Invest. 2008;118:597–607. doi: 10.1172/JCI33263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda S, Hsu LC, Liu H, Bankston LA, Iimura M, Kagnoff MF, Eckmann L, Karin M. Nod2 mutation in Crohn’s disease potentiates NF-κB activity and IL-1β processing. Science. 2005;307:734–738. doi: 10.1126/science.1103685. [DOI] [PubMed] [Google Scholar]
- Maslowski KM, Vieira AT, Ng A, Kranich J, Sierro F, Yu D, Schilter HC, Rolph MS, Mackay F, Artis D, et al. Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature. 2009;461:1282–1286. doi: 10.1038/nature08530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathur P, Murray B, Crowell T, Gardner H, Allaire N, Hsu YM, Thill G, Carulli JP. Murine peptidoglycan recognition proteins PglyrpIα and PglyrpIβ are encoded in the epidermal differentiation complex and are expressed in epidermal and hematopoietic tissues. Genomics. 2004;83:1151–1163. doi: 10.1016/j.ygeno.2004.01.003. [DOI] [PubMed] [Google Scholar]
- Mavrogiannis C, Papanikolaou IS, Elefsiniotis IS, Psilopoulos DI, Karameris A, Karvountzis G. Ulcerative colitis associated with interferon treatment for chronic hepatitis C. J Hepatol. 2001;34:964–965. doi: 10.1016/s0168-8278(01)00022-8. [DOI] [PubMed] [Google Scholar]
- Nava P, Koch S, Laukoetter MG, Lee WY, Kolegraff K, Capaldo CT, Beeman N, Addis C, Gerner-Smidt K, Neumaier I, et al. Interferon-γ regulates intestinal epithelial homeostasis through converging β-catenin signaling pathways. Immunity. 2010;32:392–402. doi: 10.1016/j.immuni.2010.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogura Y, Bonen DK, Inohara N, Nicolae DL, Chen FF, Ramos R, Britton H, Moran T, Karaliuskas R, Duerr RH, et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease. Nature. 2001;411:603–606. doi: 10.1038/35079114. [DOI] [PubMed] [Google Scholar]
- Parronchi P, Romagnani P, Annunziato F, Sampognaro S, Becchio A, Giannarini L, Maggi E, Pupilli C, Tonelli F, Romagnani S. Type 1 T-helper cell predominance and interleukin-12 expression in the gut of patients with Crohn’s disease. Am J Pathol. 1997;150:823–832. [PMC free article] [PubMed] [Google Scholar]
- Petnicki-Ocwieja T, Hrncir T, Liu YJ, Biswas A, Hudcovic T, Tlaskalova-Hogenova H, Kobayashi KS. Nod2 is required for the regulation of commensal microbiota in the intestine. Proc Natl Acad Sci USA. 2009;106:15813–15818. doi: 10.1073/pnas.0907722106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podolsky DK. Inflammatory bowel disease. N Engl J Med. 2002;347:417–429. doi: 10.1056/NEJMra020831. [DOI] [PubMed] [Google Scholar]
- Rakoff-Nahoum S, Hao L, Medzhitov R. Role of toll-like receptors in spontaneous commensal-dependent colitis. Immunity. 2006;25:319–329. doi: 10.1016/j.immuni.2006.06.010. [DOI] [PubMed] [Google Scholar]
- Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, Medzhitov R. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell. 2004;118:229–241. doi: 10.1016/j.cell.2004.07.002. [DOI] [PubMed] [Google Scholar]
- Rath HC, Schultz M, Freitag R, Dieleman LA, Li F, Linde HJ, Scholmerich J, Sartor RB. Different subsets of enteric bacteria induce and perpetuate experimental colitis in rats and mice. Infect Immun. 2001;69:2277–2285. doi: 10.1128/IAI.69.4.2277-2285.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rioux JD, Silverberg MS, Daly MJ, Steinhart AH, McLeod RS, Griffiths AM, Green T, Brettin TS, Stone V, Bull SB, et al. Genomewide search in Canadian families with inflammatory bowel disease reveals two novel susceptibility loci. Am J Hum Genet. 2000;66:1863–1870. doi: 10.1086/302913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothlin CV, Ghosh S, Zuniga EI, Oldstone MB, Lemke G. TAM receptors are pleiotropic inhibitors of the innate immune response. Cell. 2007;131:1124–1136. doi: 10.1016/j.cell.2007.10.034. [DOI] [PubMed] [Google Scholar]
- Sadler AJ, Williams BR. Interferon-inducible antiviral effectors. Nat Rev Immunol. 2008;8:559–568. doi: 10.1038/nri2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha S, Qi J, Wang S, Wang M, Li X, Kim YG, Núñez G, Gupta D, Dziarski R. PGLYRP-2 and Nod2 are both required for peptidoglycan-induced arthritis and local inflammation. Cell Host Microbe. 2009;5:137–150. doi: 10.1016/j.chom.2008.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salzman NH, Hung K, Haribhai D, Chu H, Karlsson-Sjöberg J, Amir E, Teggatz P, Barman M, Hayward M, Eastwood D, et al. Enteric defensins are essential regulators of intestinal microbial ecology. Nat Immunol. 2010;11:76–83. doi: 10.1038/ni.1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sansonetti PJ, Medzhitov R. Learning tolerance while fighting ignorance. Cell 7. 2009;138:416–420. doi: 10.1016/j.cell.2009.07.024. [DOI] [PubMed] [Google Scholar]
- Sartor RB. Targeting enteric bacteria in treatment of inflammatory bowel diseases: why, how, and when. Curr Opin Gastroenterol. 2003;19:358–365. doi: 10.1097/00001574-200307000-00006. [DOI] [PubMed] [Google Scholar]
- Shin A, Toy T, Rothenfusser S, Robson N, Vorac J, Dauer M, Stuplich M, Endres S, Cebon J, Maraskovsky E, et al. P2Y receptor signaling regulates phenotype and IFN-α secretion of human plasmacytoid dendritic cells. Blood. 2008;111:3062–3069. doi: 10.1182/blood-2007-02-071910. [DOI] [PubMed] [Google Scholar]
- Singh UP, Venkataraman C, Singh R, Lillard JW., Jr CXCR3 axis: role in inflammatory bowel disease and its therapeutic implication. Endocr Metab Immune Disord Drug Targets. 2007;7:111–123. doi: 10.2174/187153007780832109. [DOI] [PubMed] [Google Scholar]
- Smith TJ. Insights into the role of fibroblasts in human autoimmune diseases. Clin Exp Immunol. 2005;141:388–397. doi: 10.1111/j.1365-2249.2005.02824.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprenger R, Sagmeister M, Offner F. Acute ulcerative colitis during successful interferon/ribavirin treatment for chronic hepatitis. Gut. 2005;54:438–439. doi: 10.1136/gut.2004.049940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strober W, Fuss I, Mannon P. The fundamental basis of inflammatory bowel disease. J Clin Invest. 2007;117:514–521. doi: 10.1172/JCI30587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugimoto K, Ogawa A, Mizoguchi E, Shimomura Y, Andoh A, Bhan AK, Blumberg RS, Xavier RJ, Mizoguchi A. IL-22 ameliorates intestinal inflammation in a mouse model of ulcerative colitis. J Clin Invest. 2008;118:534–544. doi: 10.1172/JCI33194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tydell CC, Yuan J, Tran P, Selsted ME. Bovine peptidoglycan recognition proteinS: antimicrobial activity, localization, secretion, and binding properties. J Immunol. 2006;176:1154–1162. doi: 10.4049/jimmunol.176.2.1154. [DOI] [PubMed] [Google Scholar]
- Wang H, Gupta D, Li X, Dziarski R. Peptidoglycan recognition protein 2 (N-acetylmuramoyl-L-Ala amidase) is induced in keratinocytes by bacteria through the p38 kinase pathway. Infect Immun. 2005;73:7216–7225. doi: 10.1128/IAI.73.11.7216-7225.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Liu LH, Wang S, Li X, Lu X, Gupta D, Dziarski R. Human peptidoglycan recognition proteins require zinc to kill both Gram-positive and Gram-negative bacteria and are synergistic with antibacterial peptides. J Immunol. 2007;178:3116–3125. doi: 10.4049/jimmunol.178.5.3116. [DOI] [PubMed] [Google Scholar]
- Wang ZM, Li X, Cocklin RR, Wang M, Wang M, Fukase K, Inamura S, Kusumoto S, Gupta D, Dziarski R. Human peptidoglycan recognition protein-L is an N-acetylmuramoyl-L-alanine amidase. J Biol Chem. 2003;278:49044–49052. doi: 10.1074/jbc.M307758200. [DOI] [PubMed] [Google Scholar]
- Watanabe T, Asano N, Murray PJ, Ozato K, Tailor P, Fuss IJ, Kitani A, Strober W. Muramyl dipeptide activation of nucleotide-binding oligomerization domain 2 protects mice from experimental colitis. J Clin Invest. 2008;118:545–559. doi: 10.1172/JCI33145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe T, Kitani A, Murray PJ, Strober W. NOD2 is a negative regulator of Toll-like receptor 2-mediated T helper type 1 responses. Nat Immunol. 2004;5:800–808. doi: 10.1038/ni1092. [DOI] [PubMed] [Google Scholar]
- Watanabe T, Kitani A, Murray PJ, Wakatsuki Y, Fuss IJ, Strober W. Nucleotide binding oligomerization domain 2 deficiency leads to dysregulated TLR2 signaling and induction of antigen-specific colitis. Immunity. 2006;25:473–485. doi: 10.1016/j.immuni.2006.06.018. [DOI] [PubMed] [Google Scholar]
- Wenner WJ, Jr, Piccoli DA. Colitis associated with alpha interferon? J Clin Gastroenterol. 1997;25:398–399. doi: 10.1097/00004836-199707000-00027. [DOI] [PubMed] [Google Scholar]
- Xavier RJ, Podolsky DK. Unraveling the pathogenesis of inflammatory bowel disease. Nature. 2007;448:427–434. doi: 10.1038/nature06005. [DOI] [PubMed] [Google Scholar]
- Xu M, Wang Z, Locksley RM. Innate immune responses in peptidoglycan recognition protein L-deficient mice. Mol Cell Biol. 2004;24:7949–7957. doi: 10.1128/MCB.24.18.7949-7957.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen D, Cheung J, Scheerens H, Poulet F, McClanahan T, McKenzie B, Kleinschek MA, Owyang A, Mattson J, Blumenschein W, et al. IL-23 is essential for T cell-mediated colitis and promotes inflammation via IL-17 and IL-6. J Clin Invest. 2006;116:1310–1316. doi: 10.1172/JCI21404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zenewicz LA, Yancopoulos GD, Valenzuela DM, Murphy AJ, Stevens S, Flavell RA. Innate and adaptive interleukin-22 protects mice from inflammatory bowel disease. Immunity. 2008;29:947–957. doi: 10.1016/j.immuni.2008.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, van der Fits L, Voerman JS, Melief MJ, Laman JD, Wang M, Wang H, Wang M, Li X, Walls CD, Gupta D, Dziarski R. Identification of serum N-acetylmuramoyl-L-alanine amidase as liver peptidoglycan recognition protein 2. Biochim Biophys Acta. 2005;1752:34–46. doi: 10.1016/j.bbapap.2005.07.001. [DOI] [PubMed] [Google Scholar]
- Zheng Y, Danilenko DM, Valdez P, Kasman I, Eastham-Anderson J, Wu J, Ouyang W. Interleukin-22, a TH17 cytokine, mediates IL-23-induced dermal inflammation and acanthosis. Nature. 2007;445:648–651. doi: 10.1038/nature05505. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.